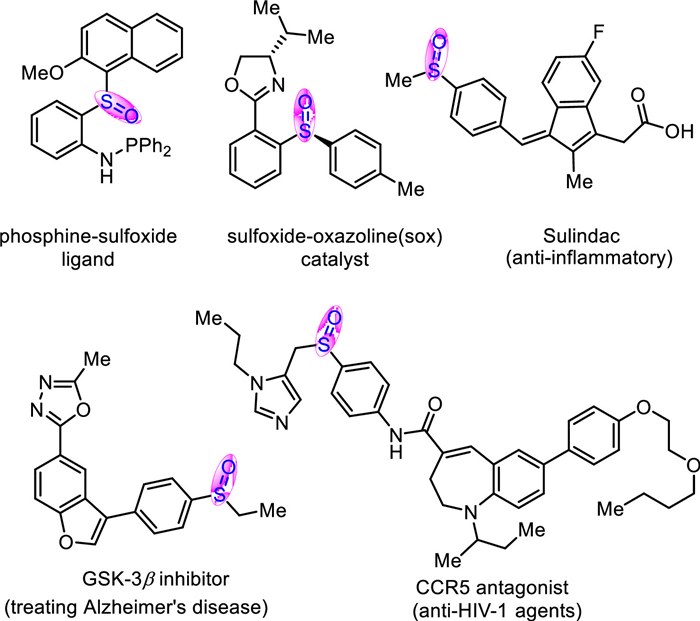
Figure 1.
Representative important sulfoxides. GSK = glycogen synthase kinase; HIV = human immunodeficiency virus.
Protecting-group-dependent chemo- and regioselective cascade rearrangement of N-arylhydroxylamines with N-thiophthalimides
Zhaoquan Guo , Wenyao Ding , Zhenguo Xi , Lin Yang , Gang Lu , Hongyin Gao

Sulfoxides are highly valuable scaffolds because of their intensive appearance and wide applications in a number of chiral ligands [1–11], bioactive pharmaceuticals [12–26] and functional materials [27] (Fig. 1). For instance, ligands contained a chiral sulfoxide unit were found to be efficient ligands in palladium-catalysed asymmetric allylic substitution reactions [1–3]. Sulindac was widely applied as an efficient anti-inflammatory drug in the treatment of pain, rheumatoid arthritis, osteoarthritis and acute gouty arthritis. In addition, sulfoxides are also extensively applied as versatile directing-groups or key intermediates in various organic transformations, such as the directed C-H functionalization, the Pummerer reaction and related rearrangement reactions [28–37].
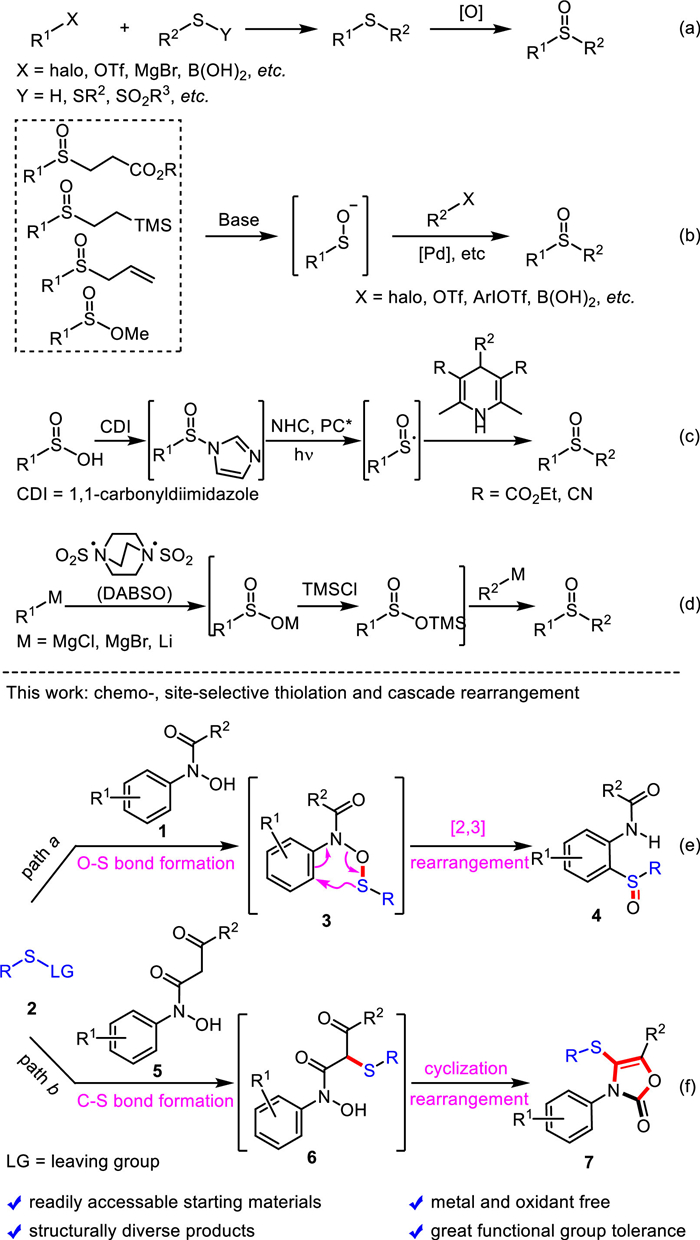
Due to the extensive applications of sulfoxides in many fields, it is not surprising that a large number of methods for the preparation of sulfoxide compounds have been developed in the past few decades [15,38–47]. For instance, the direct oxidation of sulfides, which were prepared through a variety of C-S bond formation reactions, is the most common strategy for the synthesis of sulfoxides (Scheme 1a). However, this strategy was suffering from the employment of stoichiometric amounts of oxidant, overoxidation to form sulfone products and the limited functional group compatibility [38,39,48–52]. Palladium-catalyzed cross-coupling of sulfenate anions with a variety of halides, triflates, iodonium salts or borons, was found to be an efficient approach for the construction of sulfoxide unit [53–67]. Drawbacks of these transformations are the requirement for expensive metal catalysts, ligands and often harsh conditions (Scheme 1b). Recently, a radical pathway from sulfinic acids and four-substituted Hantzsch esters in the presence of N-heterocyclic carbene and photocatalyst to sulfoxides was documented by Wu and Kuang (Scheme 1c) [68]. The sulfoxide moiety could also be prepared from organometallic reagents and DABSO in the presence of trimethylsilyl chloride (Scheme 1d) [69]. Despite these advances, more efficient, environmental-friendly and operationally simple protocols for the synthesis of structurally diverse sulfoxide compounds from readily accessible materials are still in high demand. Herein, we reported a practical and facile approach to ortho-aminoaryl sulfoxides from readily available N-arylhydroxylamines and N-thiophthalimides through a transition metal-free cascade O-thiolation and concerted N-O bond cleavage and [2,3]-sigmatropic rearrangement (Scheme 1e). Furthermore, a series of biologically useful multiple substituted oxazolone derivatives could be efficiently constructed once the protecting-group of N-arylhydroxylamines was switched from acetyl to 1,3-dicarbonyl group under very similar conditions (Scheme 1f).
At the outset, N–hydroxy-N-phenylbenzamide 1a and 2-(p-tolylthio)isoindoline-1,3–dione 2a were chosen as model substrates to start our investigation. Screening of the employment of bases such as KOAc, CsOAc, DBN, DABCO and tBuOK in DMF at −40 ℃ for 6 h, revealed that 1.0 equiv. of tBuOK was the superior option, furnishing the expected product 4a in 73% yield (Table 1, entries 1–5). Further investigation indicated that neither increasing nor decreasing the loading equivalent of tBuOK could improve the yield of 4a (Table 1, entries 6 and 7). Screening of other frequently used solvents indicated that both DMF and DME performed better than THF, CH2Cl2, toluene and ethyl acetate (Table 1, entries 5 and 8–12). The variation of different temperatures was also examined and we found that lower temperature was helpful to afford the corresponding product in higher yields albeit with relatively longer reaction time (Table 1, entries 13–16). To our delight, the desired product 4a was isolated in 82% yield in DME under −60 ℃ for 12 h in the presence of 1.2 equiv. of N-thiophthalimide 2a and 1.0 equiv. of tBuOK as base (Table 1, entry 16).
 DownLoad:
CSV
DownLoad:
CSV
 | |||||
| Entry | Solvent | Base | T (℃) | Time | Yield (%)b |
| 1 | DMF | KOAc | −40 | 6 h | 24 |
| 2 | DMF | CsOAc | −40 | 6 h | 34 |
| 3 | DMF | DBN | −40 | 6 h | 72 |
| 4 | DMF | DABCO | −40 | 6 h | trace |
| 5 | DMF | tBuOK | −40 | 6 h | 73 |
| 6c | DMF | tBuOK | −40 | 6 h | 57 |
| 7d | DMF | tBuOK | −40 | 6 h | 68 |
| 8 | THF | tBuOK | −40 | 6 h | 30 |
| 9 | CH2Cl2 | tBuOK | −40 | 6 h | 40 |
| 10 | Toluene | tBuOK | −40 | 6 h | 63 |
| 11 | EA | tBuOK | −40 | 6 h | 63 |
| 12 | DME | tBuOK | −40 | 6 h | 73 |
| 13 | DME | tBuOK | r.t. | 5 min | 21 |
| 14 | DME | tBuOK | 0 | 1 h | 34 |
| 15 | DME | tBuOK | −20 | 3 h | 64 |
| 16 | DME | tBuOK | −60 | 12 h | 82 |
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (0.24 mmol, 1.2 equiv.), base (0.2 mmol, 1.0 equiv.) in 2.0 mL solvent at a specific temperature for a certain period of time. b Isolated yield. c 0.1 mmol of base was used. d 0.4 mmol of base was used. Ac = acetyl. DBN = 1,5-diazabicyclo[4.3.0]non-5-ene. DABCO = 1,4-diazabicyclo[2.2.2]octane. DMF = N, N-dimethylformamide. DME = 1,2-dimethoxyethane. THF = tetrahydrofuran. EA = ethyl acetate. | |||||
Next, we employed the optimized reaction conditions to evaluate the generality of this cascade protocol (Scheme 2). The substrate scope of N-arylhydroxylamines 1 was first investigated by varifying the protecting group onto the nitrogen atom of the hydroxylamine, and a series of frequently-used acetyl-type protecting groups were well tolerated, giving the corresponding products 4a-4g in moderate to good yields, respectively. The compatibility of different substituents of N-arylhydroxylamines was subsequently examined. The ortho- or para-substituted N-arylhydroxylamines with varied electron-donating groups or electron-withdrawing groups were amenable to this cascade transformation, affording to the corresponding products 4h-4t in modest to high yields. Notably, this optimized conditions successfully promoted the [2,3]-sigmatropic rearrangement of para-substituted N-phenylhydroxylamines with an indole moiety (4u) and ester group carrying a furanyl ring or a long chain alkene (4v-4w), respectively. 2,3-Disubstituted N-phenylhydroxylamine was endurable to the reaction conditions, furnishing the corresponding sulfoxides 4x in 70% yield. The sterically congested products 4y could also be constructed albeit with relatively lower yield. It was not surprising that a mixture of two regioisomers 4z/4z′ were obtained when meta-methyl substituted N-phenylhydroxylamine was employed as substrate. Moreover, a variety of substituted N-naphthylhydroxylamines underwent the desired transformation to afford the corresponding sulfoxide products 4aa-4ac in moderate yields. In addition, N-hydroxybenzamide with pyridine ring structure could also react smoothly with N-thiophthalimide 2a to generate sulfoxide 4ad in 64% yield under the standard conditions.
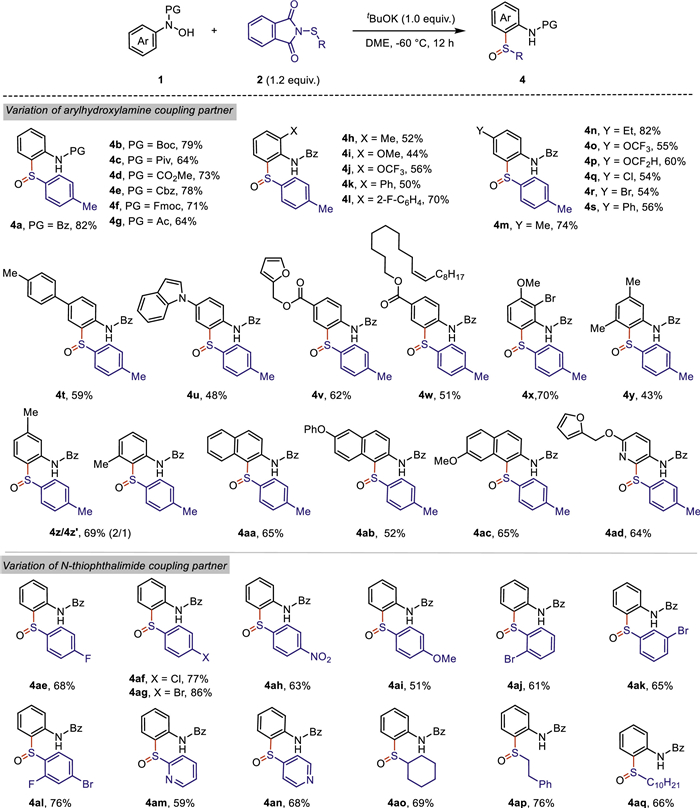
Subsequently, the scope of N-thiophthalimides 2 was evaluated, using N-phenylhydroxylamine 1a as reaction partner (Scheme 2). In general, this cascade protocol was suitable to a broad range of N-thiophthalimides bearing aryl groups with diverse electron-withdrawing and electron-donating substituents as well as sterically hindered groups, affording the corresponding sulfoxide products 4ae-4al in moderate to good yields. Notably, heteroaryl N-thiophthalimides, such as pyridinyl substituted N-thiophthalimides were also compatible with this system to furnish the desired products 4am and 4an in 59% and 68% yield, respectively. To our satisfactory, cycloalkyl and alkyl substituted N-thiophthalimides could also be efficiently introduced into this facile cascade transformation, providing the expected adducts 4ao-4aq in good yields under the optimal conditions. The X-ray analysis of compound 4k was carried out to confirm the structure of this cascade transformation.
To examine whether this cascade methodology could be expanded beyond racemic sulfoxide products, we designed the 1,3-dicarbonyl group protected N-arylhydroxylamine 5a, which might be more easily coordinated with chiral Lewis acid catalyst so that achieve the enantioselective transformation with N-thiophthalimide 2a. To our surprise, 59% yield of 3,4,5-trisubstituted oxazolone 7a, which was confirmed by X-ray analysis, was obtained rather than the expected sulfoxide product under very similar conditions followed by heating in MeCN at 80 ℃ for 10 h (Table 2, entry 1). Notably, oxazolones and their derivatives are also of great importance in organic chemistry since they were found in a wide range of bioactive molecules [70–74] and were extensively employed as alkene units in a number of transformations [75–78]. Despite great efforts have been devoted into the development of synthetic strategies for the preparation of oxazolone motif [79–84], the practical and efficient synthesis of 3,4,5-trisubstituted oxazolones under simple conditions is still challenging [85,86]. Therefore, we envisioned that the aforementioned preliminary result might open-up a new avenue for the effective preparation of highly functionalized oxazolones and we decided to reoptimize the conditions of this novel transformation. Screening of a variety of bases in DME, we found that tBuONa was superior to tBuOK, LiHMDS, KOAc, NaHCO3, Na2CO3 and K3PO4, affording to the desired product 7a in 76% yield (Table 2, entries 1–7). To further optimize the reaction, different solvents were examined in which CH2Cl2 gave the best performance, furnishing the corresponding product in 92% yield (Table 2, entries 8–13). Increasing the temperature of the first step led to lower yields of the final product (Table 2, entries 14–16). Notably, it was quite important to switch the solvent from CH2Cl2 to MeCN after warming to 25 ℃ for 1 h and then heat the system at 80 ℃ for another 10 h, otherwise resulting much longer reaction time and lower yield of the expected product (For more details, see Supporting information).
DownLoad:
CSV
 | |||
| Entry | Solvent | Base | Yield (%)b |
| 1 | DME | tBuOK | 59 |
| 2 | DME | tBuONa | 76 |
| 3 | DME | LiHMDS | 47 |
| 4 | DME | KOAc | 29 |
| 5 | DME | NaHCO3 | 22 |
| 6 | DME | Na2CO3 | 20 |
| 7 | DME | K3PO4 | 27 |
| 8 | THF | tBuONa | 72 |
| 9 | 2-Me-THF | tBuONa | 74 |
| 10 | Toluene | tBuONa | 77 |
| 11 | EA | tBuONa | 71 |
| 12 | Acetone | tBuONa | 66 |
| 13 | CH2Cl2 | tBuONa | 92 |
| 14c | CH2Cl2 | tBuONa | 61 |
| 15d | CH2Cl2 | tBuONa | 49 |
| 16e | CH2Cl2 | tBuONa | 39 |
| a Reaction conditions: step 1: 2a (0.2 mmol, 1.0 equiv.), 5a (0.24 mmol, 1.2 equiv.) and base (0.24 mmol, 1.2 equiv.) in a designated solvent (2 mL) at −60 ℃ for 10 h; step 2: stirring at 25 ℃ for 1 h; step 3: romove the original solvent and add 2 mL of MeCN and keep stirring at 80 ℃ for 10 h. b Yields of isolated products. c -40 ℃ instead of −60 ℃. d -20 ℃ instead of −60 ℃. e 0 ℃ instead of −60 ℃. | |||
Having the optimized conditions in hand, the scope and generality of this novel cascade reaction was evaluated using structurally diverse N-thiophthalimide 2 to react with N–hydroxy-3-oxo-N-arylbutanamide 5 bearing varied substituents of the aryl ring (Scheme 3). Both electron-donating groups, such as alkyl groups, and electron-withdrawing groups, such as halogens, could be efficiently introduced onto the aryl ring of N-arylhydroxylamine, leading to the corresponding oxazolone products 7a-7j in moderate to good yields. N-Naphthylhydroxylamine was tolerated and readily produced 60% yield of 3,4,5-trisubstituted oxazolone 7k under standard conditions. Gratefully, N-benzylhydroxylamine was also compatible with this system, albeit with relatively lower yield of the desired product 7l. Furthermore, N-arylhydroxylamines connected with natural product skeletons, such as cycloheptanol, 2-adamantanol, fenchol, menthol, diacetonefructose and geraniol, were also amenable to this cascade protocol, providing the corresponding products 7m-7r in moderate to good yields. Interestingly, the substituent R2 of the carbonyl group could also be diversified into ethyl, butyl, benzyl and aryl groups, generating the desired oxazolone products 7s-7w in 46%−71% yields under optimal conditions.
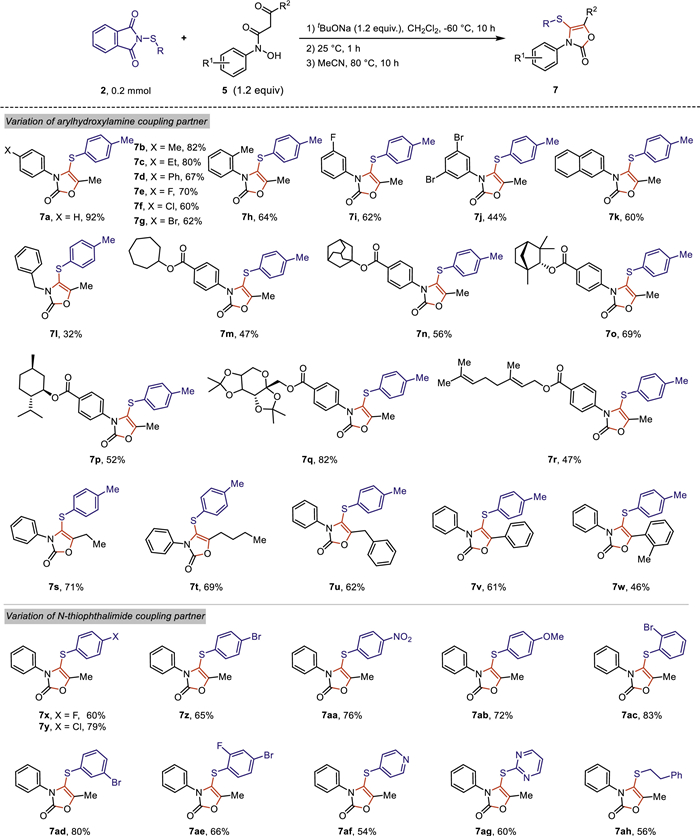
Next, the variation of N-Thiophthalimide coupling partner was investigated (Scheme 3). N-Thiophthalimides possessing -F, -Cl, -Br, -NO2 or -OMe at the para-position of the aryl ring were smoothly subjected to the optimized reaction conditions, affording aryl oxazolone 7x-7ab in moderate to good yields, respectively. The ortho-, meta- and 2,4-dihalogen substituted N-thiophthalimides also worked well, leading to the expected products 7ac-7ae in 66%−83% yields. It is worth noting that N-thiophthalimides having N-heteroaromatics, such as 2-pyridyl, 2-pyrimidinyl, could be employed in this system with moderate yields of the corresponding products, respectively (7af-7ag). In addition, alkyl substituent, such as phenylethyl, was also tolerated, affording to the desired product 7ah in 56% yield.
To gain some mechanistic insight of both cascade reactions, we set up two control experiments with TEMPO and BHT as inner additives under their standard conditions, which still afforded the corresponding products 4a and 7a in good yields, respectively (Schemes 4a and b). As a result, the radical pathways might be ruled out in both transformations. In addition, we conducted a competition experiment by employing N-thiophthalimide 2a to react with acetyl protected N-phenylhydroxylamine 1g and 1,3-dicarbonyl protected N-phenylhydroxylamine 5a, generating the expected sulfoxide product 4g in 26% yield and 3,4,5-trisubstituted oxazolone 7a in 54% yield without the formation of 4g′ (Scheme 4c). This result indicated that the reaction between 2a and 5a was relatively faster and easier than the reaction between 2a and 1g.

Based on the aforementioned experimental results, we proposed the mechanistic hypotheses for both cascade reactions. As shown in Scheme 5, acetyl protected N-phenylhydroxylamine 1g reacted smoothly with N-thiophthalimide 2a in the presence of tBuOK through nucleophilic substitution reaction (SN2) to generate the O-thiolated intermediate 3g, which underwent the subsequent [2,3]-sigmatropic rearrangement and re-aromatization to afford the final sulfoxide product 4g.
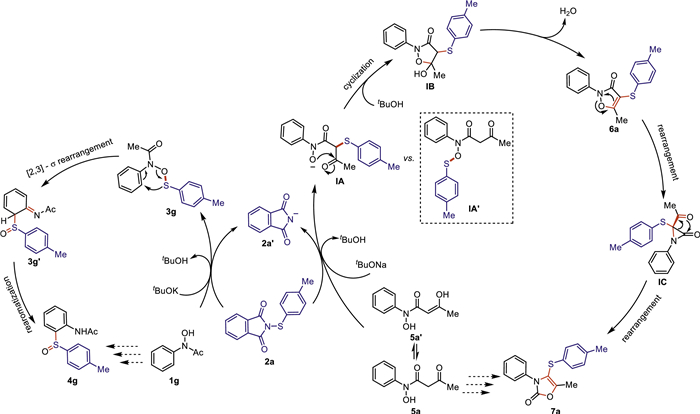
With regard to the reaction between 1,3-dicarbonyl protected N-phenylhydroxylamine 5a and N-thiophthalimide 2a in the presence of tBuONa, we assumed that the more reactive C-nucleophile, which was proved by the aforementioned competition experiment result (Scheme 4c), would attack N-thiophthalimide 2a to form C-thiolated intermediate IA rather than O-thiolated intermediate IA′, followed by the subsequent intramolecular cyclization and elimination of water to generate 6a. Notably, this type of compound could be isolated and has been unambiguously confirmed by X-ray diffraction analysis of compound 6ae (For details, see Supporting information). Subsequently, the intramolecular N-O bond cleavage/rearrangement would occur to afford aziridin-2-one species IC, followed by the cascade ring-opening/intramolecular nucleophilic O-attack of the acetyl group to carbonyl to furnish the final oxazolone product 7a (Scheme 5).
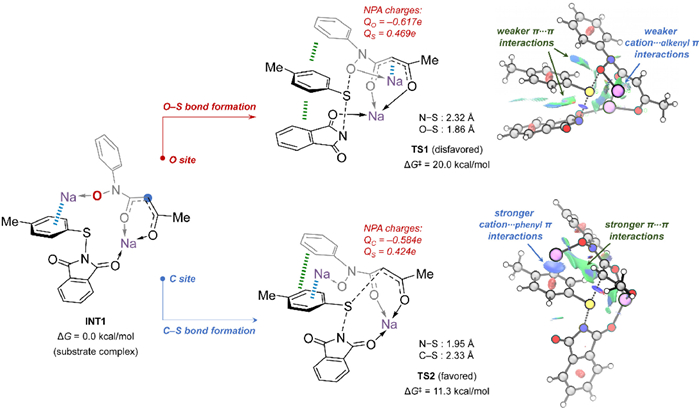
Under basic conditions, while acetyl protected hydroxylamines only have a nucleophilic O site, it is possible for 1,3-dicarbonyl protected hydroxylamines to serve as both O-nucleophile and C-nucleophile. Thus, the SN2 nucleophilic attack is critical for the observed distinguished reaction preference. DFT calculations were further performed to study these base-mediated nucleophilic substitutions of N-thiophthalimides with acetyl protected hydroxylamines and 1,3-dicarbonyl protected hydroxylamines. Since it is straightforward for the nucleophilic substitution between N-thiophthalimides and acetyl protected hydroxylamines (see details in Supporting information), we here focus on the competing O- and C-nucleophilic substitutions between N-thiophthalimides (e.g., 2a) and 1,3-dicarbonyl protected hydroxylamines (e.g., 5a). As shown in Fig. 2, these two reactants can form the complex INT1 via deprotonation in the presence of tBuONa. Subsequently, the nucleophilic substitutions proceed either at the O site (TS1, ΔG‡ = 20.0 kcal/mol) or the C site (TS2, ΔG‡ = 11.3 kcal/mol). The much lower barrier of TS2 than TS1 suggests that the 1,3-dicarbonyl protected group can introduce a more reactive C-nucleophilic site that surpasses the intrinsic O site of hydroxylamines. The NPA charges on the key O, C and S sites were first calculated to understand the origin of this chemoselectivity. However, the positive charges on the sulfur atoms (QS = 0.469e in TS1 vs. QS= 0.424e in TS2) and the negative charges on oxygen and carbon (QO = –0.617e in TS1 vs. QC = –0.584e in TS2) atoms go against the barrier difference between TS1 and TS2, which cannot explain the experimentally observed chemoselectivity. Alternatively, we evaluated the non-covalent interactions in these two transition states. The NCI plots [87,88] show that the Na···π (phenyl) interaction in TS2 (SN2 at the C site) is greater than the Na···π (alkenyl) interaction in TS1 (SN2 at the O site) (Fig. 2). In addition, TS2 also has stronger π···π interactions than those in TS1. The stronger non-covalent interactions in the favored TS2 result in the feature of early transition states, showcasing a shorter N···S distance (1.95 Å) and a longer C···S distance (2.33 Å). In contrast, the disfavored TS1 bears the feature of late transition states, having a longer N···S distance (2.32 Å) and a shorter O···S distance (1.86 Å). These results reveal that the non-covalent interactions introduced by the 1,3-dicarbonyl group play an important role in enabling the observed protecting-group-dependent chemoselectivity.
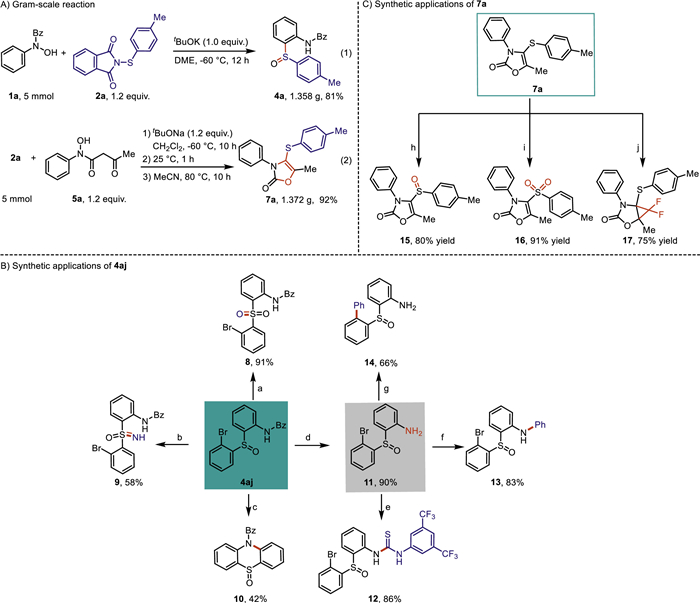
To demonstrate the reaction potential of these two types of cascade protocols, the large-scale reactions were conducted under standard conditions, both of which could obtain the corresponding adducts in good yields, respectively (Scheme 6A). To evaluate the synthetic applications of the outcome from both cascade reactions, diarylsulfoxide compound 4aj and aryl oxazolone 7a were selected as representatives and were converted into diverse functional molecules under various conditions (Schemes 6B and C). Diarylsulfoxide 4aj could be efficiently oxidated by meta-chloroperbenzoic acid (m-CPBA) to generate sulfone 8 in excellent yield (Scheme 6B, path a) [89]; the treatment of diarylsulfoxide 4aj with sodium azide in the presence of concentrated sulfuric acid afforded sulfoximine 9 in 58% yield (Scheme 6B, path b) [90]; copper-catalyzed intramolecular cyclization/C-N bond formation of diarylsulfoxide 4aj could be accomplished to furnish (5-oxido-10H-phenothiazin-10-yl)(phenyl)methanone 10 in 42% yield (Scheme 6B, path c) [91]; the protecting-group onto the nitrogen atom of diarylsulfoxide 4aj could be efficiently removed in the presence of hydrazine hydrate at 100 ℃ to obtain 2-((2-bromophenyl)sulfinyl)aniline 11 (Scheme 6B, path d) [92], which could be further transformed into 1-(3,5-bis(trifluoromethyl)phenyl)-3-(2-((2-bromophenyl)sulfin-yl)-phenyl)-thiourea 12 in excellent yield by the treatment with 1-isothiocyanato-3,5-bis(trifluoromethyl)benzene at room temperature (Scheme 6B, path e) [93], 2-((2-bromophenyl)sulfinyl)-N-phenylaniline 13 in 83% yield through the Cu(OAc)2-mediated Chan-Lam CN coupling with phenylboronic acid (Scheme 6B, path f) [94], and 2-([1,1′-biphenyl]−2-ylsulfinyl)aniline 14 via palladium-catalyzed Suzuki coupling with phenylboronic acid (Scheme 6B, path g) [95], respectively. In addition, 5-methyl-3-phenyl-4-(p-tolylthio)oxazol-2(3H)-one 7a could be chemoselectively oxidized into the corresponding sulfoxide 15 or sulfone 16 with good yields in the presence of different equivalents of meta-chloroperbenzoic acid, respectively (Scheme 6C, path h and path i) [96,97]; the C=C bond of 7a could undergo difluoromethylation with TMSCF3 and sodium iodide to gain 6,6-difluoro-1-methyl-4-phenyl-5-(p-tolylthio)-2-oxa-4-azabicyclo[3.1.0]hexan-3-one 17 in 75% yield (Scheme 6C, path j) [98].
In conclusion, we have developed two versatile and facile approaches to a wide range of structurally diverse ortho-aminoaryl sulfoxides and aryl oxazolones from N-arylhydroxylamines and N-thiophthalimides through O-S bond formation followed by [2,3]-sigmatropic rearrangement or C-S bond formation followed by series of cascade cyclization, isomerization/rearrangement process, respectively. The chemo- and regioselectivity of both transformations were controlled by the variation of the protecting groups onto the nitrogen atom of the N-arylhydroxylamines. DFT study suggested that the C-S bond formation was more favorable than the O-S bond formation when 1,3-dicarbonyl was employed as protecting-group onto the nitrogen atom of the N-arylhydroxylamine substrate. Further synthetic applications and extensions of this method to the preparation of a variety of bioactive and functional molecules are ongoing in this laboratory
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Zhaoquan Guo: Methodology, Investigation, Data curation. Wenyao Ding: Software, Data curation. Zhenguo Xi: Validation, Data curation. Lin Yang: Validation, Data curation. Gang Lu: Writing – review & editing, Supervision, Software, Investigation. Hongyin Gao: Writing – review & editing, Writing – original draft, Supervision, Investigation, Conceptualization.
Financial supports from the National Natural Science Foundation of China (Nos. 22271176, 22371171), the Taishan Scholar Project of Shandong Province (No. tsqn202306015) and Shandong University were gratefully appreciated. We thank Prof. Di Sun at Shandong University for the X-Ray diffraction and data analysis.
Supplementary material associated with this article can be found, in the online version, at doi:
J.V. Allen, J.F. Bower, J.M.J. Williams, Tetrahedron: Asymmetry 5 (1994) 1895–1898. doi: 10.1016/S0957-4166(00)86260-6
J.M.J. Williams, Synlett (1996) 705–710.
K. Hiroi, Y. Suzuki, Tetrahedron Lett. 39 (1998) 6499–6502.
M. Mellah, A. Voituriez, E. Schulz, Chem. Rev. 107 (2007) 5133–5209. doi: 10.1021/cr068440h
R. Mariz, X. Luan, M. Gatti, et al., J. Am. Chem. Soc. 130 (2008) 2172–2173. doi: 10.1021/ja710665q
M. Carmen Carreno, G. Hernandez-Torres, M. Ribagorda, et al., Chem. Commun. (2009) 6129–6144. doi: 10.1039/b908043k
C. Jiang, D.J. Covell, A.F. Stepan, et al., Org. Lett. 14 (2012) 1386–1389. doi: 10.1021/ol300063t
G. Sipos, E.E. Drinkel, R. Dorta, Chem. Soc. Rev. 44 (2015) 3834–3860. doi: 10.1039/C4CS00524D
B.M. Trost, M. Rao, Angew. Chem. Int. Ed. 54 (2015) 5026–5043. doi: 10.1002/anie.201411073
S. Otocka, M. Kwiatkowska, L. Madalinska, et al., Chem. Rev. 117 (2017) 4147–4181. doi: 10.1021/acs.chemrev.6b00517
T. Jia, M. Wang, J. Liao, Top. Curr. Chem. 377 (2019) 1–29.
P. Lindberg, A. Braendstroem, B. Wallmark, et al., Med. Res. Rev. 10 (1990) 1–54. doi: 10.1002/med.2610100102
K. Suwanborirux, K. Charupant, S. Amnuoypol, et al., J. Nat. Prod. 65 (2002) 935–937. doi: 10.1021/np010485k
R. Bentley, Chem. Soc. Rev. 34 (2005) 609–624. doi: 10.1039/b418284g
J. Legros, J.R. Dehli, C. Bolm, Adv. Synth. Catal. 347 (2005) 19–31. doi: 10.1002/adsc.200404206
M. Seto, K. Aikawa, N. Miyamoto, et al., J. Med. Chem. 49 (2006) 2037–2048. doi: 10.1021/jm0509703
K. McKeage, S.K.A. Blick, J.D. Croxtall, et al., Drugs 68 (2008) 1571–1607. doi: 10.2165/00003495-200868110-00009
I. Dini, G.C. Tenore, A. Dini, J. Nat. Prod. 71 (2008) 2036–2037. doi: 10.1021/np800237w
M. Saitoh, J. Kunitomo, E. Kimura, et al., J. Med. Chem. 52 (2009) 6270–6286. doi: 10.1021/jm900647e
M. El-Aasr, Y. Fujiwara, M. Takeya, et al., J. Nat. Prod. 73 (2010) 1306–1308. doi: 10.1021/np100105u
T.P. Wyche, J.S. Piotrowski, Y. Hou, et al., Angew. Chem. Int. Ed. 53 (2014) 11583–11586. doi: 10.1002/anie.201405990
T. Nohara, Y. Fujiwara, M. El-Aasr, et al., Chem. Pharm. Bull. 65 (2017) 209–217. doi: 10.1248/cpb.c16-00844
K.A. Scott, J.T. Njardarson, Top. Curr. Chem. 376 (2018) 1–34. doi: 10.1201/9781315378534-1
B. Schillheim, I. Jansen, S. Baum, et al., Plant Physiol. 176 (2018) 2395–2405. doi: 10.1104/pp.17.00124
Y. Li, S.A. -e. -A. Rizvi, D. Hu, et al., Angew. Chem. Int. Ed. 58 (2019) 13499–13506. doi: 10.1002/anie.201906080
C. Lutz, W. Simon, S. Werner-Simon, et al., Angew. Chem. Int. Ed. 59 (2020) 11390–11393. doi: 10.1002/anie.201914935
D. Isik, E. Quaas, D. Klinger, Polym. Chem. 11 (2020) 7662–7676. doi: 10.1039/d0py01321h
S.K. Bur, A. Padwa, Chem. Rev. 104 (2004) 2401–2432. doi: 10.1021/cr020090l
L.H.S. Smith, S.C. Coote, H.F. Sneddon, et al., Angew. Chem. Int. Ed. 49 (2010) 5832–5844. doi: 10.1002/anie.201000517
A.P. Pulis, D.J. Procter, Angew. Chem. Int. Ed. 55 (2016) 9842–9860. doi: 10.1002/anie.201601540
J.A. Fernandez-Salas, A.J. Eberhart, D.J. Procter, J. Am. Chem. Soc. 138 (2016) 790–793. doi: 10.1021/jacs.5b12579
D. Kaiser, I. Klose, R. Oost, et al., Chem. Rev. 119 (2019) 8701–8780. doi: 10.1021/acs.chemrev.9b00111
J. Yan, A.P. Pulis, G.J.P. Perry, et al., Angew. Chem. Int. Ed. 58 (2019) 15675–15679. doi: 10.1002/anie.201908319
L. Zhang, J.N. He, Y. Liang, et al., Angew. Chem. Int. Ed. 58 (2019) 5316–5320. doi: 10.1002/anie.201900434
D. Wang, C.G. Carlton, M. Tayu, et al., Angew. Chem. Int. Ed. 59 (2020) 15918–15922. doi: 10.1002/anie.202005531
M.H. Aukland, M. Siauciulis, A. West, et al., Nat. Catal. 3 (2020) 163–169. doi: 10.1038/s41929-019-0415-3
M. Chen, Y. Liang, T. Dong, et al., Angew. Chem. Int. Ed. 60 (2021) 2339–2345. doi: 10.1002/anie.202010740
C. Bolm, Coord. Chem. Rev. 237 (2003) 245–256. doi: 10.1016/S0010-8545(02)00249-7
E. Wojaczynska, J. Wojaczynski, Chem. Rev. 110 (2010) 4303–4356. doi: 10.1021/cr900147h
G.E. O'Mahony, A. Ford, A.R. Maguire, J. Sulfur Chem. 34 (2013) 301–341. doi: 10.1080/17415993.2012.725247
J. Zhu, W.C. Yang, X.D. Wang, et al., Adv. Synth. Catal. 360 (2018) 386–400. doi: 10.1002/adsc.201701194
H. Yu, Z. Li, C. Bolm, Org. Lett. 20 (2018) 7104–7106. doi: 10.1021/acs.orglett.8b03046
Z. Cheng, P. Sun, A. Tang, et al., Org. Lett. 21 (2019) 8925–8929. doi: 10.1021/acs.orglett.9b03192
V.D. Nguyen, G.C. Haug, S.G. Greco, et al., Angew. Chem. Int. Ed. 61 (2022) e202210525. doi: 10.1002/anie.202210525
F. Saito, Angew. Chem. Int. Ed. 61 (2022) e202213872. doi: 10.1002/anie.202213872
Y.J. Hou, Y. Li, Z.W. Zhao, et al., Org. Lett. 25 (2023) 517–521. doi: 10.1021/acs.orglett.2c04238
S. Xing, Y.Y. Zhu, W. Liu, et al., Org. Lett. 24 (2022) 3378–3383. doi: 10.1021/acs.orglett.2c01151
X. Gu, X. Li, Y. Chai, et al., Green Chem. 15 (2013) 357–361. doi: 10.1039/c2gc36683e
J.J. Boruah, S.P. Das, S.R. Ankireddy, et al., Green Chem. 15 (2013) 2944–2959. doi: 10.1039/c3gc40304a
H. Hussain, I.R. Green, I. Ahmed, Chem. Rev. 113 (2013) 3329–3371. doi: 10.1021/cr3004373
A. Yoshimura, K.C. Nguyen, S.C. Klasen, et al., Chem. Commun. 51 (2015) 7835–7838. doi: 10.1039/C5CC02009C
T. Nevesely, E. Svobodova, J. Chudoba, et al., Adv. Synth. Catal. 358 (2016) 1654–1663. doi: 10.1002/adsc.201501123
J. O'Donnell, A. Schwan, J. Sulfur Chem. 25 (2004) 183–211. doi: 10.1080/1741599042000220761
G. Maitro, S. Vogel, G. Prestat, et al., Org. Lett. 8 (2006) 5951–5954. doi: 10.1021/ol062315a
G. Maitro, G. Prestat, D. Madec, et al., J. Org. Chem. 71 (2006) 7449–7454. doi: 10.1021/jo061359u
G. Maitro, S. Vogel, M. Sadaoui, et al., Org. Lett. 9 (2007) 5493–5496. doi: 10.1021/ol702343g
E. Bernoud, G. Le Duc, X. Bantreil, et al., Org. Lett. 12 (2010) 320–323. doi: 10.1021/ol902620t
F. Gelat, A.C. Gaumont, S. Perrio, J. Sulfur Chem. 34 (2013) 596–605. doi: 10.1080/17415993.2013.795226
F. Izquierdo, A. Chartoire, S.P. Nolan, ACS Catal. 3 (2013) 2190–2193. doi: 10.1021/cs400533e
L. Zong, X. Ban, C.W. Kee, et al., Angew. Chem. Int. Ed. 53 (2014) 11849–11853. doi: 10.1002/anie.201407512
T. Jia, A. Bellomo, S. Montel, et al., Angew. Chem. Int. Ed. 53 (2014) 260–264. doi: 10.1002/anie.201307172
T. Jia, M. Zhang, H. Jiang, et al., J. Am. Chem. Soc. 137 (2015) 13887–13893. doi: 10.1021/jacs.5b08117
T. Jia, M. Zhang, I.K. Sagamanova, et al., Org. Lett. 17 (2015) 1168–1171. doi: 10.1021/acs.orglett.5b00092
H. Jiang, T. Jia, M. Zhang, et al., Org. Lett. 18 (2016) 972–975. doi: 10.1021/acs.orglett.6b00073
T. Jia, M. Zhang, S.P. McCollom, et al., J. Am. Chem. Soc. 139 (2017) 8337–8345. doi: 10.1021/jacs.7b03623
L. Wang, M. Chen, P. Zhang, et al., J. Am. Chem. Soc. 140 (2018) 3467–3473. doi: 10.1021/jacs.8b00178
M. Suzuki, K. Kanemoto, Y. Nakamura, et al., Org. Lett. 23 (2021) 3793–3797. doi: 10.1021/acs.orglett.1c01292
X. Wang, Y. Tang, S. Ye, et al., Org. Lett. 24 (2022) 2059–2063. doi: 10.1021/acs.orglett.2c00657
D.C. Lenstra, V. Vedovato, E. Ferrer Flegeau, et al., Org. Lett. 18 (2016) 2086–2089. doi: 10.1021/acs.orglett.6b00712
C. Puig, M.I. Crespo, N. Godessart, et al., J. Med. Chem. 43 (2000) 214–223. doi: 10.1021/jm991106b
N.H. Nam, Y. Kim, Y.J. You, et al., Bioorg. Med. Chem. Lett. 11 (2001) 3073–3076. doi: 10.1016/S0960-894X(01)00622-9
I. Nomura, C. Mukai, Org. Lett. 4 (2002) 4301–4304.
S.P. Fearnley, C. Thongsornkleeb, J. Org. Chem. 75 (2010) 933–936. doi: 10.1021/jo902172r
L. Salerno, V. Pittala, M.N. Modica, et al., Eur. J. Med. Chem. 85 (2014) 716–726. doi: 10.1016/j.ejmech.2014.08.023
I. Nomura, C. Mukai, J. Org. Chem. 69 (2004) 1803–1812. doi: 10.1021/jo0356816
S.V. D'Andrea, J.P. Freeman, J. Szmuszkovicz, J. Org. Chem. 55 (1990) 4356–4358. doi: 10.1021/jo00301a028
T. Choshi, H. Fujimoto, E. Sugino, et al., Heterocycles 43 (1996) 1847–1854. doi: 10.3987/COM-96-7532
S. Hoshimoto, H. Matsunaga, M. Wada, et al., Chem. Pharm. Bull. 50 (2002) 435–438. doi: 10.1248/cpb.50.435
G.R. Lenz, C. Costanza, J. Org. Chem. 53 (1988) 1176–1183. doi: 10.1021/jo00241a011
H. Aichaoui, J.H. Poupaert, D. Lesieur, et al., Tetrahedron 47 (1991) 6649–6654. doi: 10.1016/S0040-4020(01)82317-6
C.A. Marques, M. Selva, P. Tundo, et al., J. Org. Chem. 58 (1993) 5765–5770. doi: 10.1021/jo00073a041
M. Yamashita, S. -H. Lee, G. Koch, et al., Tetrahedron Lett. 46 (2005) 5495–5498.
F.M. Istrate, A.K. Buzas, I.D. Jurberg, et al., Org. Lett. 10 (2008) 925–928. doi: 10.1021/ol703077g
Z. Lu, W. Cui, S. Xia, et al., J. Org. Chem. 77 (2012) 9871–9877. doi: 10.1021/jo301794w
Z. Lu, F. Luo, L. Wang, et al., J. Org. Chem. 78 (2013) 10894–10901. doi: 10.1021/jo4018793
J. Hu, J. Ma, Z. Zhang, et al., Green Chem. 17 (2015) 1219–1225. doi: 10.1039/C4GC02033B
E.R. Johnson, S. Keinan, P. Mori-Sánchez, et al., J. Am. Chem. Soc. 132 (2010) 6498–6506. doi: 10.1021/ja100936w
J. Contreras-García, E.R. Johnson, S. Keinan, et al., J. Chem. Theory Comput. 7 (2011) 625–632. doi: 10.1021/ct100641a
B.K. Peters, T. Zhou, J. Rujirawanich, et al., J. Am. Chem. Soc. 136 (2014) 16557–16562. doi: 10.1021/ja5079877
C.R. Johnson, M. Haake, C.W. Schroeck, J. Am. Chem. Soc. 92 (1970) 6594–6598. doi: 10.1021/ja00725a035
X. Rui, Y. Zhu, R. Dai, et al., Asian J. Org. Chem. 10 (2021) 793–798. doi: 10.1002/ajoc.202100097
X. Yang, G. Shan, Y. Rao, Org. Lett. 15 (2013) 2334–2337. doi: 10.1021/ol400437a
K. Xu, W. Li, S. Zhu, et al., Angew. Chem. Int. Ed. 58 (2019) 17625–17630. doi: 10.1002/anie.201910049
C. Sambiagio, S.P. Marsden, A.J. Blacker, et al., Chem. Soc. Rev. 43 (2014) 3525–3550. doi: 10.1039/C3CS60289C
S. Bag, S. Jana, S. Pradhan, et al., Nat. Commun. 12 (2021) 1393. doi: 10.1038/s41467-021-21633-2
K.C. Nicolaou, P. Maligres, T. Suzuki, et al., J. Am. Chem. Soc. 114 (1992) 8890–8907. doi: 10.1021/ja00049a022
J. Wang, S. Zhang, C. Xu, et al., Angew. Chem. Int. Ed. 57 (2018) 6915–6920. doi: 10.1002/anie.201802540
F. Wang, T. Luo, J. Hu, et al., Angew. Chem. Int. Ed. 50 (2011) 7153–7157. doi: 10.1002/anie.201101691

Figure 1 Representative important sulfoxides. GSK = glycogen synthase kinase; HIV = human immunodeficiency virus.

Scheme 2 Scope of ortho-aminoaryl sulfoxides. All reactions were carried out with: 1 (0.2 mmol, 1.0 equiv.), 2 (0.24 mmol, 1.2 equiv.), tBuOK (0.2 mmol, 1.0 equiv.) in 2.0 mL DME at −60 ℃ for 12 h. Yields were determined by the isolated products.

Scheme 3 Scope of aryl oxazolones. Reaction conditions: 2 (0.2 mmol, 1.0 equiv.), 5 (0.24 mmol, 1.2 equiv.), tBuONa (0.24 mmol, 1.2 equiv.) in 2.0 mL CH2Cl2 at −60 ℃ for 10 h; then slowly warm up to 25 ℃ for 1 h, remove CH2Cl2 and then add 2 mL of MeCN and keep heating at 80 ℃ for 10 h. Yields were determined by the isolated products.

Figure 2 Competing SN2 nucleophilic substitutions of N-thiophthalimides with 1,3-dicarbonyl protected hydroxylamines.

Scheme 6 Gram-scale reactions and synthetic applications. Reaction conditions: (a) m-CPBA, DCM, 25 ℃, 4 h; (b) NaN3, CHCl3, H2SO4 (con.), 40 ℃, 12 h; (c) CuBr2, DMEDA, toluene, 120 ℃, 6 h; (d) hydrazine hydrate, sealed tube, 100 ℃, 3 h; (e) 1-isothiocyanato-3,5-bis(trifluoromethyl)benzene, DCM, 25 ℃, 12 h; (f) phenylboronic acid, Cu(OAc)2, TEA, DCM, 25 ℃, 20 h; (g) phenylboronic acid, Pd(PPh3)2Cl2, K2CO3, DMF/H2O (4:1), N2, 90 ℃, 5 h; (h) m-CPBA (1.5 equiv.), DCM, 25 ℃, 4 h; (i) m-CPBA (4.0 equiv.), DCM, 25 ℃, 8 h; (j) TMSCF3, NaI, THF, N2, 65 ℃, sealed tube.
Table 1. Optimization study.a
| |||||
| Entry | Solvent | Base | T (℃) | Time | Yield (%)b |
| 1 | DMF | KOAc | −40 | 6 h | 24 |
| 2 | DMF | CsOAc | −40 | 6 h | 34 |
| 3 | DMF | DBN | −40 | 6 h | 72 |
| 4 | DMF | DABCO | −40 | 6 h | trace |
| 5 | DMF | tBuOK | −40 | 6 h | 73 |
| 6c | DMF | tBuOK | −40 | 6 h | 57 |
| 7d | DMF | tBuOK | −40 | 6 h | 68 |
| 8 | THF | tBuOK | −40 | 6 h | 30 |
| 9 | CH2Cl2 | tBuOK | −40 | 6 h | 40 |
| 10 | Toluene | tBuOK | −40 | 6 h | 63 |
| 11 | EA | tBuOK | −40 | 6 h | 63 |
| 12 | DME | tBuOK | −40 | 6 h | 73 |
| 13 | DME | tBuOK | r.t. | 5 min | 21 |
| 14 | DME | tBuOK | 0 | 1 h | 34 |
| 15 | DME | tBuOK | −20 | 3 h | 64 |
| 16 | DME | tBuOK | −60 | 12 h | 82 |
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (0.24 mmol, 1.2 equiv.), base (0.2 mmol, 1.0 equiv.) in 2.0 mL solvent at a specific temperature for a certain period of time. b Isolated yield. c 0.1 mmol of base was used. d 0.4 mmol of base was used. Ac = acetyl. DBN = 1,5-diazabicyclo[4.3.0]non-5-ene. DABCO = 1,4-diazabicyclo[2.2.2]octane. DMF = N, N-dimethylformamide. DME = 1,2-dimethoxyethane. THF = tetrahydrofuran. EA = ethyl acetate. | |||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Condition optimization.a
| |||
| Entry | Solvent | Base | Yield (%)b |
| 1 | DME | tBuOK | 59 |
| 2 | DME | tBuONa | 76 |
| 3 | DME | LiHMDS | 47 |
| 4 | DME | KOAc | 29 |
| 5 | DME | NaHCO3 | 22 |
| 6 | DME | Na2CO3 | 20 |
| 7 | DME | K3PO4 | 27 |
| 8 | THF | tBuONa | 72 |
| 9 | 2-Me-THF | tBuONa | 74 |
| 10 | Toluene | tBuONa | 77 |
| 11 | EA | tBuONa | 71 |
| 12 | Acetone | tBuONa | 66 |
| 13 | CH2Cl2 | tBuONa | 92 |
| 14c | CH2Cl2 | tBuONa | 61 |
| 15d | CH2Cl2 | tBuONa | 49 |
| 16e | CH2Cl2 | tBuONa | 39 |
| a Reaction conditions: step 1: 2a (0.2 mmol, 1.0 equiv.), 5a (0.24 mmol, 1.2 equiv.) and base (0.24 mmol, 1.2 equiv.) in a designated solvent (2 mL) at −60 ℃ for 10 h; step 2: stirring at 25 ℃ for 1 h; step 3: romove the original solvent and add 2 mL of MeCN and keep stirring at 80 ℃ for 10 h. b Yields of isolated products. c -40 ℃ instead of −60 ℃. d -20 ℃ instead of −60 ℃. e 0 ℃ instead of −60 ℃. | |||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: