Figure 1.
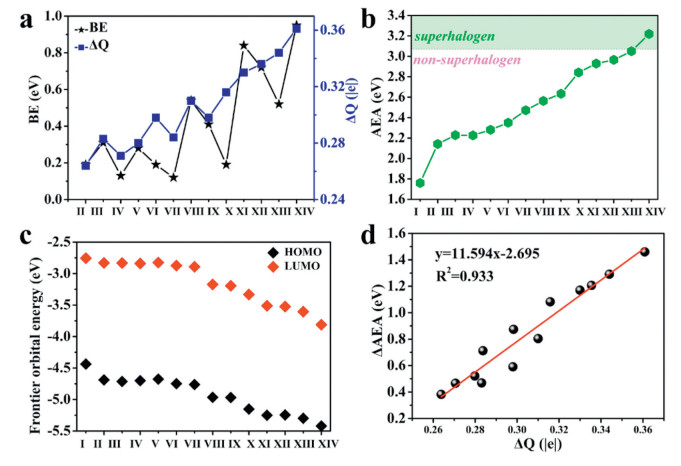
The calculated (a) BEs and ΔQ of the inner-core clusters, (b) AEA values, (c) energy levels of the frontier MOs, and (d) relationship between the ΔAEA and ΔQ in PAl12(BLAs) clusters.

The electron affinity (EA) and ionization potential (IP) are fundamental electronic properties of an element, which are influenced by its position within the periodic table. These properties suggest that elements, when forming new compounds, tend to gain or lose a specific number of electrons to achieve a stable closed-shell configuration. It is worth noting that the quantum confinement effect leads to the condensation of electron states within compact clusters into electron shells analogous to those of atoms, thereby giving rise to the emergence of superatoms with electron properties akin to single atoms [1–3]. This concept marks the beginning of a new era in the field of cluster science. Superatoms not only extend the traditional periodic table into three-dimensional (3D) space but also provide a diverse array of alternative building blocks for the assembly and design of functional materials [4–8]. This advancement presents novel opportunities for the precise control of innovative devices.
Over the past few decades, with ongoing refinement of theoretical concepts and advancements in experimental techniques, a variety of superatoms with unique physicochemical properties and valence electronic configurations have been successively proposed and confirmed. These superatoms encompass various categories such as superhalogens [9–15], superalkalis [16–20], magnetic superatoms [21,22], isovalent superatoms [23], and rare-earth superatoms [24]. Superhalogen, one of the most clearly defined categories within the superatom family, was initially conceptualized by Gutsev and Boldyrev [9] in the early 1980s and later experimentally verified by Wang et al. in 1999 [10]. Possessing an AEA higher than that of halogens, superhalogens hold significant potential for applications in perovskite solar cells [25,26], light-emitting diodes [27], hydrogen energy storage [28], and other fields [29]. Nevertheless, conventional strategies for the superhalogen design predominantly depend on various electron counting rules, including the octet rule [30], the 18-electron rule [31], the Jellium model [32], the Wade-Mingos rule [33], and the aromatic rule [34]. These traditional design principles are heavily dependent on the intrinsic properties of clusters and exhibit apparent limitations. For instance, the Al13 cluster, a typical superhalogen with 39 valence electrons, can easily gain an additional electron to fill the 2P orbital, achieving a 40e closed-shell configuration. The B12H122- cluster is a classic case that adheres to the Wade-Mingos rule. Pathak et al. designed the CB11H12 [33] superhalogen by substituting one boron atom in closo‑borane with a carbon atom. However, the precise manipulation of electron filling of cluster shells within cluster shells or the target replacement of specific elements within clusters remains a long-standing challenge in the field of controlled chemical synthesis. Thus, a series of key and fundamental questions arise: Is it possible to regulate the electronic properties of clusters solely by manipulating their external environments without altering their internal electronic shell filling? Is there any possibility for us to surpass the most famous Jellium model to transform clusters with magic number valence electrons into those exhibiting strong oxidizing or reducing properties? This would challenge traditional electron counting rules, as clusters with magic number electrons are considered stable species that do not typically gain (strong oxidizing power) or lose (strong reducing power) electrons. Thus, addressing these inquiries could expedite the further development and application of superatoms, as it offers a feasible and straightforward means to modulate the characteristics of synthesized clusters, without changing their constituent components to create new structures. In 2018, Khanna et al. showed that ligand attachment can significantly reduce the ionization energy of metallic clusters to superalkali levels without compromising their electronic shell structure [35]. This seminal contribution left a critical question concerning the inverse effect: whether a cluster's AEA can similarly be increased to the superhalogen range while preserving its shell electronic configuration. Despite the significance of this question, no successful strategies employing suitable ligands to increase AEA into superhalogen regime have been reported in the seven years since Khanna et al.’s work, underscoring the persistent challenge of achieving this outcome.
Consequently, this long-standing scientific challenge has motivated us to explore the glimmer for overcoming the abovementioned limitations of the classical Jellium model in superatom construction. Here, we established that the ligand field can be utilized for the targeted design of superhalogens without altering their constituent components, irrespective of the cluster's electronic shell being open or closed. Various organic Lewis acids were employed as ligands and were evaluated for their impact on the geometry and electronic characteristics of the PAl12 cluster, a typical superalkali. Here, the selection basis of boron Lewis acid ligands stems from the following advantages: (1) Moderate electron-withdrawing capabilities, (2) minimal perturbation to cluster electronic structures compared to halogen/superhalogen ligands, (3) preservation of cluster stability after ligation, and (4) versatility in liquid-phase experiments and catalytic systems. Unlike conventional ligands, these Lewis acids, while engaging in local charge transfer with the cluster's core, preserve the effective valence electrons of the metallic cluster. The attachment of a B(C6F5)3 ligand on the superalkali PAl12 cluster results in its surprising transformation into a superhalogen. Furthermore, by connecting B(C6F5)3 ligands to the bare Al13, CAl12, and PAl12 clusters, which possess distinct 39, 40, and 41 valence electrons, respectively, we observed that successive ligand attachment not only maintains the superatomic states of the metallic clusters but also significantly enhances their AEA values, resulting in the formation of superhalogen species. This finding undoubtedly demonstrates the efficacy of the ligand field strategy in superhalogen design, bypassing the constraints of the traditional Jellium model, which requires changes in the shell filling of the cluster.
We first optimized the lowest energy structures of the neutral and charged PAl12 clusters (Figs. S1 and S3 in Supporting information), from which the AIP of PAl12 was calculated to be about 5.25 eV, exhibiting a superalkali characteristic. As shown in Fig. S3, the gain or lose one electron does not alter the whole structural framework, the icosahedral geometry, of PAl12. These structural and electronic characteristics are consistent with a previous study [36], validating the accuracy of the present theoretical level. Our current objective is to utilize the PAl12 superalkali as a model cluster to demonstrate the modulation of AEA through ligand attachment, with the goal of transforming the cluster from superalkali to superhalogen. Boron Lewis acids (BLAs), known for their electron-withdrawing properties, may possess considerable potential. To explore this possibility, we optimized the BLAs to obtain stable conformations (Fig. S2 in Supporting information), and then tried various attachment sites to determine the neutral and anionic ground-state structures of the ligated clusters, which are shown in Figs. S1 and S3, respectively. The ligand arrangement in Fig. S2 was systematically and rigorously considered by evaluating spatial size and fluorination levels. This order not only highlights variations in electron-accepting ability of boron Lewis acids ligands but also effectively reveals a strong positive correlation between ligand electronic effects and enhanced cluster AEA. As depicted in Fig. S1, all ligated clusters maintain their metallic icosahedral cores, with Al-B bond lengths ranging from 2.34 Å to 2.77 Å in PAl12(BLAs) clusters. Notably, the average Al-Al and P-Al bond lengths remain nearly constant during ligation process, indicating that the bonding characteristics within the clusters were largely unaffected. Given the electron acceptor nature of BLAs, we performed a Hirshfeld charge analysis for the PAl12(BLAs) ground states. Fig. 1a demonstrates that the charge accumulation (ΔQ) within the inner-core PAl12 cluster increased monotonically from 0.26 |e| to 0.36 |e| with the addition of BLAs ligands. To further explore the binding strength between the BLAs ligands and PAl12, the binding energies (BEs), shown in Fig. 1a, were computed according to the following equation:
|
|
(1) |
where E presents the total energy of the corresponding system. The calculated BEs are in the range of 0.12–0.95 eV, which are comparable to those of the N-ethyl-2-pyrrolidone ligand (0.48–1.05 eV) [35]. Subsequently, the AEAs of PAl12(BLAs) were calculated to evaluate the effect of ligands on the electronic properties of the ligated clusters. As depicted in Fig. 1b, an enhancement in the electron-withdrawing capacity of the BLA ligands is accompanied by a consistent increase in AEA values, with only a few data points exhibiting minor deviations. Despite the relatively low BEs of the ligands, they exert a significant influence on the electronic properties of PAl12. Taking the B(C6F5)3 ligand as an example, upon the attachment, the AEA of PAl12 increased from 1.76 eV to 3.22 eV, a significant change that is noteworthy because it transforms the traditional superalkali into superhalogen without altering the cluster's components. To explain this phenomenon, we examined the energy levels of the frontier MOs of the clusters. Fig. 1c illustrates that the introduction of ligands results in a shift of the HOMO and LUMO orbitals of PAl12(BLAs) to lower energy levels, consistent with the observed increase in AEA. Interestingly, the HOMO-LUMO gap in clusters with various BLAs ligands remains stable at 1.61–1.88 eV, close to the 1.68 eV of the bare cluster. Further analysis revealed a positive correlation between the ligand's electron-withdrawing ability and the cluster's AEA, a relationship that is well-described by the linear equation y = 11.594x - 2.695 (Fig. 1d). This suggests that the local charge transfer between ligands and clusters leads to a reduction in the electronic energy levels of the clusters, thereby enhancing the clusters’ AEA. This finding indicates that the use of BLAs as ligands offers an effective strategy for the development of new superhalogens.
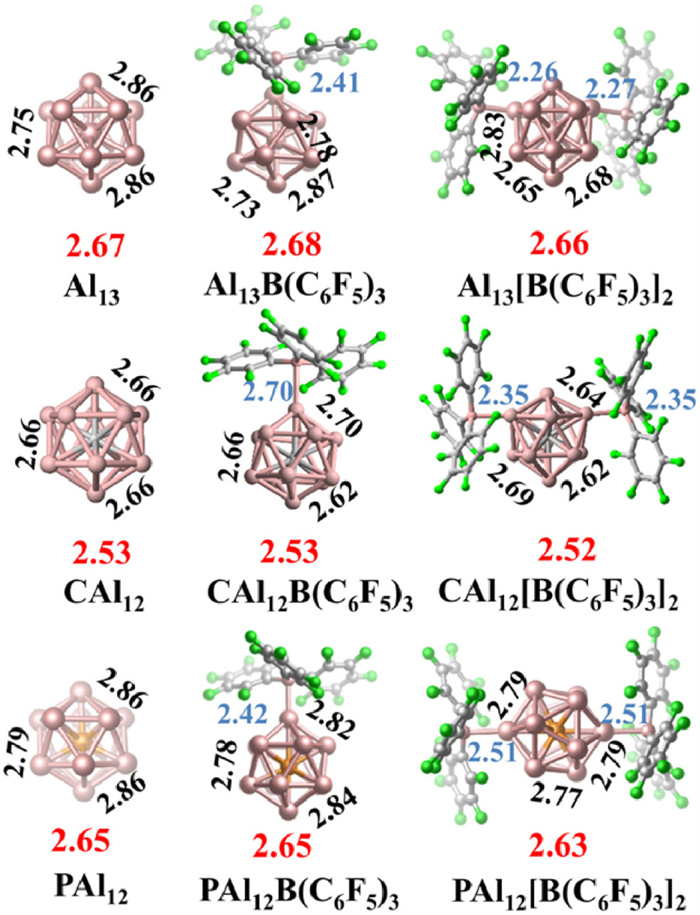
Recognizing the significant influence of BLAs ligands on the electronic properties of PAl12, we further investigate the questions raised in the introductory section: Can the ligand strategy overcome the shell filling effect to modulate the electronic properties of clusters while retaining their superatomic states and creating superhalogens? The B(C6F5)3 ligand appears to offer a breakthrough opportunity, as it demonstrates the most pronounced enhancement in the AEA of PAl12. To assess this potential, we examined all possible ligand binding sites to identify the neutral and anionic ground states of MAl12[B(C6F5)3]n clusters (M = Al, C, and P; n = 0–2), as depicted in Fig. 2 and Fig. S4 (Supporting information), respectively. Owing to variations in shell electrons, the three bare clusters exhibit different symmetrical icosahedral geometries, and accordingly, the M-Al bond lengths and optimal binding sites of B(C6F5)3 in the ligated clusters also differ. As shown in Fig. 2, when a second B(C6F5)3 ligand is combined to MAl12[B(C6F5)3], PAl12[B(C6F5)3]2 tends to form a para-position structure, while Al13[B(C6F5)3]2 and CAl12[B(C6F5)3]2 tend to form a meta-position geometry. The successive attachment of ligands leads to distortions in the metallic cluster core structure. For instance, the Al-Al bond length in PAl12[B(C6F5)3]n ranges from 2.68 Å to 2.80 Å. Notably, the average bond length between the central atom M and the surface Al atoms remains relatively unchanged, indicating that the bonding characteristics within the clusters were not significantly affected by ligation. Hirshfeld charge analysis revealed the charge transfer of the metallic cores during the bonding process (Fig. 3a). With an increasing number of ligands, the metallic cores of the ligated clusters continuously lose electrons. When two ligands are combined, the charge amounts on the three metallic cores are nearly identical, suggesting insensitivity of the charge transfer to the cluster's shell electron count. To further visualize the charge distribution, we analyzed the electrostatic potential (ESP) of MAl12[B(C6F5)3]n (Fig. 3b). Evidently, the deeper red hue of the metallic cores with increasing ligand quantities corresponds to the electron accumulating on the ligands, aligning with the findings from the charge transfer analysis. To probe the interactions between the B(C6F5)3 ligand and these aluminum-based clusters, we computed the BEs illustrated in Fig. 3c using the following formula:
|
|
(2) |

where M = Al, C, and P; n = 1–2. Owing to the superalkali nature of PAl12, its first ligand BE exceeds those of the other clusters. The addition of a second ligand results in nearly equivalent BEs across the three clusters, with values ranging from 0.45 eV to 0.64 eV. Furthermore, the variation in the AEAs of ligated Al clusters was calculated to evaluate the effect of ligands on their electronic properties, which are shown in Fig. 3d. As ligands are progressively attached, their AEA values continuously rise. Remarkably, the attachment of a single ligand significantly enhanced the AEA values of the Jellium closed-shell superatomic CAl12 cluster and PAl12 superalkali cluster to 3.25 and 3.22 eV, respectively, entering the superhalogen range. The addition of a second ligand further elevated their AEA values to 3.46 and 3.93 eV, which are 1.88 and 2.23 times than those of the corresponding bare clusters. Although the classical Al13 superhalgon cluster showed less pronounced changes in AEA values compared to the other two, its value still increased by approximately 0.62 eV. These findings unambiguously demonstrate that, regardless of their electronic shell filling state (closed or open), B(C6F5)3 exhibits a considerable capacity to modulate the electronic properties of these clusters.
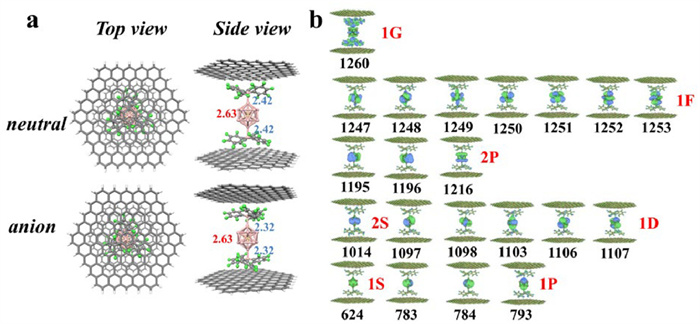
We subsequently explored the influence of the B(C6F5)3 ligation on the superatomic states of these aluminum-based clusters, which is the focus of this investigation. Taking PAl12 as an example, it can be seen that PAl12 possesses the 41e superatomic MOs (1S)2(1P)6(2S)2(1D)10(2P)6(1F)14(1G)1 (Fig. S5c in Supporting information) and the ligation process does not disrupt the shell filling, as all superatomic MOs of PAl12 are retained in the PAl12[B(C6F5)3]n (n = 1–2) clusters (Fig. 4a). Analogous situations also exist in the Al13[B(C6F5)3]n (n = 1–2) and CAl12[B(C6F5)3]n (n = 1–2) clusters, with (1S)2(1P)6(1D)10(2S)2(2P)6(1F)13 and (1S)2(1P)6(2S)2(1D)10(2P)6(1F)14 configurations, respectively (Figs. S6a and S7a in Supporting information). Therefore, the B(C6F5)3 ligand can significantly alter the electronic properties, i.e., AEA, of the clusters without changing their effective valence electron count and the superatomic states, resulting in the formation of superhalogens, which is significantly different from the famous Jellium model. To further investigate the microscopic reasons behind the remarkable increase in the AEAs of these ligated clusters, we have examined their one-electron energy levels. Taking PAl12[B(C6F5)3]n as an example (Fig. 4b), the degeneracy of the superatomic states in the clusters is progressively lifted with the continuous addition of ligands, and the LUMO orbitals gradually shifted towards deeper energy levels, specifically from −2.76 eV to −4.41 eV. It is well-accepted that the LUMO determines the capacity of a molecule to gain additional electrons. The decrease in the LUMO enhances the species’ tendency to accept extra electrons, thereby stabilizing the formation of anions. Consequently, this observation correlates with the AEAs of PAl12[B(C6F5)3]n increasing from the initial 1.76 eV to 3.93 eV. Similar trends are also observed in Al13[B(C6F5)3]n and CAl12[B(C6F5)3]n (Figs. S6b and S7b in Supporting information). Therefore, it is reasonable to conclude that the remarkable increase of AEAs of these ligated clusters, leading to the formation of superhalogens, can be attributed to the downwards shift of the electronic energy spectrum due to the ligation effect. Note that the ligand effects of B(C6F5)3 on aluminum-based clusters exhibit opposite charge transfer characteristics compared to the C6H11NO ligand used by Khanna et al. [35]. This discrepancy results in the electronic spectra of the aluminum clusters shifting in opposite directions under the influence of the two different ligands, ultimately leading to the formation of superatoms with distinctly different electronic properties. Moreover, the energetic stability of the ligated clusters was further assessed by calculating their HOMO-LUMO gaps (Fig. S8 in Supporting information). It can be seen from the figure that the H-L gaps in all clusters are larger than 1.42 eV, signifying their remarkable stability. Additionally, the thermodynamic stability of PAl12[B(C6F5)3]n (n = 1–2) was examined through AIMD simulations (Fig. S9 in Supporting information). The results revealed minimal energy fluctuations and the maintenance of structural integrity over a 10 ps simulation duration, thereby demonstrating the cluster's stability.

To deepen our understanding of the interactions between the MAl12 (M = Al, C, and P) superatomic states and B(C6F5)3, we employed charge decomposition analysis (CDA), a method based on fragment orbital concepts, to decompose the charge transfer between molecular fragments into their orbital contributions. The superatomic states were delineated by delocalization across the MAl12 framework. Observations from Fig. 4c indicate that, due to the superalkali nature of PAl12, the 1G and 1F superatomic MOs near the HOMO of PAl12B(C6F5)3 exhibit partial participation of ligand molecular orbitals, with contributions of 36 % and 17 %, respectively. The PAl12 deep-level 2P and 1D superatomic MOs transfer charge to the ligand, demonstrating strong interactions. Notably, the 2S superatomic MO contributes 26 % and 68 % respectively, interacting with the ligand to form the fully occupied bonding and anti-bonding states. In Al13B(C6F5)3 and CAl12B(C6F5)3 (Figs. S6c and S7c in Supporting information), the 1F superatomic MO near the HOMO exhibit minimal interaction with the ligand, suggesting that the MOs of Al13 and CAl12 do not participate in the charge transfer to the ligand. For these two clusters, interaction energies with the ligand are primarily concentrated in the 2S and 1P superatomic MOs, with the 2S superatomic MOs being similar in nature to the PAl12 metal center. The aforementioned CDA analyses highlight that the interactions between the deep superatomic states of these three clusters and the ligand facilitate the charge rearrangement through the Al-B bond. This phenomenon is quite intriguing, as it suggests that the interaction mode between the metal core and the ligand remains relatively invariant regardless of shell occupancy.
Furthermore, to ascertain the applicability and versatility of the BLAs ligand strategy in the superhalogen construction, we selected the superalkali Al3 [17], the multiple valence superatom Al7 [37], the planar Au8 [38], and the pyramidal Au20 [39] as extended clusters, all of which were synthesized via gas-phase mass spectrometry. The B(C6F5)3 ligand was utilized to connect these clusters and modulate their electronic properties. The neutral and anionic ground-state structures of both the naked and ligated clusters are depicted in Fig. S10 (Supporting information). As anticipated, Fig. S10 shows that the geometric configurations and interatomic bond lengths of the clusters underwent minimal changes following B(C6F5)3 binding, suggesting that the ligation process had a negligible impact on the metal clusters’ bonding characteristics. Nonetheless, computational analyses revealed a substantial alteration in the AEAs upon the ligand attachment. Table S1 (Supporting information) indicates that, upon addition of a single B(C6F5)3 ligand, the AEAs of the Al7, Au8, and Au20 clusters increased by 1.45, 0.40, and 0.67 eV, respectively, conferring the superhalogen properties of these systems. Despite the Al3B(C6F5)3 cluster not transformed into a superhalogen, its AEA also increased by approximately 1.24 eV. Hirshfeld charge analysis indicated that the ligand adsorption resulted in the transfer of 0.45, 0.26, 0.10, and 0.10 e from the cluster metal cores (Al3, Al7, Au8, and Au20) to the ligands, consequently lowering the LUMO orbital energies. The formation of these superhalogens is ascribed to the charge transfer complexes that form between the ligand and the clusters. Apparently, the aforementioned findings clearly indicate that BLAs ligands, particularly the B(C6F5)3 ligand, can significantly enhance the AEAs of clusters independently of their composition. This ligand field-driven regulatory effect contrasts sharply with the traditional superhalogen construction strategy that depends on electron counting rules. In addition, it is also worth noting that, given the extensive application of ligands in cluster chemical synthesis, the BLAs ligand field strategy is poised to open up new possibilities for the synthesis of both liquid and solid-state superhalogens, potentially broadening the practical applications of superatoms considerably. Thus, in conjunction with the work of Khanna et al. [35], our findings demonstrate that judicious ligand selection enables precise modulation of the electronic properties of metal clusters, facilitating the formation of superatoms with distinct electron-tuning capabilities, i.e., the superalkali and superhalogen. These results provide a critical advancement in understanding the construction of superalkalis and superhalogens without modifying the shell configurations of the parent clusters.
The proposed ligand strategy demonstrates improved experimental control over traditional superatomic design methods, i.e., the Jellium model, which relies on the electronic shell filling of clusters. However, significant progress is required to achieve the “designable and fabricable” paradigm for superatomic devices. Recent advances in atomic manufacturing [40], intercalation stripping [41], and nano-confinement [42,43] have facilitated the uniform distribution of atoms or functional units within confined spaces, leading to the development of efficient single-atom catalysts, superatoms, and functional heterostructure materials. Building upon these achievements, we propose a potential approach: integrating ligated clusters into 2D materials to facilitate the precise manipulation of superatoms. To assess this approach, a model of MAl12[B(C6F5)3]n (M = Al, C, and P; n = 0, 2) on a hydrogen-saturated graphene nanoplate (2.2 nm × 2.4 nm) was constructed, denoted as MAl12[B(C6F5)3]n@graphene. Fig. 5a and Figs. S11-S13 (Supporting information) display the optimized neutral and anionic structures of MAl12[B(C6F5)3]n@graphene. The theoretical AEA values of MAl12[B(C6F5)3]2@graphene (M = Al, C, and P) are 4.08, 3.45, and 3.79 eV, respectively, which are comparable to those of the isolated MAl12[B(C6F5)3]2 clusters. This similarity suggests that MAl12[B(C6F5)3]2 retain their superhalogen characteristics under confinement. Furthermore, the effect of intercalation on the superatomic states of MAl12[B(C6F5)3]n (M = Al, C, and P; n = 0, 2) has also been investigated, which constitutes the central theme of this study. Fig. 5b and Fig. S14 (Supporting information) illustrate that graphene nanosheets exert little influence on the superatomic states of the confined PAl12[B(C6F5)3]2, maintaining an electron configuration identical to that of the gas-phase PAl12 cluster (1S)2(1P)6(2S)2(1D)10(2P)6(1F)14(1G)1. Similar observations are depicted in Figs. S15 and S16 (Supporting information) for the MAl12[B(C6F5)3]n@graphene (M = Al and C; n = 0, 2) systems. This finding demonstrates that the B(C6F5)3 ligand can preserve the capacity to construct superhalogens under intercalation confinement, independent of cluster shell filling, thus offering a feasible route for the development of stable superatomic-based devices with unique properties.
In summary, we propose a ligand strategy that transcends traditional and well-known electronic counting rules, allowing for the modulation of the AEA of the metal core without changing the electron shell filling of the clusters. Our finding suggests that the increase in the AEA of the cluster may stem from charge transfer complexes formed between the ligand and the metal core, which lower the LUMO energy level through electrostatic Coulomb potential. Specifically, the BLAs ligands have shown their unique regulatory capabilities: in the PAl12(BLAs) system, we have confirmed that the AEA can be tuned by the electron-withdrawing ability of the BLAs ligands, with the electronic spectra shift correlating to the extent of local charge transfer. Furthermore, in MAl12[B(C6F5)3]n clusters, the superhalogens induced by the B(C6F5)3 ligand maintain the superatomic state of the cluster, unaffected by electron shell filling, which differs from traditional thiol and halogen ligands. Additionally, the efficacy of the B(C6F5)3 ligand in constructing superhalogens in gold and aluminum clusters has also been verified, indicating its potential universality in controlling the oxidation state of clusters. These findings have broadened the horizons for the design of novel superhalogens, substantially increasing the diversity of functional clusters. Considering the core role of ligands in the synthesis of liquid or condensate phase clusters, we anticipate that the BLAs ligand strategy can inspire theorists and experimentalists to explore innovative methods for synthesizing superhalogens across multiple phases, thereby accelerating the progress of superatoms in catalysis, energy, and functional device fields.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jun Li: Writing – original draft, Investigation, Formal analysis, Data curation. Shi-Hu Du: Validation, Formal analysis. Yao Zhang: Validation, Formal analysis. Jia Liu: Validation, Formal analysis. Jing Chen: Validation, Funding acquisition, Formal analysis. Shi-Bo Cheng: Writing – review & editing, Supervision, Project administration, Funding acquisition, Conceptualization.
This study is based upon work supported by the National Natural Science Foundation of China (NSFC, Nos. 12474274, 92161101), the Innovation Project of Jinan Science and Technology Bureau (No. 2021GXRC032), the Natural Science Foundation of Shandong Province (No. ZR2024MA091). The scientific calculations in this paper have been done on the HPC cloud platform of Shandong University and the HPC platform for theoretical and computational chemistry of the School of Chemistry and Chemical Engineering of Shandong University.
Supplementary material associated with this article can be found, in the online version, at doi:
W.D. Knight, K. Clemenger, W.A. de Heer, et al., Phys. Rev. Lett. 52 (1984) 2141–2143.
S.N. Khanna, P. Jena, Phys. Rev. B 51 (1995) 13705–13716.
X. Roy, C.H. Lee, A.C. Crowther, et al., Science 341 (2013) 157–160. doi: 10.1126/science.1236259
S.N. Khanna, P. Jena, Phys. Rev. Lett. 69 (1992) 1664–1667.
D.E. Bergeron, P.J. Roach, A.W. Castleman Jr., et al., Science 307 (2005) 231–235. doi: 10.1126/science.1105820
Z.X. Luo, A.W. Castleman Jr., Acc. Chem. Res. 47 (2014) 2931–2940. doi: 10.1021/ar5001583
P. Jena, Q. Sun, Chem. Rev. 118 (2018) 5755–5870. doi: 10.1021/acs.chemrev.7b00524
X.B. Liu, W. Tiznado, L.J. Cui, et al., J. Am. Chem. Soc. 146 (2024) 16689–16697. doi: 10.1021/jacs.4c03977
G.L. Gutsev, A.I. Boldyrev, Chem. Phys. 56 (1981) 277–283.
X.B. Wang, C.F. Ding, L.S. Wang, et al., J. Chem. Phys. 110 (1999) 4763–4771.
D.E. Bergeron, A.W. Castleman Jr., T. Morisato, et al., Science 304 (2004) 84–87.
M. Willis, M. Götz, A.K. Kandalam, et al., Angew. Chem. Int. Ed. 49 (2010) 8966–8970. doi: 10.1002/anie.201002212
Y. Gao, S. Bulusu, X.C. Zeng, J. Am. Chem. Soc. 127 (2005) 15680–15681. doi: 10.1021/ja055407o
M.M. Wu, H. Wang, Y.J. Ko, et al., Angew. Chem. Int. Ed. 50 (2011) 2568–2572. doi: 10.1002/anie.201007205
S. Giri, S. Behera, P. Jena, Angew. Chem. Int. Ed. 53 (2014) 13916–13919. doi: 10.1002/anie.201408648
G.L. Gutsev, A.I. Boldyrev, Chem. Phys. Lett. 92 (1982) 262–266.
T. Zhao, Q. Wang, P. Jena, Nanoscale 9 (2017) 4891–4897.
J. Li, H.C. Huang, J. Wang, et al., Nanoscale 11 (2019) 19903–19911. doi: 10.1039/c9nr05613k
R. Parida, G.N. Reddy, A. Ganguly, et al., Chem. Commun. 54 (2018) 3903–3906. doi: 10.1039/c8cc01170b
V. Chauhan, S. Sahoo, S.N. Khanna, J. Am. Chem. Soc. 138 (2016) 1916–1921. doi: 10.1021/jacs.5b10986
J.U. Reveles, P.A. Clayborne, A.C. Reber, et al., Nat. Chem. 1 (2009) 310–315. doi: 10.1038/nchem.249
V.M. Medel, J.U. Reveles, S.N. Khanna, et al., Proc. Natl. Acad. Sci. U. S. A. 108 (2011) 10062–10066. doi: 10.1073/pnas.1100129108
S.J. Peppernick, K.D.D. Gunaratne, A.W. Castleman Jr., Proc. Natl. Acad. Sci. U. S. A. 107 (2010) 975–980. doi: 10.1073/pnas.0911240107
S.B. Cheng, C. Berkdemir, A.W. Castleman Jr., Proc. Natl. Acad. Sci. U. S. A. 112 (2015) 4941–4945. doi: 10.1073/pnas.1504714112
H. Fang, S. Wang, J. Liu, et al., J. Mater. Chem. A 5 (2017) 13373–13381.
H. Fang, P. Jena, Proc. Natl. Acad. Sci. U. S. A. 114 (2017) 11046–11051. doi: 10.1073/pnas.1704086114
Q. Yao, H. Fang, K. Deng, et al., Nanoscale 8 (2016) 17836–17842.
P.A. Berseth, A.G. Harter, R. Zidan, et al., Nano Lett. 9 (2009) 1501–1505. doi: 10.1021/nl803498e
Y. Gao, M.H. Wu, P. Jena, Nat. Commun. 12 (2021) 1331.
S. Smuczyńska, P. Skurski, Inorg. Chem. 48 (2009) 10231–10238. doi: 10.1021/ic901253r
H.J. Zhai, J. Li, L.S. Wang, J. Chem. Phys. 121 (2004) 8369–8374.
W. Ekardt, Phys. Rev. B 29 (1984) 1558–1564.
B. Pathak, D. Samanta, R. Ahuja, et al., ChemPhysChem 12 (2011) 2423–2428. doi: 10.1002/cphc.201100320
B.Z. Child, S. Giri, S. Gronert, et al., Chem. Eur. J. 20 (2014) 4736–4745. doi: 10.1002/chem.201305057
V. Chauhan, A.C. Reber, S.N. Khanna, Nat. Commun. 9 (2018) 2357.
M. Akutsu, K. Koyasu, J. Atobe, et al., J. Phys. Chem. A 110 (2006) 12073–12076. doi: 10.1021/jp065161p
J.U. Reveles, S. Khanna, P. Roach, et al., Proc. Natl. Acad. Sci. U. S. A. 103 (2006) 18405–18410. doi: 10.1073/pnas.0608781103
W. Huang, R. Pal, L.M. Wang, et al., J. Chem. Phys. 132 (2010) 054305.
J. Li, X. Li, H.J. Zhai, et al., Science 299 (2003) 864–867.
K. Zhang, C. Wang, M. Zhang, et al., Nat. Nanotechnol. 15 (2020) 1019–1024. doi: 10.1038/s41565-020-00778-z
R. Yang, L. Mei, Z. Lin, et al., Nat. Rev. Chem. 8 (2024) 410–432.
Q. Fu, X.H. Bao, Chem. Soc. Rev. 46 (2017) 1842–1874.
Q. Fu, X.H. Bao, Nat. Catal. 2 (2019) 834–836. doi: 10.1038/s41929-019-0354-z

Figure 1 The calculated (a) BEs and ΔQ of the inner-core clusters, (b) AEA values, (c) energy levels of the frontier MOs, and (d) relationship between the ΔAEA and ΔQ in PAl12(BLAs) clusters.

Figure 2 Ground state structures of MAl12[B(C6F5)3]n (M = Al, C, and P; n = 0–2). Selected Al-Al bond lengths (in Å) are shown in black, the average bond lengths of interior M atom to surface Al atoms are given in red, while the Al-B bond lengths are given in light blue.

Figure 3 Calculated (a) accumulated charge of the MAl12 metallic cores, (b) electrostatic potential, (c) binding energies, and (d) AEA of MAl12[B(C6F5)3]n (M = Al, C, and P; n = 0–2) clusters.

Figure 4 Theoretical (a) isosurface maps of the α occupied MOs of PAl12[B(C6F5)3]n (n = 1–2), the capital letters and numbers in red signify the angular momentum quantum number and the principal quantum number of the superatom, respectively, (b) one-electron energy levels of PAl12[B(C6F5)3]n (n = 0–2) clusters. The occupied and unoccupied states are represented by solid and dashed lines. The red dashed lines indicate the LUMOs. The levels are also marked with their angular character, (c) CDA analysis of PAl12B(C6F5)3 with PAl12 and B(C6F5) as fragments, the numbers marked beside the red lines indicate the contribution from the fragmental MO to the MOs of PAl12B(C6F5)3. The purple numbers in (a) and (c) indicate the orbital numbers. States marked with star symbol in (c) present the anti-bonding states. All the isosurface graphs are plotted with an isosurface value of 0.02.

Figure 5 (a) The optimized geometries of the neutral and anionic PAl12[B(C6F5)3]2@graphene. The average bond lengths (in Å) of interior P atom to surface Al atoms are given in red text, while the Al-B bond lengths are given in light blue text, and (b) isosurface maps of the occupied MOs of PAl12[B(C6F5)3]2@graphene, where the capital letters and numbers in red signify, respectively, the angular momentum quantum number and the principal quantum number of the superatom.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: