Citation:
Hao Hu, Zhengxin Ye, Lei Zhang, Kejun Dong, Bei Yan, Longjie Li, Wei Zhang, Xianjin Xiao. A PAM-free and universal CRISPR-Cas12a activation model for ultra-sensitive DNA methylation detection[J]. Chinese Chemical Letters,
2026, 37(1): 111184.
doi:
10.1016/j.cclet.2025.111184
A PAM-free and universal CRISPR-Cas12a activation model for ultra-sensitive DNA methylation detection
English
A PAM-free and universal CRISPR-Cas12a activation model for ultra-sensitive DNA methylation detection
Received Date:
28 October 2024 Accepted Date:
07 April 2025 Revised Date:
06 February 2025 Available Online:
15 January 2026
Abstract:
DNA methylation is an important promising biomarker for cancer diagnosis and monitoring. Therefore, the assessment of DNA methylation levels is helpful for the prognosis and diagnosis of cancer. However, it is still a huge challenge to sensitively and accurately quantify the levels of DNA methylation in clinical sample. In this work, we proposed a protospacer adjacent motif (PAM)-free mediated CRISPR-Cas12a ultra-sensitive and quantitative DNA methylation detection method. Through recognizing the dsDNA with toehold region, CRISPR-Cas12a not only got rid of the limitation of PAM, but also improved its distinction ability for single CpG site methylation, nearly 5-fold that of conventional PAM-containing dsDNA. We further introduced assist-strand and design an artificial mismatch to greatly improve the ability to distinguish single CpG methylation site. Our results showed that the discrimination factor was > 200. Then, we constructed toe-dsDNA by using "heating and freezing", which made our method universally applicable and feasible. In addition, we greatly simplified the difficulty of primer design. Our method detected four highly methylated genes acyl carrier protein (ACP), CLV3/ESR-related (CLE), Disabled (DAB) and Homeobox (HOX) with a detection limit of 0.01% and excellent linearity in DNA methylation standards. Then, we verified the clinical utility of this method in 29 hepatocellular carcinomas, 11 ovarian cancers and 4 health people. In conclusion, we have successfully constructed a PAM-free CRISPR-Cas12a DNA methylation quantification method, which achieves high congruence in sensitivity, specificity and universality, fully demonstrating its significant clinical application value.
DNA methylation is one of the earliest discovered and most thoroughly studied mechanisms of epigenetic regulation [1,2]. DNA methylation refers to the addition of a methyl group (-CH3) to a DNA molecule under the action of DNA methyltransferase, usually at the cytosine residue of a CpG dinucleotide [2]. The biological effects of DNA methylation depend not only on its presence in the genome but also on its location [3]. Abnormal DNA methylation in promoter regions is associated with various pathologies, such as cancer [4], diabetes [5] and neurological disorders [6]. High methylation of tumor suppressor gene promoters or low methylation of oncogenes can lead to different types of cancer [4]. Studies have confirmed that the promoter regions of NEBL, ACP-1 and Septin-9 genes are abnormally methylated in tumor tissue of hepatocellular carcinoma (HCC) [7-9]. Moreover, non-CpG methylation may play a role in maintaining the pluripotency of stem cells or in regulating neuronal gene expression. Gene body methylation is generally associated with active transcription and play a role in regulating alternative splicing or suppressing intragenic promoters. It is worth mentioning that methylation also occurs in intergenic regions and within repetitive DNA elements, such as transposons. This type of methylation suppresses the activity of transposable elements, which can otherwise disrupt genomic integrity by inserting themselves into genes. Therefore, accurately locating the methylation of specific regions of different genes and determining their level of methylation is of great significance for early diagnosis and treatment of tumors.
In recent years, biological detection technology has been greatly developed, and more and more advanced technologies are being used to detect various biomarkers [10-15]. So far, scientists have proposed a variety of methods for detecting DNA methylation. These techniques can be roughly divided into as follows: bisulfite conversion based-methods [16], methylation-sensitive restriction endonucleases based-methods (MSRE) [17], bisulfite-free direct detection method [18], and new biosensor methods [19]. Among them, the method based on sodium bisulfite conversion is considered the gold standard for DNA methylation detection. Its principle is to use bisulfite to convert unmethylated cytosine (C) in the DNA sequence into uracil (U), while methylated cytosine remains unchanged [16]. Bisulfite sequencing polymerase chain reaction (PCR) (BSP) is a simple and rapid method that can provide verification results [20]. However, due to the presence of allele mixing and heterogeneous methylation patterns in the DNA sequence, the sequencing results can be very noisy. More importantly, methylated C only make up 0.1% of the total DNA bases [21], therefore the sensitivity of BSP cannot meet the clinical needs for early cancer screening. PCR is the most common and stable method of DNA amplification and can effectively increase the concentration of the target nucleic acid [22]. Methylation-specific PCR (MSP) and Methylight can quantify the methylated or unmethylated state of the sequence [23]. However, their primers are complicated, usually involving several methylation sites, which leads to complex and cumbersome detection system design. In addition, their sensitivities are not high enough [24]. MSRE uses enzyme to digest unmethylated DNA without requiring complicated chemical treatments or expensive sequencing equipment [25]. However, only a few MSREs are currently available, which cannot be used for large-scale screening of DNA methylation sites. In addition, incomplete digestion of unmethylated DNA by MSRE may lead to false positives [26]. Bisulfite-free direct detection methods, such as ten-eleven translocation (TET)-assisted pyridine borane sequencing (TAPS), face challenges related to limited conversion efficiency and insufficient detection sensitivity [18]. These issues constrain their broader application and adoption. In recent years, a variety of novel biosensing technologies have been developed for DNA methylation detection, such as electrochemistry [27], colorimetry [28] and Raman spectroscopy [19]. However, these methods are expensive, operationally complex and require professional training, which is not conducive to large-scale clinical promotion [29]. Therefore, there is an urgent need to develop an easy-to-use, fast, highly sensitive and specific quantitative method for DNA methylation detection to provide an effective clinical means for early cancer diagnosis and prognosis evaluation.
In recent years, CRISPR-Cas12a has developed into a powerful tool for nucleic acid diagnosis including virus, gene mutation, enzyme and miRNA [30-32]. However, researches on DNA methylation detection method based on CRISPR-Cas12a are still relatively rare. The main reason lies in the two major limitations faced by CRISPR-Cas12a: (1) The system requires a protospacer adjacent motif (PAM) sequence, therefore it is not universal; (2) Its specificity for low abundance samples is poor, with insufficient sensitivity [33,34]. Some studies have attempted to combine MSRE with CRISPR-Cas12a to detect DNA methylation, but this requires the presence of a PAM sequence in the methylation region [35,36]. In addition, incomplete digestion by MSRE could also lead to false-positive results. To avoid the limitation of PAM sequences, some studies have tried to single-strand the dsDNA, such as using rolling circle amplification (RCA).
However, CRISPR-Cas12a's specificity for single-stranded DNA is very poor and cannot effectively distinguish between methylated and unmethylated DNA [37]. In addition, RCA is prone to "leakage", leading to false positives. The latest research attempts to combine methylation-dependent DNA endonuclease (GlaI) with CRISPR-Cas12a for DNA methylation detection [21]. However, the EXPAR in this method has a serious "leakage" problem and its sensitivity is only 0.1%, which cannot meet clinical needs. Therefore, although CRISPR-Cas12a has very high sensitivity and has the potential to realize simple and low-cost quantitative detection of ultra-low abundance DNA methylation, to achieve this goal, we still need to overcome the two key issues of "insufficient specificity" and "low sequence universality".
In this study, we innovatively proposed a PAM-free CRISPR-Cas12a hyper-sensitive DNA methylation quantification method. Based on our previous work, we found that CRISPR-Cas12a can recognize a type of double-stranded DNA with a toehold region (toe-dsDNA). CRISPR-Cas12a's ability to recognize single-base mismatches in toe-dsDNA was higher than in regular dsDNA containing a PAM sequence. However, this discrimination ability is far from sufficient for detecting DNA methylation, especially for single CpG methylation site. To further enhance the ability of CRISPR-Cas12a to detect single methylation site, we have improved the toe-dsDNA and CRISPR-Cas12a system in this work: (1) Introducing an assisted-strand; (2) designing artificial mismatches in crRNA. These improvements significantly enhanced the ability of CRISPR-Cas12a to distinguish single CpG methylation site. Then, we constructed toe-dsDNA in PCR products by using the "heating and freezing" method and successfully developed a novel and ultra-sensitive DNA methylation detection method. This new method not only achieved quantitative detection of DNA methylation, but also significantly simplified primer design and improved detection efficiency. We selected methylation genes frequently found in HCC and ovarian cancer (such as ACP-1, DAB, HOX and CLE) as target models. The results showed that the detection sensitivity of this method can reach 0.01% in DNA methylation standard. Finally, we successfully tested the blood samples from 29 HCCs, 11 ovarian cancer and 4 normal people. The results of our detection were consistent with Methylight. In conclusion, we successfully constructed a PAM-free CRISPR-Cas12a DNA methylation quantification method, which achieved high congruence in sensitivity, specificity and universality, fully demonstrating its significant clinical application value.
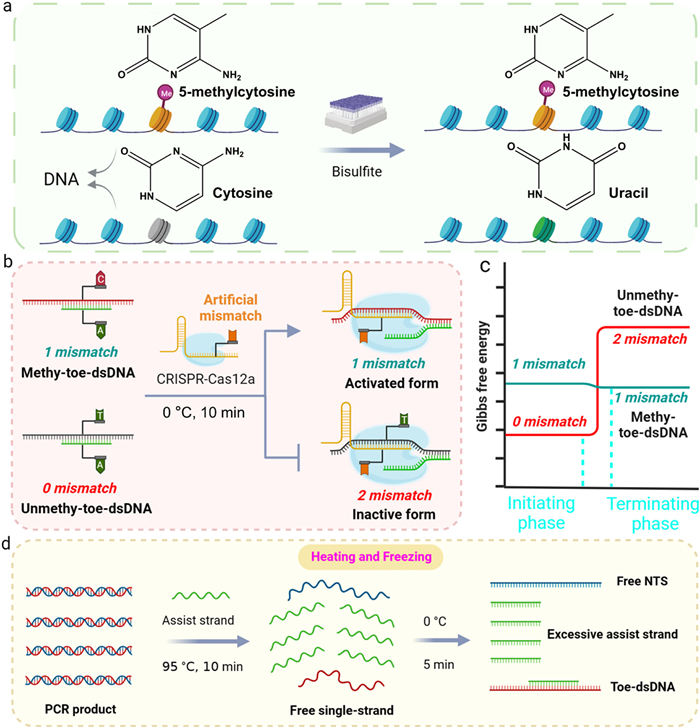
The principle of our proposed PAM-free CRISPR-Cas12a hyper-sensitive DNA methylation detection method was shown in Fig. 1. First, we need treat methylated and unmethylated DNA with bisulfite. Bisulfite can convert unmethylated C to U, but it cannot convert methylated C. In the subsequent PCR process, U is treated as thymine (T), while methylated C is still recognized as C. Therefore, after bisulfite treatment, the original methylated C and unmethylated C appear as C and T differences in sequence (Fig. 1a). In our previous research, we found that CRISPR-Cas12a can recognize toe-dsDNA and its ability to recognize single-base mismatches in toe-dsDNA is higher than in traditional dsDNA containing a PAM sequence [38]. However, this discrimination ability is far from sufficient for detecting DNA methylation, especially for single CpG methylation site. To further enhance the ability of CRISPR-Cas12a to distinguish single methylation site, we improved toe-dsDNA and CRISPR-Cas12a respectively (Fig. 1b): (1) Introducing an assisted-strand that fully matches with unmethylated DNA but mismatches with methylated DNA; (2) designing an artificial mismatch on the crRNA, which is adjacent to the methylation position. According to the thermodynamics of DNA hybridization reaction, because the number of mismatches between methylated toe-dsDNA (methy-toe-dsDNA) and CRISPR-Cas12a remains unchanged during DNA strand displacement reaction, the reaction between the crRNA and methy-toe-dsDNA was thermodynamically balanced (ΔG ≈ 0) and therefore very sensitive to sequence alterations [39]. Then, the methy-toe-dsDNA binds to CRISPR-Cas12a and activates its trans-cleavage activity, thereby cleaving the free DNA probe to produce fluorescent signal. However, the number of mismatches increased from 0 to 2 between unmethy-toe-dsDNA and CRISPR-Cas12a, so the strand displacement reaction cannot occur due to the thermodynamically balance is broken (ΔG > 0) (Fig. 1c) [40]. Therefore, the assisted-strand and the artificial mismatch significantly enhance CRISPR-Cas12a to recognize single CpG methylation site and greatly improve the ability to quantify the level of DNA methylation. Next, we use the "heating and freezing" method to construct methy-toe-dsDNA and unmethy-toe-dsDNA (Fig. 1d). At the high temperature of 95 ℃, the long dsDNA products generated by PCR are dissociated into long ssDNA strands.
Figure 1
Figure 1.
Principle of PAM-free CRISPR-Cas12a hyper-sensitive DNA methylation detection method in this work. (a) Mechanism of bisulfite conversion for methylated and unmethylated DNA. (b) Principle of introducing an assist-strand and designing an artificial mismatch to improve CRISPR-Cas12a's specificity. (c) ΔG of the strand displacement reaction between methy-toe-dsDNA/unmethy-toe-dsDNA and CRISPR-Cas12a. (d) Process of "heating and freezing".
Then, rapidly cool to 0 ℃, due to the ssDNA strands are too long, its recovery kinetics to form dsDNA is inhibited at 0 ℃. However, the assisted-strand is short and its concentration is far higher than that of the long ssDNA strand, so it can quickly bind to the long ssDNA strand at 0 ℃ to form toe-dsDNA. Interestingly, CRISPR-Cas12a can also bind to Toe-dsDNA and activate its trans-cleavage activity under low temperature conditions [41]. Through the reaction principle, we can see that toe-dsDNA activates CRISPR-Cas12a without the limitation of PAM sequence and improves the specificity of its methylation recognition. More importantly, by constructing toe-dsDNA of any sequence through the "heating and freezing" method, it greatly broadens the application range of CRISPR-Cas12a in DNA methylation detection.
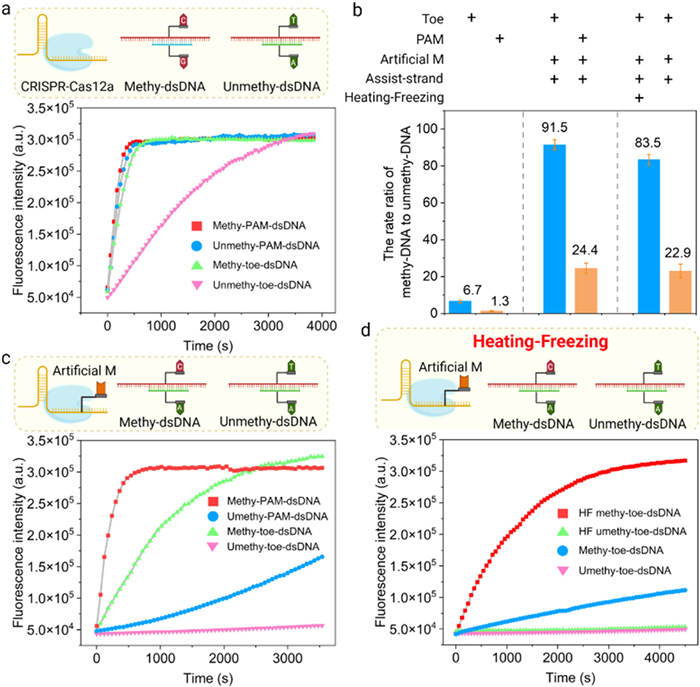
Next, we selected the hotspot of methylated-gene in HCC as target to verify the above principle. With the ACP-1 gene as the target model, we first set the toehold region length of methy-toe-dsDNA to be 5-nt and only contained a single CpG methylation site (ACP-Me-1 and ACP-Me-2). Subsequently, we synthesized the methylated and non-methylated ACP-1 sequences that had been treated with bisulfite and crRNA (ACP-crRNA-1). As shown in Figs. 2a and b, the discrimination factor (the ratio of fluorescence signal rate between methylation and non-methylation) of CRISPR-Cas12a for the single CpG methylation site of toe-dsDNA reached 6.7. Compared with conventional dsDNA with PAM, the discrimination factor nearly increased by 5-fold. However, this discriminating ability is far from sufficient to detect single methylation site. To enhance the discrimination ability of CRISPR-Cas12a for single CpG methylation site, we designed a crRNA (ACP-crRNA-2) containing an artificial mismatch and the corresponding assisted-strand (Assisted strand-1). The results showed that the discrimination factor of CRISPR-Cas12a for single CpG methylation site of toe-dsDNA reached 91.5, while the discrimination factor of conventional dsDNA with PAM was only 24.4 (Figs. 2b and c). To further validate DNA strand displacement reaction, we conducted polyacrylamide gel electrophoresis. As shown in Fig. S1 (Supporting information), lane 7 has a darker crRNA band compared to lane 3, surfacing that it can bind to the MT-assist strand by strand migration. When Cas12a was added, the trans-cleavage activity was activated, which in turn digested the ssDNA in the system (lane 9). Comparatively, the activity of Cas12a was not activated in the MT group, which is consistent with the fluorescence results (lane 10). These results fully confirmed that the toe-dsDNA can activate CRISPR-Cas12a without the need for PAM sequence, and significantly enhances the specificity of CRISPR-Cas12a for single CpG methylation site.
Figure 2
Figure 2.
Establishment and verification of the principle of our method. (a) Fluorescence curve of CRISPR-Cas12a for the single CpG methylation site in toe-dsDNA and conventional dsDNA with PAM. (b) Discrimination factor of CRISPR-Cas12a for the single CpG methylation site in toe-dsDNA and conventional dsDNA with PAM. Discrimination factor means the rate ratio of methy-toe-dsDNA and unmethy-toe-dsDNA. (c) Fluorescence curve for the single CpG methylation site using artificial mismatch and assist-strand. (d) Fluorescence curve for the single CpG methylation site by "heating and freezing". HF means heating and freezing. Reaction setup: 100 nmol/L methy-toe-dsDNA and unmethy-toe-dsDNA, 1000 nmol/L assist-strand, 20 nmol/L LbCas12a, 10 nmol/L crRNA, 200 nmol/L ssDNA FQ probe. n = 3.
Since the PCR products of genomic DNA cannot naturally form toe-dsDNA, with the help of the assisted-strand, we used the "heating-freezing" method to convert dsDNA product into toe-dsDNA. First, we treated the methylated and non-methylated standards (0.2 µg) with bisulfite. The conversion rate of the unmethylated C to U in the synthetic sequence by bisulfite treatment reached about 99% (Fig. S2 in supporting information). Then, we obtained the dsDNA products through simple PCR (ACP-FP-1 and ACP-RP-1). Next, we added a 10-fold assisted-strand to the dsDNA products and heated it at 95 ℃ for 10 min.
Then, we immediately transferred it to 0 ℃ for freezing treatment and maintained it for 5 min. After that, we added CRISPR-Cas12a to the reaction system and continued to maintain it at 0 ℃ for another 10 min. Finally, we detected the fluorescent signal in the machine. The results showed that the discrimination factor of CRISPR-Cas12a for single CpG methylation site reached 83.5 in the methylated standards (Figs. 2b and d). However, if there was no "heating and freezing" process, the differentiation factor was only 22.9. Theoretically, there should be no signal generated without the heating and annealing process. We speculated that this phenomenon may be due to asymmetric PCR amplification (the concentration of FP is higher than that of RP), which leads to the generation of target single strands and the subsequent formation of some toe-dsDNA. The results of polyacrylamide gel electrophoresis further confirmed that PCR combined with the "heating and freezing" process could realize the detection of single CpG methylation site on genomic DNA (Fig. S3 in Supporting information). Building on this foundation, we further examined the reaction time after Cas12a was introduced into the system. The results showed that a 10-min reaction time at 0 ℃ yielded the optimal outcome (Fig. S4 in Supporting information). In addition, changes in the position of methylation sites may lead to differences in the crRNA binding efficiency and further Cas12a trans-cleavage activity. We designed single methylation sites at different positions, and the results showed that the ability to distinguish between positions 8~10 was good, while the ability to distinguish between positions 2, 4, and 18 was poor (Fig. S5 in Supporting information). Therefore, the single methylation site should be designed in the middle of the detection sequence as much as possible. In conclusion, we achieved excellent DNA methylation detection performance by using the "heating-freezing" method to form Toe-dsDNA and activate CRISPR-Cas12a.
Then, we optimized the reaction conditions and the ratio of each component. First, we adjusted the length of the Toe-dsDNA. We designed toe-dsDNAs with different toehold lengths (Assisted strand-1~5) and found that the distinction between methylated and unmethylated DNA was best when the toehold length was 5-nt (Fig. S6 in Supporting information). It should be noted that DNA methylation mainly occurs on CpG islands and usually involves multiple consecutive sites in cancer. To verify the performance of our method in detecting the methylation of multiple consecutive sites, we targeted ACP-1 and designed toe-dsDNAs containing 1, 2, 3 and 4 methylation sites, respectively (Fig. 3a, ACP-Me-3~6 and ACP-crRNA-3~6). The results showed that the method's ability to distinguish methylated DNA increases with the number of methylated sites (Fig. 3b). Then, we improved the design principle of PCR primers. We targeted 2 methylation sites and designed 0, 1, 2, 3 and 5 methylation sites on the primer sequence, respectively (Fig. 3c, ACP-Me-7~11). Then, using our method to detect the above PCR products, the results showed that the number of methylation sites on the primer hardly affect the detection of DNA methylation (Fig. 3d). Compared with MSP and Methylight technology, our detection method has lower requirements for primer, so the method is simpler in primer design and has a wider adjustment range. Then, we adjusted the concentration of the assisted-strand. We aimed to detect 2 methylation sites, the results showed that the distinction between methylated and unmethylated DNA was best when the concentration of the assisted-strand was 15-fold that of DNA, which reached 209.3 (Fig. S7 in Supporting information). Then, we optimized the concentration of CRISPR-Cas12a and the best effect was achieved when the CRISPR-Cas12a concentration is 5 nmol/L (Fig. S8a in Supporting information). Optimizing probe concentration is crucial for minimizing background noise and reducing resource consumption. As demonstrated in Fig. S8b (Supporting information), a probe concentration of 200 nmol/L not only achieves a high signal-to-noise ratio but also reduces the overall probe usage. In addition, we also investigated whether the detection system can detect target DNA with stem-loop structure. The results show that the system is not affected by the secondary structure of the target sequence (Fig. S9 in Supporting information). Finally, we optimized the reaction temperature. To investigate the activation effect of toe-dsDNA on CRISPR-Cas12a at different temperatures, we designed strand migration reaction temperatures of 0 ℃ and 37 ℃, respectively. The results showed that compared to the strand migration reaction at 37 ℃, it has a better strength at 0 ℃ (Fig. S10 in Supporting information).
Figure 3
Figure 3.
Conditional optimization of our method. (a) Sequences of the methy-toe-dsDNA. The methylation positions are marked in red and toehold region in green. (b) Discrimination factor for the single methylation site between methy-toe-dsDNA and unmethy-toe-dsDNA. (c) Methylation positions and numbers in FP and RP of ACP. The methylation positions are marked in red. (d) Discrimination factor for the single methylation site using different primers. Reaction setup: 100 nmol/L toe-dsDNA or PCR products, 1000 nmol/L assist-strand, 20 nmol/L LbCas12a, 10 nmol/L gRNA, 200 nmol/L ssDNA FQ probe. n = 3.
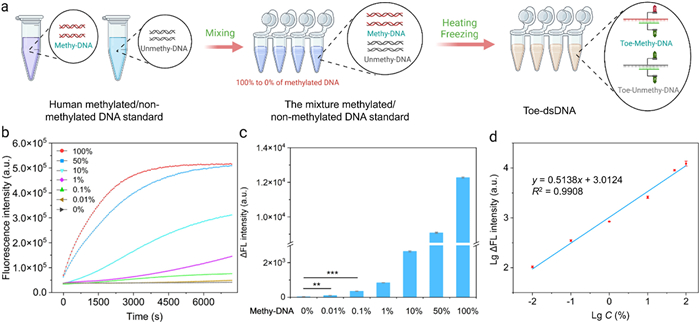
As mentioned above, the abundance of DNA methylation is extremely low in total DNA bases. Therefore, in the presence of a large amount of unmethylated DNA, it becomes crucial to accurately quantify the level of methylated DNA. For this purpose, we used the above optimized conditions to explore the sensitivity of this method. After analyzing the gene sequence of ACP-1 methylation standard, we designed partial sequences containing a single and four CpG methylation sites as targets (ACP-Me-1; ACP-Me-6) and synthesized the corresponding crRNAs (ACP-crRNA-2; ACP-crRNA-6). We used methylated and unmethylated DNA standards to prepare mixed samples of different proportions (0.2 µg in total), including 0%, 0.01%, 0.1%, 1%, 10%, 50% and 100% methylated DNA. Then, we performed bisulfite conversion on these mixed samples and used this method to detect their methylation levels. The results showed that the sensitivity of the 4 methylation sites reached 0.01% (Figs. 4a and b), and a linear relationship in the range of 0.01%−100% (Fig. 4c). The relevant equation is Y = 0.5138X + 3.0124 (R2 = 0.9908), where Y represents the measured signal rate and X represents the measured methylation level (Fig. 4d). The method also has a high sensitivity to single CpG methylation site, reaching 0.1% (Figs. S11a and b in Supporting information). The above results demonstrate that our method has high sensitivity and specificity in DNA methylation detection and can accurately quantify the level of DNA methylation.
Figure 4
Figure 4.
Analysis of sensitivity and specificity of this method for DNA methylation detection. (a) Schematic diagram of our method to detect mixed samples of different proportions. (b) Fluorescence curve of four methylation sites in ACP methylated standard detecting by our method. (c) Statistical analysis of four methylation sites in ACP methylated DNA standard. (d) Relationship between the rise rate of the fluorescence intensity and the concentration of ACP. LgΔFL intensity means the logarithm of the rise rate of the fluorescence intensity. LgC means the logarithm of target concentration. Student's t-tests. ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001. n = 3.
The biggest advantage of this method is that it can construct toe-dsDNA for any DNA sequence, overcoming the problem of CRISPR-Cas12a's PAM limitation on the sequence. To further confirm that this method has no sequence limitation, we chose the methylation genes CLV3/ESR-related (CLE), disabled (DAB) and homeobox (HOX), which are highly associated with HCC and ovarian cancer obtained from previous screening. We designed CLE, DAB and HOX sequences containing different methylation sites and the corresponding crRNAs, respectively (CLE-crRNA-1, DAB-crRNA-1, HOX-crRNA-1, ACP-Me-12~14 and Assisted strand-7~9). Then, we used this method to test CLE, DAB, and HOX. The discrimination factors of CLE, HOX and DAB was 108,222 and 138.9, respectively (Figs. S12-S14 in Supporting information). Next, we further explored its sensitivity in methylated standards. We still used the methylated and unmethylated DNA standards to configure mixed samples of different proportions, specifically 0%, 0.01%, 0.1%, 1%, 10%, 50% and 100%. After bisulfite conversion and detection, the results showed that the sensitivity of CLE, HOX, and DAB all reached 0.01% (Figs. S15-S17 in Supporting information). The linear relationship of CLE was in the range of 0.01%−100% and the equation was Y = 0.4625X + 3.2761 (R2 = 0.9923) (Fig. S18 in Supporting information). For HOX, the linear relationship was in the range of 0.01%−100% and the equation was Y = 0.5422X + 3.5302 (R2 = 0.988) (Fig. S19 in Supporting information). For DAB, the linear relationship was in the range of 0.01%−100% and the equation was Y = 0.4933X + 3.1892 (R2 = 0.9934) (Fig. S20 in Supporting information). Compared with other methods (Table S8 in Supporting information), our method has demonstrated the advantages of high sensitivity, universality and quantitative analysis in detecting DNA methylation. It should be noted that although the PCR post-processing steps of this method methylation detection, which is of great significance for expanding the application of CRISPR-Cas12a.
To evaluate the potential applications of our method in clinical sample detection, we collected serum samples from 11 ovarian cancers and 29 HCCs and performed test using this method. The study was approved by the Ethics Committee of Tongji Medical College, Huazhong University of Science and Technology: No. [2022] IEC (A131). We selected DAB, HOX and CLE as the targets for HCC and HOX for ovarian cancer. The actual sample detection process was shown in Fig. 5a: First, we extracted circulating DNA from the serum and performed conversion with bisulfite (100 ng in total); next, we used the previously mentioned primers to amplify ACP-1, HOX and CLE, obtaining the amplified PCR products, respectively; then, we performed "heating and freezing" treatment on the PCR products; finally, we added CRISPR-Cas12a at 0 ℃ for 10 min and then detected the fluorescent signal. Then, we calculated the methylation percentage of clinical samples by standard curves. The test results showed our method detected methylation abundance ranging from 0.03% to 78.03% in clinical samples (Figs. 5b and c, and Fig. S21 in Supporting information). In addition, we collected leukocyte samples from 4 healthy people (approved No. [2022] IEC (A131)). As was shown in Fig. S22 (Supporting information), the methylation level of the 4 healthy people was 0% in the leukocyte. To verify the accuracy of the method, we used the Methylight to test the above samples, the results showed that the detection results of our method were consistent with the Methylight (Figs. 5b and c, and Fig. S21 in Supporting information). It should be noted that due to the limited sensitivity of the Methylight method, it cannot detect low abundance methylation levels. Compared with Methylight, our method has excellent feasibility, stability and reliability in clinical applications, fully demonstrating its clinical application value.
Figure 5
Figure 5.
(a) Schematic workflow for DNA methylation detection in real clinical samples by our method. (b) Comparison of the abundances of CLE, HOX and DAB methylation in 20 HCC samples reported by our method and Methylight. (c) Comparison of the abundances of HOX mutation in 11 ovarian cancer samples reported by our method and next-generation sequencing (NGS). + means positive, - means negative, / means no signal detected.
In conclusion, we have proposed a PAM-free mediated CRISPR-Cas12a ultra-sensitive and quantitative DNA methylation detection method. Compared with traditional methods, this method has the advantages of high sensitivity, strong specificity and wide versatility for DNA methylation detection. By constructing a novel recognition mode of CRISPR-Cas12a—toe-dsDNA, we had successfully overcome the limitation that "the specificity and universality of DNA methylation detection based on CRISRP-Cas12a were difficult to balance". We introduced assisted-strand and design an artificial mismatch on crRNA sequence and significantly improved the ability to distinguish single CpG site methylation. Combining toe-dsDNA, PCR and "heating and freezing" technologies, we have developed a quantitative and sensitive DNA methylation detection method. Using our method detected four highly methylated genes ACP-1, CLE, DAB, and HOX in DNA methylation standards, the sensitivity reached 0.01% and with linear relationship in the range of 0.01%−100%. Importantly, we successfully detected clinical samples from 29 HCCs, 11 ovarian cancers and 4 normal people (approved No [2022] IEC (A131)). Our results are highly consistent with Methylight. This demonstrated our method have excellent reliability and feasibility in the detection of human genomic DNA. In summary, we constructed a PAM-free mediated CRISPR-Cas12a DNA methylation quantitative detection method, which revealed its great potential in building a methylation detection platform.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was financially supported by the Natural Science Foundation of Wuhan City (Chenguang Project) (No. 2024040801020331), the Natural Science Foundation of Hubei Province of China (No. 2023AFB402), the National Key Research and Development Program of China (No. 2023YFE0210200) and Interdisciplinary Research Program of HUST. We are very grateful for the clinical samples provided by the precision medicine center, Yangtze Delta region institute of Tsinghua university, Jiaxing, China. We are grateful to Biorender (https://app.biorender.com/) for offering the online illustration platform.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111184.
Figure 1
Principle of PAM-free CRISPR-Cas12a hyper-sensitive DNA methylation detection method in this work. (a) Mechanism of bisulfite conversion for methylated and unmethylated DNA. (b) Principle of introducing an assist-strand and designing an artificial mismatch to improve CRISPR-Cas12a's specificity. (c) ΔG of the strand displacement reaction between methy-toe-dsDNA/unmethy-toe-dsDNA and CRISPR-Cas12a. (d) Process of "heating and freezing".
Figure 2
Establishment and verification of the principle of our method. (a) Fluorescence curve of CRISPR-Cas12a for the single CpG methylation site in toe-dsDNA and conventional dsDNA with PAM. (b) Discrimination factor of CRISPR-Cas12a for the single CpG methylation site in toe-dsDNA and conventional dsDNA with PAM. Discrimination factor means the rate ratio of methy-toe-dsDNA and unmethy-toe-dsDNA. (c) Fluorescence curve for the single CpG methylation site using artificial mismatch and assist-strand. (d) Fluorescence curve for the single CpG methylation site by "heating and freezing". HF means heating and freezing. Reaction setup: 100 nmol/L methy-toe-dsDNA and unmethy-toe-dsDNA, 1000 nmol/L assist-strand, 20 nmol/L LbCas12a, 10 nmol/L crRNA, 200 nmol/L ssDNA FQ probe. n = 3.
Figure 3
Conditional optimization of our method. (a) Sequences of the methy-toe-dsDNA. The methylation positions are marked in red and toehold region in green. (b) Discrimination factor for the single methylation site between methy-toe-dsDNA and unmethy-toe-dsDNA. (c) Methylation positions and numbers in FP and RP of ACP. The methylation positions are marked in red. (d) Discrimination factor for the single methylation site using different primers. Reaction setup: 100 nmol/L toe-dsDNA or PCR products, 1000 nmol/L assist-strand, 20 nmol/L LbCas12a, 10 nmol/L gRNA, 200 nmol/L ssDNA FQ probe. n = 3.
Figure 4
Analysis of sensitivity and specificity of this method for DNA methylation detection. (a) Schematic diagram of our method to detect mixed samples of different proportions. (b) Fluorescence curve of four methylation sites in ACP methylated standard detecting by our method. (c) Statistical analysis of four methylation sites in ACP methylated DNA standard. (d) Relationship between the rise rate of the fluorescence intensity and the concentration of ACP. LgΔFL intensity means the logarithm of the rise rate of the fluorescence intensity. LgC means the logarithm of target concentration. Student's t-tests. ns, P > 0.05; *, P < 0.05; **, P < 0.01; ***, P < 0.001. n = 3.
Figure 5
(a) Schematic workflow for DNA methylation detection in real clinical samples by our method. (b) Comparison of the abundances of CLE, HOX and DAB methylation in 20 HCC samples reported by our method and Methylight. (c) Comparison of the abundances of HOX mutation in 11 ovarian cancer samples reported by our method and next-generation sequencing (NGS). + means positive, - means negative, / means no signal detected.

 DownLoad:
DownLoad:



 下载:
下载:






 下载:
下载:
