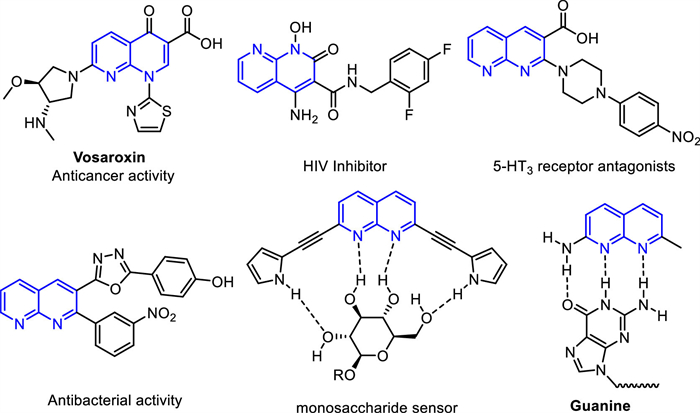
Scheme 1.
Representative 1,8-naphthyridine derivatives.
Construction of effective and recyclable non-noble metal coordination polymers and their multiple applications in water
Jiahao Li , Bin Pan , Anruo Mao , Likui Wang , Dawei Wang

As the ubiquitous building blocks in organic synthesis, naphthyridines are one of the most important nitrogen heterocycles [1,2]. Among them, 1,8-naphthyridine and their derivatives are not only universal in natural products but also endowed with antibacterial, anticancer, HIV inhibitor, and depression-relieving activities, etc. (Scheme 1) [3-5]. In addition, the two nitrogen atoms of 1,8-naphthyridine each have a lone pair of electrons, which makes 1,8-naphthyridine and its derivatives widely used as ligands, as molecular sensors and recognitions, and in optoelectronic devices such as blue organic light-emitting diodes [6,7].
The broad utility of 1,8-naphthyridine derivatives has led to a number of routes being developed for their fabrication, with a focus now being placed on more sustainable, greener, and cleaner methods (Scheme 2) [8]. The Friedlander reaction is a classical method to access quinolines and 1,8-naphthyridine through the condensation of 2-aminobenzaldehydes or 2-aminonicotinaldehydes with another carbonyl compound in the presence of base or Lewis acid [9]. In the following decades, many researchers improved the Friedlander reaction by using various metal catalysts, such as Ir, Ru, Cu, and Mn, and organic solvents [10-13]. In addition, a novel metal-catalyzed Friedlander reaction of 2-aminonicotinaldehydes with terminal alkynes leading to 3-substituted 1,8-naphthyridine was reported in recent years, which has different regioselectivity compared to traditional Friedlander reaction [14]. However, most of the above synthetic approaches often use highly genotoxic, unstable, and easily self-condensed 2-aminonicotinaldehydes as precursor and suffer harsh reaction conditions [15,16]. Later, an improved copper-catalyzed protocol for the synthesis of 1,8-naphthyridine via oxidative cyclization of oxime acetates with 2-amino-3-pyridinemethanol was further developed [17].
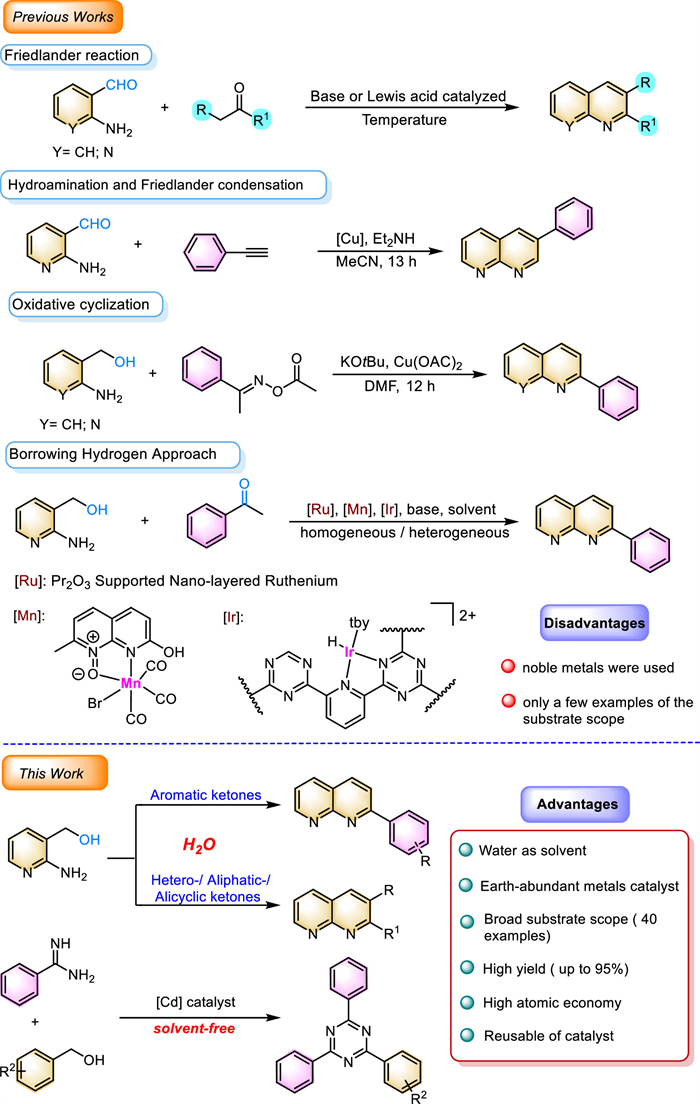
Along this line, innovative catalytic methodologies have emerged, commonly referred to as the borrowing hydrogen (BH) and acceptorless dehydrogenative coupling (ADC) pathway [18-23], where alcohols are dehydrogenated to form more active intermediates, followed by condensation and reduction processes [24-29]. In comparison to traditional Friedlander reaction pathways, condensation of 2-amino-3-pyridinemethanol with carbonyl compounds is more competitive due to the use of the more widely available and less toxic alcohol as the starting material, while hydrogen, as the only by-product, has atom efficiency [30-33].
Several catalytic dehydrogenative coupling syntheses of 1,8-naphthyridine from 2-amino-3-pyridinemethanol and ketones have been reported. In 2019, Chaudhari and Nagaoka [34] described an efficient pathway for the synthesis of 1,8-naphthyridine from 2-amino-3-pyridinemethanol and acetophenone by Pr2O3 supported Ru nanolayers catalysts. In 2022, Li group [35] developed the dehydrogenative coupling of 2-amino-3-pyridinemethanol with acetophenone to 1,8-naphthyridine catalyzed by iridium(Ⅲ) terpyridine complex. Bera and co-workers [36] found the use of visible-light-driven photocatalysts for the synthesis of quinolines and 1,8-naphthyridine from 2-amino-3-pyridinemethanol as starting materials. Despite significant advances, these approaches still suffer more or less of limitations, such as narrow substrate scope, the use of noble metals, and harsh reaction conditions. Therefore, the development of environmentally friendly catalytic systems remains a challenging task, particularly highly efficient and recyclable catalysts using water as the solvent [37,38].
The construction of coordination polymers (CPs) is currently of tremendous interest not only owing to their great structural diversities but also due to potential functions such as gas storage and separation, chemical sensing, magnetic and luminescent materials [39-41]. Noteworthy, one of the especially critical applications for CPs is that related to their heterogeneous catalysis [42-44]. Along this line, some functional CPs have been developed as highly efficient heterogeneous catalysts capable of catalyzing a variety of organic reactions. Interestingly, the greatest advantage of CPs over some of the more traditional catalyst materials is their chemical customizability and versatility [45-47]. Compared to non-toxic metal, Cd(Ⅱ) with a d10 electronic configuration are high active in catalytic kinetics. Moreover, incorporating Cd(Ⅱ) formation of typical electron donor (ED) and electron acceptor (EA) interactions between electron-deficient organic ligand and electron-rich aromatic carboxylates within the CPs [48,49]. Based on the above reasons, Cd(Ⅱ)-coordination polymers have been widely designed, which is beneficial for fabricating many highly active polymers in the field of catalysis.
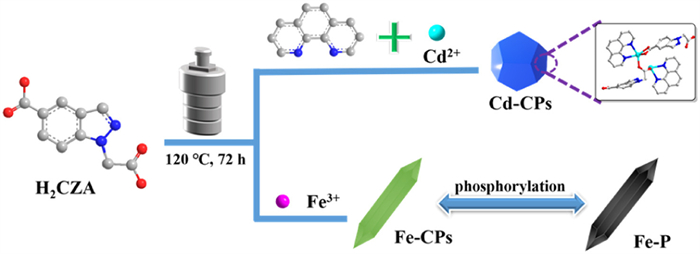
In the past, our group developed several novel metal catalysts based on nitrogen-containing heterocycles skeleton ligands, which could realize numerous universal acceptorless dehydrogenation and borrowing hydrogen reactions [50-52]. However, these noble catalysts are either expensive or show no reactivity in water, which limited the practical industrial applications and would be a great waste for modern organic synthesis [53-55]. Motivated by the prospects mentioned above, we engineered a new O-donor 1H-indazole-5-carboxylic acid (H2CZA) ligand and a N-donor 1,10-phenanthroline to afford a rare #-like” architecture CPs, [Cd2(CZA)2(phen)2]n (denoted as Cd–CPs). Thanks to the possible π-π stacking interaction between 1,10-phenanthroline and pyridine ring from H2CZA, the stability of the catalyst structure is enhanced [56]. Meanwhile, Due to the Lewis acidity provided by Cd(Ⅱ) ions and Lewis basicity supported by the coordinated CZA-anions, Cd-CPs exhibits good catalytic activity for the synthesis of 1,8-naphthyridine derivatives by the reaction of 2-amino-3-pyridinemethanol with ketones in water through acceptorless dehydrogenative coupling. Meanwhile, we found that Cd–CPs can also achieve efficient synthesis of 1,3,5-triazine derivatives in a solvent-free condition. In additional, we engineered innovative coordination polymers [Fe(CZA)2]n (denoted as Fe–CPs). The spindle-like structures of Fe-P is derived from Fe–CPs through phosphorylation, which showed a good OER performance and outstanding working stability.
Herein, H2CZA was synthesized via a two-step procedure according to previous literature with slight modification [57]. Then, we elaborately designed a novel coordination polymers Cd-CPs, which was constructed by a one-pot solvothermal route between H2CZA, Cd(OAc)2·2H2O, and 1,10-phenanthroline. For coordination polymers such as Fe-CPs, FeCl3 and H2CZA were mixed into MeCN/H2O in a Teflon-lined stainless-steel autoclave at 120 ℃ for 72 h. To synthesize Fe-P, a facile phosphorylation route was employed (Scheme 3). More detailed experimental steps were described in Supporting information.
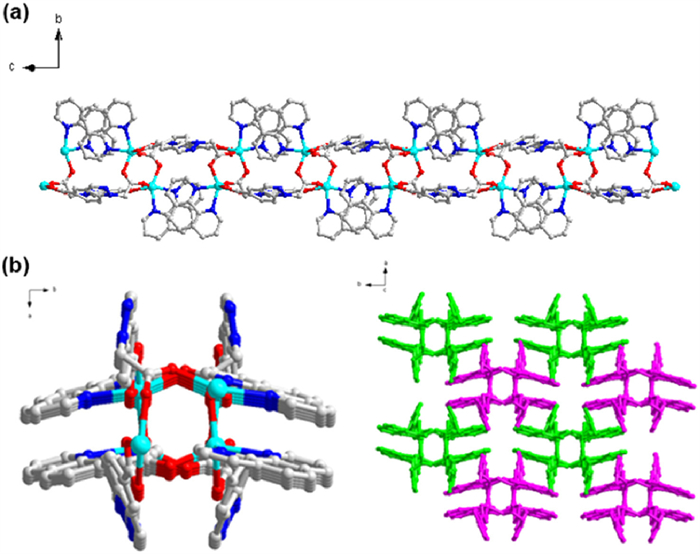
Single-crystal X-ray diffraction analysis (SC-XRD) demonstrates that Cd-CPs crystallizes in the P21/c space group with cell parameters of a = 14.5128(5) Å, b = 16.3515(7) Å, and c = 18.2577(7) Å at 298 K (Fig. S1 in Supporting information). The asymmetric unit of complex is composed of two Cd(Ⅱ) ions, two H2CZA linkers, and two 1,10-phenanthroline ligands. Each of the Cd(Ⅱ) centers has a six-coordinated octahedral geometry in which four coordination sites are occupied by oxygen atoms from two different H2CZA ligands and another two nitrogen atoms from a 1,10-phenanthroline molecule. Two carboxylate groups from two H2CZA ligands link two Cd2+ centers to form a [Cd2(COO)2] unit (CCDC: 2293907). Meanwhile, as bridging ligands, H2CZA ligands link adjacent Cd(Ⅱ) cations via carboxylate oxygen and pyridine nitrogen atoms to produce a highly ordered 1D chain structure (Fig. 1a). Fig. 1b is a close-up view of the 1D chain structure of Cd-CPs along the c axis. Interestingly, this 1-D architecture exhibits a novel “#-like” architecture, which is very rarely observed in coordination polymer.
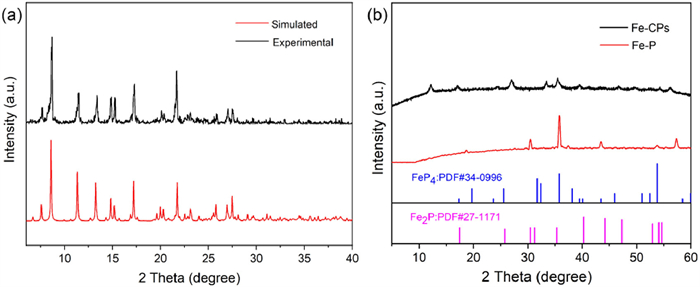
Besides the SC-XRD technique, the crystalline nature of as-synthesized polymers was demonstrated by powder X-ray diffraction (PXRD). The PXRD pattern of Cd-CPs exhibits good agreement between all major peak positions of the experimental PXRD and those of the simulated pattern from the SC-XRD date, affirming that Cd-CPs has a highly pure phase (Fig. 2a). Moreover, with phosphorylation, the corresponding Fe-P was obtained. Phosphorylation refers to a gas-phase reaction approach, as the annealing temperature is elevated, the decomposition of NaH2PO2·H2O is accompanied by release of phosphine (PH3) and H2O vapor, which subsequently react with Fe-CPs to form Fe phosphate [58-60]. From the PXRD pattern of the Fe-P, the peaks attributed to the Fe-CPs precursor are not detected, indicating that the complete phosphorylation of the Fe-CPs. Furthermore, the diffraction peaks at 19.2°, 24.7°, 35.7°, 37.8° and 43.7° can be indexed to the FeP4 (PDF #34–0996) [61]. Meanwhile, a series of diffraction peaks at 30.6°, 35.1°, 41.1°, and 47.8° were found, corresponding to the Fe2P (PDF #27–1171) [62]. The above results indicate that the Fe-CPs is transformed into a FeP4 and Fe2P heterostructure during the phosphorylation treatment (Fig. 2b).
In order to investigate the micromorphological structure of the as-prepared materials, scanning electron microscope (SEM) and transmission electron microscope (TEM) characterization were referred to for the finished analysis. As depicted by the SEM images of the Cd-CPs in Figs. 3a and b. The Cd-CPs have a diameter of 77 nm and nearly spheroidal shape. Besides, Fe-CPs consists of uniform spindle-like particles (length = ~0.9 µm) as presented in Figs. 3c and d. After phosphorylation, the SEM images of Fe-P clearly reveal that the overall morphology of original spindle-like structures remains unchanged, with slight shrinkage in particle size owing to the combustion of the organic linkers during this transformation (Figs. 3e and f) [63]. Meanwhile, we can see that the Fe-P is composed by nanoparticles and there are many voids among the nanoparticles. which is expected to greatly facilitate the diffusion of electrolyte and improve mass transport during the OER [64-68]. Both Fe3+ and Cd2+ have the similar six-coordination behavior, and the morphology of the two polymers is different from that of SEM, possibly due to the following reasons: (1) the nature of metal ions. The coordination atoms of Cd2+ with a d10 electronic configuration, while Fe3+ electronic configuration is d5 state. The electronic configuration of a metal ion affects the spatial arrangement of the organic ligand linkers and thus determines the pore size and shape of the porous structure. (2) The differences of organic ligands. The coordination polymer Cd-CPs used two ligands, H2CZA and 1,10-phenanthroline, while the Fe-CPs were synthesized using only one H2CZA ligand. Organic ligands are the bridging ligands of metal ion centers and act as the “skeleton” in CPs. 1,10-Phenanthroline, a molecule with a strong electron-donating ability was doped into Cd-CPs as co-ligands. By changing the bridging, the host structure becomes more stable, and the morphology of the CPs is also changed. (3) The difference of solvent and pH in the preparation process. For the preparation of Cd-CPs, we adjusted the pH of the reaction solution to an alkaline state of pH 10–11 with Et3N, while using water as the solvent. However, in the preparation of Fe-CPs, MeCN/H2O was used as the mixed solvent, and Et3N was not used to adjust the pH of the solution [69,70].

Subsequently, the internal structures of Cd-CPs, Fe-CPs and Fe-P were analysed by TEM. As shown in Figs. 3g–l. The TEM images of all samples formed (Cd-CPs, Fe-CPs and Fe-P) are consistent with the above SEM result. In addition, from the TEM images of Fe-P (Figs. 3k and l), the fabrication Fe-P become porous with rough surface, which are different from the original Fe-CPs precursor with a smooth surface. The HRTEM image of Fe-P (Fig. 3o) displays distinct lattice fringes with spacing of 0.298 nm and 0.250 nm, consistent with the (110) crystal planes of Fe2P phase and (131) planes of the FeP4 phase, respectively, further testifying the polycrystalline nature of the material. It is expected that such spindle-like structure with highly porous structure will facilitate exposure of catalytic sites and generate internal electric fields to accelerate electron transfer, thereby promoting the catalytic activity to a higher condition [71-76]. Further, the energy-dispersive spectroscopy (EDS) mapping of Cd-CPs exhibits uniform spatial distribution of Cd, N, and O elements in the whole Cd-CPs sample (Fig. 3m). Similarly, as depicted in Fig. 3n. The EDS mapping of Fe-P is fully proven that fabrication Fe-P is composed of C, N, and P elements, and that P atom is successfully introduced into the material during the phosphorylation process. The amount of P in the Fe-P was further confirmed by ICP-AES, which indicates the amount of P doping was 24.6 wt%.
To further investigate chemical states and elemental compositions of Cd-CPs, Fe-CPs and Fe-P, X-ray photoelectron spectroscopy (XPS) data were collected, as shown in Figs. S2 and S3 (Supporting information), respectively. The C 1s spectrum of the Cd-CPs was deconvoluted into four peaks at 284.8, 286.3, 287.3, and 288.8 eV (Fig. S2a), corresponding to C—C/C=C, C—N, C=N, and C=O in the organic ligands [77]. Additionally, to further investigate “N” chemical states of Cd-CPs, the XPS of corresponding monomer structure (N-donors 1,10-phenanthroline and O-donor 1H-indazole-5-carboxylic acid (H2CZA) ligand) data were collected (Fig. S2b), The N 1s spectra of 1,10-phenanthroline consist of a peak with binding energy (BE) ≈ 398.2 eV, which usually attributed to pyridine-N [78]. For H2CZA, its N 1s XPS spectrum can be fitted by two different nitrogen species, pyrazole C—N, 398.7 eV, and pyrazole C=N, 400.4 eV. As a contrast, the high-resolution N 1s spectrum of Cd-CPs can be deconvoluted into pyrazole C—N and coordinated pyrazole nitrogen (Cd-N, 400.1 eV) species [79]. The O 1s spectrum of Cd-CPs was shown in Fig. S2c, where the peaks near 530.9, 531.7 and 533.3 eV were correlated with O—Cd, O—H and C=O, respectively [80]. These results demonstrate that Cd-CPs is coordinated by cadmium ions and organic ligands, in which the Cd atom coordinates with N and O atoms. The doublet observed in high-resolution spectrum for the Cd 3d was ascribed to the binding states of Cd 3d5/2 and Cd 3d3/2, respectively. The characteristic peaks at around 411.9 and 405.2 eV are indexed to Cd(Ⅱ) [81], suggesting the existence of Cd(Ⅱ) in Cd-CPs (Fig. S2d).
Meanwhile, The C 1s spectrum of Fe-CPs and Fe-P could be divided into four peaks corresponding to C—C/C=C, C—N, C=N, and C=O. The presence of C—N and C=N verified that N was doped into the polymer (Fig. S3a). The O 1s spectrum could be decomposed into three characteristic peaks: Fe-O (529.5 eV), O—H (531.3 eV) and C=O (533.1). After phosphorylation, the contents of O-Fe decreased (Fig. S3b). The characteristic peaks at both Fe 2p3/2 and Fe 2p1/2 can be decomposed into six peaks, which are located near 710.1, 712.4, 717.9, 723.4, 726.1 and 732.1 eV, respectively. The peaks concerted at 710.1 and 723.4 eV are consistent with the binding energy of Fe(Ⅲ). By contrast, the peaks at 712.4 and 726.1 eV correspond to the binding energy of Fe(Ⅱ), and the peak near 717.9 and 732.1 eV are attributed to satellite peaks of Fe 2p3/2 and Fe 2p1/2 (Fig. S3c), which is caused by slight oxidation of the sample surface [82]. Compared with Fe-CPs, the Fe 2p spectrum has a shift toward higher binding energy in Fe-P, indicating that there is a strong interfacial interaction between Fe and P [83,84]. Additionally, the peak at near 707.1 eV is assigned to Fe-P. The high-resolution P 2p spectrum of Fe-P (Fig. S3d) consists of two major peaks at 133.2, and 129.3 eV belonging to P-O, and Fe-P, respectively, demonstrating the interaction of P atoms with the matrix and Fe phosphorylation [85,86].
The chemical structure of Cd-CPs was ascertained using solid-state 13C NMR spectroscopy and FT-IR. In the solid-state 13C NMR, the presence of two obvious peaks at δ 180 ppm was attributed to the carbon atom of carbonyl group. The peaks around δ 150 ppm correspond to the carbon atom near nitrogen atom in the pyrazole and pyridine unit. Moreover, some peaks in the range of δ 110–140 ppm could be assigned to the aromatic carbons from phenyl rings (Fig. S4 in Supporting information). Again, structural details of Cd-CPs were further confirmed by FT-IR (Fig. S8 in Supporting information), the characteristic absorption band at 3400~3500 cm−1 is attributed to O—H skeleton stretching vibrations of the -OH groups. In additional, the strong absorption bands of Cd-CPs in the ~1700 cm−1 were attributed to the stretching vibration of C=O groups. Besides, we can observe characteristic peaks at ~1400–1500 cm−1 which are attributed to the fundamental vibrations of phenyl rings.
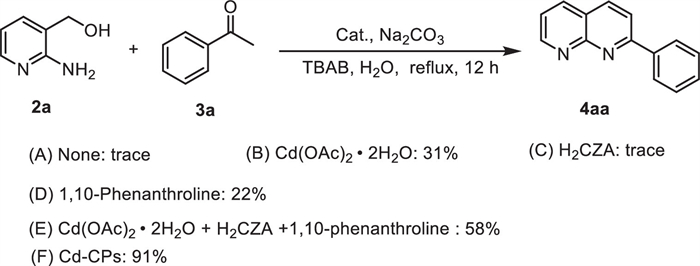
In the initial experiment, we focused on developing a more efficient catalytic system for the dehydrogenation coupling of 2-amino-3-pyridinemethanol (2a) and acetophenone (3a) as a model system (Table 1). The control experiments showed that no reaction occurred in the presence of Cd-CPs and Na2CO3 alone (Table 1, entries 1 and 2). Different bases were then further screened using Cd-CPs as the catalyst in water (Table 1, entries 3–8). As evidenced from the results, the use of Na2CO3 (1.0 equiv.) led to the optimal yield of 1,8-naphthyridine (4aa) in 76%, while other inorganic bases could only afford 1,8-naphthyridine (4aa) in the yield ranging between 49% and 61%. Therefore, in order to further improve the reaction yield, some phase transfer catalysts (TBAB, TBAI and TEAB) were added to this reaction system (Table 1, entries 9–11). To our delight, the yield can be improved significantly when the addition of phase transfer catalysts, with TBAB performing best in 91% yield. Furthermore, the yield of desired product (4aa) has decreased under nitrogen and oxygen atmospheres (Table 1, entries 12 and 13). Only 76% yield of 1,8-naphthyridine (4aa) was obtained using Fe-CPs as catalyst under the same condition (Table 1, entry 14). When Fe-P were used as catalysts for this transformation, no desired product was detected, indicating that the coordination polymer structure has undergone changes after phosphorylation (Table 1, entry 15). Next, decreasing the reaction temperature from 100 ℃ to 80 ℃ leads to a lower yield (69%) of the desired product (Table 1, entries 16 and 17). Moreover, extending or shortening the reaction time (8 or 12 h), the yield of 4aa was decreased slightly, suggesting a saturation point in the reaction kinetics. (Table 1, entries 18 and 19).
 DownLoad:
CSV
DownLoad:
CSV
 |
|||||
| Entry | Catalyst | Additive | Temp (℃) | Time (h) | Yield (%)b |
| 1 | ̶ | Na2CO3 | Reflux | 12 | NR |
| 2 | Cd-CPs | – | Reflux | 12 | NR |
| 3 | Cd-CPs | Et3N | Reflux | 12 | Trace |
| 4 | Cd-CPs | DBU | Reflux | 12 | Trace |
| 5 | Cd-CPs | NaOMe | Reflux | 12 | 61 |
| 6 | Cd-CPs | NaHCO3 | Reflux | 12 | 58 |
| 7 | Cd-CPs | Na2CO3 | Reflux | 12 | 76 |
| 8 | Cd-CPs | K2CO3 | Reflux | 12 | 49 |
| 9c | Cd-CPs | Na2CO3 | Reflux | 12 | 91 |
| 10d | Cd-CPs | Na2CO3 | Reflux | 12 | 85 |
| 11e | Cd-CPs | Na2CO3 | Reflux | 12 | 81 |
| 12c, f | Cd-CPs | Na2CO3 | Reflux | 12 | 83 |
| 13c, g | Cd-CPs | Na2CO3 | Reflux | 12 | 42 |
| 14c | Fe-CPs | Na2CO3 | Reflux | 12 | 76 |
| 15c | Fe-P | Na2CO3 | Reflux | 12 | NR |
| 16 c | Cd-CPs | Na2CO3 | 80 | 12 | 69 |
| 17 c | Cd-CPs | Na2CO3 | 120 | 12 | 88 |
| 18 c | Cd-CPs | Na2CO3 | Reflux | 8 | 76 |
| 19 c | Cd-CPs | Na2CO3 | Reflux | 14 | 90 |
| a Reagents and conditions: 2a (0.5 mmol), 3a (0.75 mmol), additive (1.0 equiv.), catalyst (2 mol% Cd or Fe), H2O (2 mL). b Yields of isolated product. c TBAB (0.5 equiv.). d TBAI (0.5 equiv.). e TEAB (0.5 equiv.). f Oxygen atmosphere. g Nitrogen atmosphere. |
|||||
Having established the reaction conditions, various aryl ketones (3) were subjected to react with 2-amino-3-pyridinemethanol (2a) in order to investigate the reaction scope and the results are summarized in Scheme 4. To our delight, regardless of the identity of electron-donating groups or electron-withdrawing groups attached to the phenyl ring, the acetophenone could couple with 2-amino-3-pyridinemethanol (2a) efficiently and produce desired compounds with good to excellent yields in the presence of Cd-CPs and Na2CO3. Meanwhile, it was found that with electron-withdrawing substituent acetophenone afforded the desired products in higher yields than those with electron-donating substituents. Gratifyingly, the effect of the steric hindrance on this transformation was insignificant. Compared to para-substituted substrates, the desired product obtained by ortho-substituted substrates only decreased slightly (Scheme 4, 4ag, 4am and 4as). Interestingly, an excellent yield could still be maintained by enhancing the electronic effect (Scheme 4, 4av, 4aw). For example, 2′, 5′-dichloroacetophenone and 2′, 4′-difluoroacetophenone were transformed into desired products in 88% and 87% isolated yield, respectively.

Encouraged by the excellent results achieved by aryl ketones, the scope of various ketone derivatives was further investigated (Scheme 5). In general, various aliphatic ketones were smoothly transformed into the desired 1,8-naphthyridines derivatives (4ba−4bc) in good to excellent yield. Additionally, nonmethyl cyclo-ketones (cyclopentanone, cyclohexanone, cycloheptanone, cyclooctanone, α-tetralone, and 1-methyl-4-piperidone) were also well-compatible with this transformation, furnished the desired products (4bd-4bi) in excellent yields (4bd: 89%; 4be: 94%; 4bf: 90%; 4bg: 87%; 4bh: 85%; 4bi: 86%).
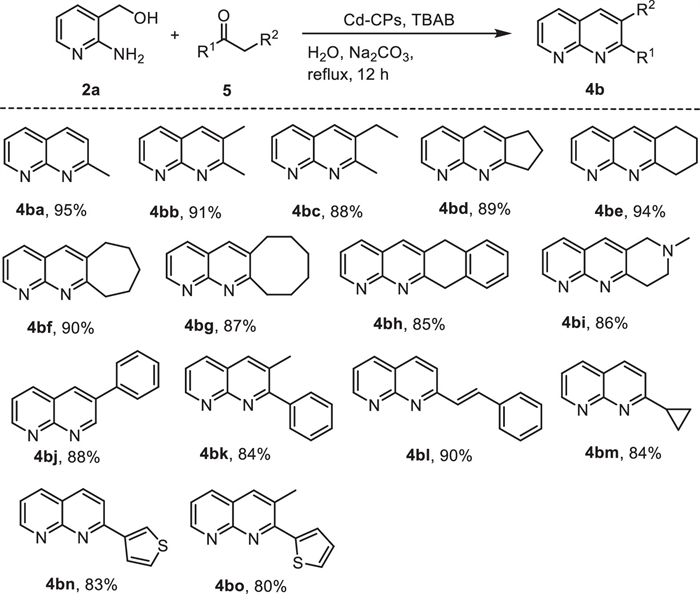
It is worth mentioning that the product 6,7,8,9,10,11-hexahydrocycloocta[b][1,8]naphthyridine (4bg) is a crucial skeleton in drugs, possessing inhibitor activity [87]. Interestingly, the reaction of 2-amino-3-pyridinemethanol (2a) with phenylacetaldehyde (5j) afforded the corresponding product 4bj in 88% isolated yields. Nonmethyl ketones (propiophenone, 5k) was also found to be suitable coupling partners, furnishing the desired 1,8-naphthyridines derivative (4bk) in moderate yield. Moreover, when the substrate contains easily reducible functional group (such as alkene), it also exhibited good reaction efficiency and afforded the corresponding product (4bl, 90%). To further test the substrate scope, heterocyclic ketones were also employed. To our delight, the reaction of cyclopropyl methyl ketone (5m), 3-acetylthiophene (5n) and 2-propionylthiophene (5o) afforded the corresponding product in moderate and good yield.
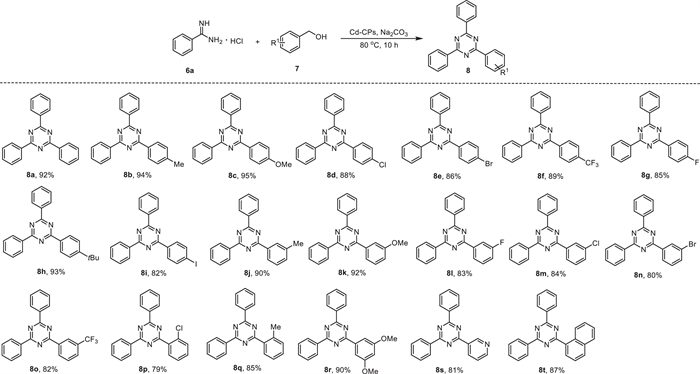
1,3,5-Triazines and its derivatives represent a privileged skeleton in both biologically active pharmaceuticals and natural molecules owing to their multifarious pharmacological properties [88]. Previous reports for the synthesis of 1,3,5-triazine derivatives typically required harsh conditions, such as organic solvents, chemical oxidants, and highly genotoxic alkyl halide as starting materials [89]. Therefore, the development of new catalytic methodologies using sustainable, readily available, and eco-friendly alcohols as reagents under solvent-free conditions has drawn considerable attention [90-92]. Considering such outstanding activities of Cd-CPs, we next explored Cd-CPs to the reaction between benzamidine hydrochloride (6a) and benzyl alcohols (7) under solvent-free conditions, as shown in Scheme 6. Reactions proceeded smoothly under optimized conditions with various substituted benzyl alcohols bearing both electron-donating and electron-withdrawing groups, delivering the corresponding 1,3,5-triazine derivatives in moderate to good yields (8a-8q). Besides, steric hindrance has a slight effect on this transformation. For example, when methyl substituted benzyl alcohols were chosen, para- and meta-substituted substrates gave higher yields than those with ortho-substituted substrates (8b, 8j, and 8q). To our delight, the heteroaryl substituted alcohols also yielded the desired 8s and 8t with 81%, and 87% isolated yields, respectively.
The electrocatalytic activities of the as-prepared catalysts for the OER were subsequently tested in a three-electrode measurement apparatus with 1.0 mol/L KOH as the electrolyte. Linear sweep voltammograms (LSVs) for all the samples were measured as shown in Fig. 4a. The results demonstrated that Fe-CPs exhibited a better OER catalytic activity than Cd-CPs, the overpotential of Fe-CPs and Cd-CPs was 365 and 408 mV at a current density of 10 mA/cm2, respectively, which indicated significant effect of the morphological structure on electrocatalytic OER performances of the catalyst. Then, we chose the Fe-CPs and Cd-CPs to calcine and investigated the electrocatalytic OER activity at 400 ℃, namely, Fe-400 and Cd-400. As displayed in Fig. 4a, in comparison with electrocatalysts Fe-CPs and Cd-CPs, the calcined materials Fe-400 and Cd-400 can greatly increase the catalytic OER performance by reducing the overpotential of 365 and 408 mV to 320 and 345 mV at the same conditions. Furthermore, in situ P-doped samples were obtained by the pyrolysis of NaH2PO2·H2O to improve its electrocatalytic OER performance. To our delight, the overpotential could be further reduced to 279 mV by phosphorylation, which was also significantly better than that of commercial IrO2 (310 mV). Meanwhile, it was about 86 mV less than that of the Fe-CPs catalyst, revealing more efficient intrinsic activity boosting. The dopants of P can regulate the electron environment of catalyst. P-doped carbon could lead to a negative charge density in the C atoms because of the lower electronegativity of P (2.19) compared to C (2.55), which may also regulate the adsorption/desorption and transmission process of the OER-relevant intermediates and products [93-95]. Notably, we found the LSV curve is relatively smooth when the current density reaches over 100 mA/cm². Therefore, we consulted relevant literature and analyzed experimental results to explain this phenomenon, as follows: (1) The electrolyte is stirred during the reaction. (2) The superaerophobicity of the spindle-like Fe-P. To confirm the effect of superaerophobicity on the extraction of bubbles, a control experiment was conducted. When the GCE (with spindle-like Fe-P) were immersed in H2O2 solution, oxygen bubbles quickly evolved from the surface. The tiny oxygen bubbles were released from the electrode surface, which left the surface of sample quickly before they grew larger. In summary, due to the spindle-like structure, the contact between the electrolyte and the electrode is enhanced on the electrode surface, while the superaerophobic surface facilitates bubble extraction and reduces the electrode dead zone caused by bubble coverage [96,97].

In addition, the excellent electrocatalytic activities of the above-mentioned catalysts for OER could be further confirmed by its small Tafel slope, and several results were plotted and analysed in Fig. 4b. The Tafel slope of the spindle-like Fe-P (64.9 mV/dec) is the smallest in comparison with Fe-CPs (98.0 mV/dec), Fe-400 (84.8 mV/dec), Cd-CPs (147.1 mV/dec), Cd-400 (115.2 mV/dec), and Cd-P (91.2 mV/dec). In general, the low Tafel slope value for Fe-P suggested the boosted intrinsic kinetics and optimal electrocatalyst for OER. Moreover, the electrochemically active surface area (ECSA) is a crucial parameter for an electrocatalyst, indicating the ability of the electrocatalyst to provide the active sites for electrochemical reaction. Here, the ECSA was calculated based on the electrochemical double-layer capacitance (Cdl), which can be obtained by the CVs scan curves at different scan rates in a non-faradaic potential region. Fig. S10 (Supporting information) shows the CVs scan curves of Fe-CPs, Fe-400, and Fe-P at different scan rates (10–100 mV/s). As displayed in Fig. 4d, the Cdl values of Fe-CPs, Fe-400, and Fe-P were 0.64, 0.67, and 3.0 mF/cm2, respectively. The Cdl of Fe-P is nearly 4.48 times higher than that of Fe-400 and is about 4.74 times higher than that pristine Fe-CPs, and the significantly increased ECSA of Fe-P is attributed to the introduction of P and the rough surface morphology, which lead to more exposed active sites toward OER. The charge transfer resistance (Rct) can be obtained from the electrochemical impedance spectroscopy (EIS) to test the catalytic kinetics. As shown in Nyquist plots (Fig. 4e), the Rct of Fe-400 was 3.3 Ω, which was the smaller than that of Fe-400 (4.6 Ω), and Fe-CPs (11.7 Ω). Consequently, Fe-P electrode offers faster charge transfer between the electrolyte interface and electrode during the OER process. Finally, the catalytic stability of Fe-P was evaluated by comparing the OER polarization curve of the Fe-P before and after the 1000 cycles. As presented in Fig. 4f, the polarization curve of Fe-P after cycling overlapped well, indicating the excellent stability and practical application prospect of Fe-P. In addition, Table S2 (Supporting information) show a comparison of the overpotential and Tafel slope values of various Fe-based transition metal phosphide electrocatalysts. The results indicate that OER performance of Fe-P is found to be comparable to those of previously reported transition metal phosphide, surpassing the majority of mono-/bimetallic catalysts reported in recent studies.
In order to gain a preliminary understanding of the reaction mechanism, a set of initial verifying experiments were performed in Scheme 7. Firstly, the reaction was not affected in the presence of free radical scavenger such as 2,6-di-tert-butyl-4-methylphenol (BHT), 2,2,6,6-tetramethylpiperidin-1-oxyl (TEMPO), affording the product 4aa in 90% and 88% isolated yields, respectively (Scheme 7a). Therefore, this transformation does not follow a single-electron transfer or free radical pathway.

To better understand the effect of different substituents on the reactivity for this transformation, the control experiment was subsequently performed. When 2a reacted with acetophenone containing electron-donating group (4-tert-butylacetophenone, 3e) or electron-withdrawing group (4-trifluoromethyl acetophenone, 3j) for 2 h under optimized catalytic conditions, the experimental results indicated that the molar ratio of two desired products (4ae vs. 4aj) was observed to be 1:2.4, suggesting that the reaction was more favorable for acetophenone containing electron-withdrawing group (Scheme 7b). This phenomenon may be attributed to the enhanced electrophilicity due to the electron-withdrawing substituent on the phenyl ring of the acetophenones.
Furthermore, it was accepted that the capture of key intermediate was an efficient approach to explain the reaction process. Firstly, 2-amino-3-pyridinecarboxaldehyde (2a’) was formed in 67% isolated yield when 2a went through this reaction in the absence of 3a under standard conditions. Meanwhile, control experiments indicated that both the catalyst and the base were necessary for this transformation, since a significant decrease in the formation of 2a’ (26%, 48% yields respectively) was observed in the absence of Cd-CPs or base (Scheme 7c, (ⅰ)). These experimental observations suggest that the dehydrogenation step catalyzed by Cd-CPs was assisted by Na2CO3, whose Cd sites acted as an active site of dehydrogenation steps of this transformation. Subsequently, the second step of this transformation involves Friedlander condensation reaction of 2a’ with 3a under standard condition to form 4aa in 95% (Scheme 7c, (ⅱ)). Moreover, without Cd-CPs, 75% of desired product 4aa was observed and only 21% of 4aa was obtained in the absence of Na2CO3, demonstrating the crucial role of the base in the Friedlander condensation and cyclization steps (Scheme 7c, (ⅲ)).
To better show the catalytic performence of Cd-CPs, the reaction of 2a and 3a was conducted compared with different metal salt catalysts. As expected, only a trace amount of desired product 4aa was detected when the model reaction was conducted without any catalyst (Scheme 8A). Besides, With the use of a commercially available simple cadmium salts (Cd(OAc)2·2H2O) to replace Cd-CPs as the catalyst, 4aa was isolated in 31% yield (Scheme 8B). Additionally, control experiments were conducted in the presence of only organic linker (H2CZA and 1,10-phenanthroline), and the product 4aa was obtained with low yield. (Schemes 8C and D). Interestingly, it was observed that the mixture of Cd(OAc)2·2H2O and organic linker, affording desired product 4aa in moderate yield (58%) (Scheme 8E). These experiments suggested that the polymer Cd-CPs catalyst has played a significant role during this transformation.
Building upon our above-mentioned experiment results and existing literature [34-36], we proposed a plausible reaction mechanism depicted in Scheme 9. The initial step involved the formation of an alkoxy cadmium intermediate (A) by the reaction of Cd-CPs with 2a and Na2CO3. Accompanied by the β-hydride elimination of alkoxide intermediate complex A, [Cd]-H species and 2-amino-3-pyridinecarboxaldehyde (2a’) were generated. Furthermore, base-promoted condensation between resulting carbonyl compound 2a’ and acetophenone 3a occurred to give ketimine-aldehydes (B), which underwent intramolecular condensation to afford 4aa as desired products. Alternatively, 4aa may also be produced via the intramolecular cyclodehydration of α,β-unsaturated ketones, which are formed by the 2a’ couples with 3a in the presence of base and catalyst. Meanwhile, [Cd-H] released hydrogen gas and was regenerated to complete the catalytic cycle.
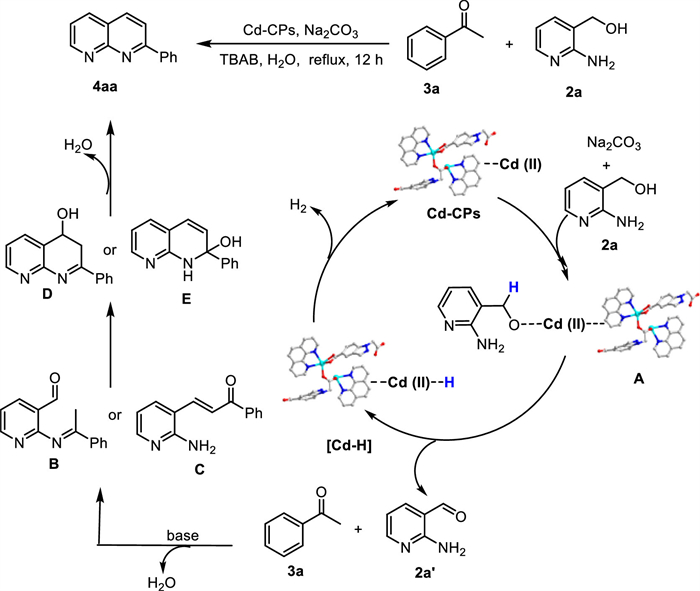
To demonstrate the potential practical industrial applicability of this pathway in organic synthesis, gram-scale reactions and product derivatizations were performed (Scheme 10a). The gram-scale reaction between 2a and 3j under the standard reaction conditions afforded the desired product 4aj (19.8 g) in 89% yield. Subsequently, a biologically active inhibitor 4bg was prepared on a gram scale with 83% yield, indicating the practicability of this catalyst for large-scale synthesis. Additionally, 6a could be coupled with 3-pyridinemethanol (7s), yielding product 8s in 77% yield under solvent-free conditions.
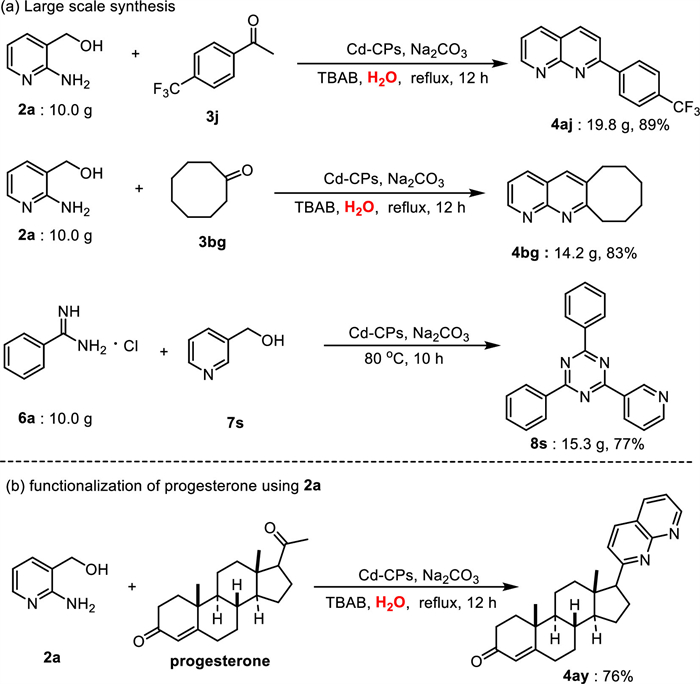
To further confirm the synthetic versatility of this protocol, representative products derivatizations were carried out (Scheme 10b). We applied it to the functionalization of the bioactive steroid progesterone, which possesses carbonyl groups and has a wide range of applications in hormone replacement therapy [98]. Surprisingly, 2a can be smoothly converted to a valuable functional progesterone derivatives 4ay in 76% yield through acceptorless dehydrogenative coupling.
In order to clarify the good performance of Fe-CPs in OER reaction and to identify the active intermediates produced by coordinating polymer Fe-CPs during the OER process, we attempted to analyze the true active species in the OER by increasing the amount of water in the initial reaction system to obtain a single crystal structure. Ultimately, we successfully obtained a new structural compound generated through the reaction of Fe-CPs and excess water (Fig. S11 in Supporting information). The structure of the cultivated single crystal (denoted as Fe–CPs-mono) is a 0 D mononuclear motif, in which the complex contains one coordinated Fe3+ and two H2CZA ligand (CCDC: 2267044). Meanwhile, it was indicated that Fe atom is coordinated with two nitrogen atoms from two H2CZA ligand, and four oxygen atoms from two water molecules and two oxygen atoms from the ligand. Based on the above exploration and literature reports, it is considered that the 0-dimensional structure Fe–CPs-mono may be the active intermediates of Fe-CPs during the OER process [99].
The reusability of Cd-CPs for acceptorless dehydrogenation coupling of 2-amino-3-pyridinemethanol with ketones under water conditions was evaluated by determining the activity after different cycles. Typically, after each cycle, the catalyst was subjected to centrifugation and washed three times with methanol and anhydrous ethanol before being dried 8 h under vacuum at 80 ℃ for subsequent cycle testing. As shown in Fig. 5, the activity of Cd-CPs was maintained at 80% after five rounds, exhibiting excellent reusability for this transformation. This can be attributed to the inherent stability of the nearly spheroidal shape Cd-CPs structure, which boosts the overall catalyst stability.
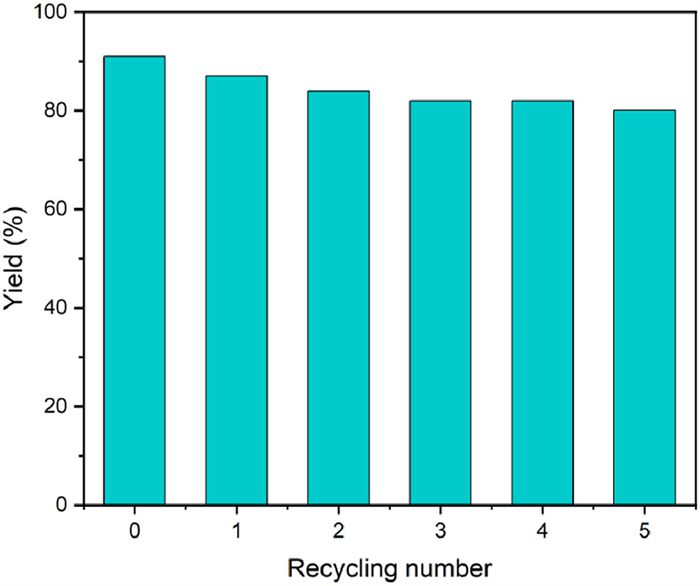
In addition, to verify the structural change of the Cd-CPs, before and after this transformation, the recovered catalyst was characterized (Figs. S5–S7 in Supporting information). The SEM and TEM morphology of the recovered Cd-CPs resembles a nearly spheroidal shape morphology similar to the pristine catalyst. Moreover, abreaction-corrected HAADF-STEM mapping images of recovered Cd-CPs showed that Cd, C, N, and O elements were uniformly dispersed on catalyst surface after five cycles, providing evidence for the catalyst without any significant change in the chemical structure during the reaction. Subsequently, the Cd contents in the Cd-CPs after five rounds were analysed by ICP-AES. The results indicated that the Cd contents of the fresh and recovered catalyst were 3.02 wt% and 2.94 wt%, confirming the good stability of catalyst and that the decrease in catalystic activity is due to the loss of Cd during the reaction.
Subsequently, the structural changes of the catalyst after the reaction via FT-IR, solid 13C NMR, and XPS. Compared with the fresh catalyst, the spent catalyst peak was decreasing with the increase of recycle time, which indicated the fact that the catalyst efficiency gradually decreased when the recycled catalyst was used (Figs. S4 and S8 in Supporting information). Moreover, the catalyst Cd-CPs recovered from the cycle was analyzed by XPS. As shown in Fig. S9 high-resolution C 1s, N 1s, Cd 3d, and O 1s XPS of Cd-CPs, exhibiting the same binding energy as before. These results suggest that the good stability and the potential applicability of Cd-CPs.
In summary, by developing two novel coordination polymers cadmium and iron catalyst, we successfully applied cadmium catalyst to establish an efficient acceptorless dehydrogenation coupling of 2-amino-3-pyridinemethanol with ketones in water, which enables direct construction of 1,8-naphthyridine and their derivatives together with the merits of broad substrate, functionality tolerance and easy applicability for the large-scale synthesis and fabrication of bioactive molecules. Moreover, the developed catalytic transformation features high atom-efficiency, water as the solvent and reusable catalyst, which offers a practical and sustainable pathway for the synthesis of 1,8-naphthyridine derivatives with structural diversity. In addition, we have demonstrated an iron phosphide Fe-P with spindle-like structures derived from the phosphorylation of coordinating polymer Fe-CPs that can serve as an effective candidate catalyst for oxygen evolution reaction. Specifically, Fe-P required a low overpotential, as low as 279 mV, to offer the current density of 10 mA/cm2, and small Tafel slope of 64.9 mV/dec with good stability.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jiahao Li: Writing – original draft, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Bin Pan: Methodology, Investigation, Formal analysis. Anruo Mao: Writing – original draft, Formal analysis, Data curation. Likui Wang: Visualization, Software, Methodology, Formal analysis. Dawei Wang: Writing – review & editing, Software, Resources, Project administration, Funding acquisition, Conceptualization.
We gratefully acknowledge financial support of this work by the National Natural Science Foundation of China (No. 21861039), Postgraduate Research & Practice Innovation Program of Jiangsu Province (No. KYCX24_2530), and the Fundamental Research Funds for the Central Universities. We also thank the characterizations supported by the Central Laboratory, School of Chemical and Material Engineering, Jiangnan University.
Supplementary material associated with this article can be
found, in the online version, at doi:
C.F.H. Allen, Chem. Rev. 47 (1950) 275–305. doi: 10.1021/cr60147a004
H. Jia, Z. Tan, M. Zhang, Acc. Chem. Res. 57 (2024) 795–813. doi: 10.1021/acs.accounts.4c00009
X.Z. Zhao, S.J. Smith, M. Metifiot, et al., J. Med. Chem. 57 (2014) 5190–5202. doi: 10.1021/jm5001908
A.F. Eweas, N.M. Khalifa, N.S. Ismail, M.A. Al-Omar, A.M.M. Soliman, Med. Chem. Res. 23 (2013) 76–86.
M. Ahmed, S.O. Kelley, ACS Chem. Biol. 12 (2017) 2563–2569. doi: 10.1021/acschembio.7b00540
J.M. Fang, S. Selvi, J.H. Liao, et al., J. Am. Chem. Soc. 126 (2004) 3559–3566. doi: 10.1021/ja039237w
S. Deolka, R. Govindarajan, E. Khaskin, et al., Angew. Chem. Int. Ed. 60 (2021) 24620–24629. doi: 10.1002/anie.202109953
V.F. Batista, D.C.G.A. Pinto, A.M.S. Silva, ACS Sustain. Chem. Eng. 4 (2016) 4064–4078. doi: 10.1021/acssuschemeng.6b01010
G. Koller, Ber. Dtsch. Chem. Ges. (A and B Ser.) 60 (2006) 407–410.
J. Marco-Contelles, E. Pérez-Mayoral, A. Samadi, M.C. Carreiras, E. Soriano, Chem. Rev. 109 (2009) 2652–2671. doi: 10.1021/cr800482c
A.K. Panday, R. Mishra, A. Jana, T. Parvin, L.H. Choudhury, J. Org. Chem. 83 (2018) 3624–3632. doi: 10.1021/acs.joc.7b03272
A. Das, N. Anbu, P. Varalakshmi, A. Dhakshinamoorthy, S. Biswas, New J. Chem. 44 (2020) 10982–10988. doi: 10.1039/d0nj01891k
S.W. Maurya, H. Sagir, M.D. Ansari, I.R. Siddiqui, ChemistrySelect 6 (2021) 13601–13608. doi: 10.1002/slct.202102127
B. Li, S. Nguyen, J. Huang, et al., Tetrahedron Lett. 57 (2016) 1958–1962. doi: 10.1016/j.tetlet.2016.03.070
E.C. Anderson, H.F. Sneddon, C.J. Hayes, Green Chem. 21 (2019) 3050–3058. doi: 10.1039/c9gc00408d
S.S. Choudhury, S. Jena, D.K. Sahoo, et al., ACS Omega 6 (2021) 19304–19313. doi: 10.1021/acsomega.1c02798
Y.Y. Liu, Y. Wei, Z.H. Huang, Y. Liu, Org. Biomol. Chem. 19 (2021) 659–666. doi: 10.1039/d0ob02348e
A.E.F. Denjean, A. Nova, D. Balcells, ACS Catal. 14 (2024) 11332–11342. doi: 10.1021/acscatal.4c02182
Y. Liu, P. Ji, G. Zou, et al., Angew. Chem. Int. Ed. 63 (2024) e202410351. doi: 10.1002/anie.202410351
S. Mullick, A. Ghosh, D. Banerjee, Chem. Commun. 60 (2024) 4002–4014. doi: 10.1039/d4cc00003j
A. Alexandridis, T. Rancon, A. Halliday, A. Kochem, A. Quintard, Org. Lett. 26 (2024) 5788–5793. doi: 10.1021/acs.orglett.4c01969
A. Samanta, A. Chaubey, D. Pal, K. Majhi, D. Srimani, Chem. Commun. 60 (2024) 10398–10401. doi: 10.1039/D4CC03407D
B. Zhu, H. Tian, Z. Zhang, et al., J. Catal. 435 (2024) 115569. doi: 10.1016/j.jcat.2024.115569
Y. Gao, G. Hong, Y. Zhao, Chem. Soc. Rev. 52 (2023) 5541–5562. doi: 10.1039/d3cs00424d
Y. Liu, H. Diao, G. Hong, et al., J. Am. Chem. Soc. 145 (2023) 5007–5016. doi: 10.1021/jacs.2c09958
K. Yu, Q. Nie, Q. Chen, W. Liu, Nat. Commun. 15 (2024) 6798. doi: 10.1038/s41467-024-51188-x
M. Subaramanian, C. Gouda, T.K. Roy, et al., ACS Catal. 14 (2024) 8294–8309. doi: 10.1021/acscatal.3c06091
Z. Ma, Z. Wu, C. Kreyenschulte, et al., Green Chem. 26 (2024) 11140–11146. doi: 10.1039/d3gc04436j
N. Wang, Y. Li, Z. Chen, C. Zhao, Z. Ke, ACS Catal. (2024) 18032–18044. doi: 10.1021/acscatal.4c05200
S. Das, R. Mondal, G. Chakraborty, et al., ACS Catal. 11 (2021) 7498–7512. doi: 10.1021/acscatal.1c00275
J.L. Sun, H. Jiang, P.H. Dixneuf, M. Zhang, et al., J. Am. Chem. Soc. 146 (2024) 11289–11298.
Q. Liu, C. Ci, H. Zhao, et al., Green Chem. 25 (2023) 678–683. doi: 10.1039/d2gc03878a
M. Maji, K. Chakrabarti, D. Panja, S. Kundu, J. Catal. 373 (2019) 93–102. doi: 10.1016/j.jcat.2019.03.028
C. Chaudhari, K. Sato, Y. Ogura, S.I. Miayahara, K. Nagaoka, ChemCatChem 12 (2020) 2198–2202. doi: 10.1002/cctc.201902311
X. Chen, Y. Ai, D. Liu, et al., Mater. Chem. Front. 6 (2022) 1228–1235. doi: 10.1039/d2qm00077f
K. Patra, A. Bhattacherya, C. Li, J.K. Bera, H.S. Soo, ACS Catal. 12 (2022) 15168–15180. doi: 10.1021/acscatal.2c05086
D. Pathak, B.K. Kalita, A. Sarmah, et al., Green Chem. 25 (2023) 7642–7652. doi: 10.1039/d3gc02172f
T. Nakayama, S. Fujiki, T. Enda, et al., Org. Biomol. Chem. 22 (2024) 759–766. doi: 10.1039/d3ob01815f
J.Q. Liu, Z.D. Luo, Y. Pan, et al., Coord. Chem. Rev. 406 (2020) 213145. doi: 10.1016/j.ccr.2019.213145
Z. Liu, S. Liu, B. Liu, et al., Chin. Chem. Lett. 35 (2024) 109626. doi: 10.1016/j.cclet.2024.109626
L. Lu, Q. Li, J. Du, W. Shi, P. Cheng, Chin. Chem. Lett. 33 (2022) 2928–2932. doi: 10.1016/j.cclet.2021.10.090
J.A. Johnson, X. Zhang, T.C. Reeson, Y.S. Chen, J. Zhang, J. Am. Chem. Soc. 136 (2014) 15881–15884. doi: 10.1021/ja5092672
S. Khan, D. Markad, S.K. Mandal, Inorg. Chem. 62 (2023) 275–284. doi: 10.1021/acs.inorgchem.2c03369
A. Bavykina, N. Kolobov, I.S. Khan, et al., Chem. Rev. 120 (2020) 8468–8535. doi: 10.1021/acs.chemrev.9b00685
K. Wang, X.L. Lv, D. Feng, et al., J. Am. Chem. Soc. 138 (2016) 914–919. doi: 10.1021/jacs.5b10881
H.L. Jiang, D. Feng, K. Wang, et al., J. Am. Chem. Soc. 135 (2013) 13934–13938. doi: 10.1021/ja406844r
W. Jiang, J. Yang, Y.Y. Liu, S.Y. Song, J.F. Ma, Inorg. Chem. 56 (2017) 3036–3043. doi: 10.1021/acs.inorgchem.6b03174
J. Zhang, J. Wu, G. Tang, et al., Sens. Actuators B: Chem. 272 (2018) 166–174. doi: 10.1016/j.snb.2018.05.121
K. Liu, W. Shi, P. Cheng, Dalton Trans. 40 (2011) 8475–8490. doi: 10.1039/c0dt01578d
J. Li, W. Zeng, L. Wang, G. Shi, D. Wang, Chem. Eng. J. 474 (2023) 145642. doi: 10.1016/j.cej.2023.145642
J. Li, A. Mao, W. Yao, H. Zhu, D. Wang, Green Chem. 24 (2022) 2602–2612. doi: 10.1039/d2gc00190j
W. Hu, Y. Zhang, H. Zhu, D. Ye, D. Wang, Green Chem. 21 (2019) 5345–5351. doi: 10.1039/c9gc02086a
W. Yao, Y. Zhang, H. Zhu, C. Ge, D. Wang, Chin. Chem. Lett. 31 (2020) 701–705. doi: 10.1016/j.cclet.2019.08.049
J. Li, L. Chen, L. Wang, G. Shi, D. Wang, J. Catal. 429 (2024) 115205. doi: 10.1016/j.jcat.2023.115205
G. Zhu, Z.C. Duan, H. Zhu, D. Ye, D. Wang, Chin. Chem. Lett. 33 (2022) 266–270. doi: 10.1016/j.cclet.2021.06.060
J. Fu, Z. Tang, X. Feng, Y. Wen, Sci. China Chem. 53 (2010) 1060–1067. doi: 10.1007/s11426-010-0052-9
J. Li, A. Mao, X. Hu, et al., Dalton Trans. 53 (2024) 5064–5072. doi: 10.1039/d3dt04221a
J. Li, Q. Zhou, C. Zhong, et al., ACS Catal. 9 (2019) 3878–3887. doi: 10.1021/acscatal.9b00293
Y. Li, C. Zhao, ACS Catal. 7 (2017) 2535–2541. doi: 10.1021/acscatal.6b03497
T. Zhai, L. Wan, S. Sun, et al., Adv. Mater. 29 (2017) 1604167. doi: 10.1002/adma.201604167
X. Zhang, M.I. Alvarado-Ávila, Y. Liu, et al., Electrochim. Acta 438 (2023) 141582. doi: 10.1016/j.electacta.2022.141582
Z. Huang, C. Lv, Z. Chen, et al., Nano Energy 12 (2015) 666–674. doi: 10.1016/j.nanoen.2015.01.027
P. Li, W. Li, R. Chen, Y. Lin, ACS Sustain. Chem. Eng. 8 (2020) 9206–9216. doi: 10.1021/acssuschemeng.0c03333
T. Wu, H. Hu, Y. Wu, et al., Chem. Eng. J. 500 (2024) 157023. doi: 10.1016/j.cej.2024.157023
Y. Jia, X. Yao, Acc. Chem. Res. 56 (2023) 948–958. doi: 10.1021/acs.accounts.2c00809
Y. Zhao, H. Wang, J. Li, et al., Adv. Funct. Mater. 33 (2023) 2305268. doi: 10.1002/adfm.202305268
W. Li, C. Wang, X. Lu, Coord. Chem. Rev. 464 (2022) 214555. doi: 10.1016/j.ccr.2022.214555
L. Peng, J. Yang, Y. Yang, et al., Adv. Mater. 34 (2022) e2202544. doi: 10.1002/adma.202202544
G. Cai, P. Yan, L. Zhang, H.C. Zhou, H.L. Jiang, Chem. Rev. 121 (2021) 12278–12326. doi: 10.1021/acs.chemrev.1c00243
Q. Huang, A. Zeb, Z. Xu, et al., Coord. Chem. Rev. 494 (2023) 215335. doi: 10.1016/j.ccr.2023.215335
Z.W. Seh, J. Kibsgaard, C.F. Dickens, et al., Science 355 (2017) eaad4998. doi: 10.1126/science.aad4998
J. Jiang, C. Zhang, L. Ai, Electrochim. Acta 208 (2016) 17–24. doi: 10.1016/j.electacta.2016.05.008
S. Swathi, R. Yuvakkumar, P. Senthil Kumar, et al., Fuel 308 (2022) 122051. doi: 10.1016/j.fuel.2021.122051
T. Zhai, H. Niu, Y. Yan, et al., J. Alloys Compd. 853 (2021) 157353. doi: 10.1016/j.jallcom.2020.157353
C. Lin, X. He, H. Li, et al., CrystEngComm 23 (2021) 7090–7096. doi: 10.1039/d1ce01015h
J. Gu, J. He, H. Zheng, C. Sun, New J. Chem. 47 (2023) 8507–8514. doi: 10.1039/d3nj00887h
J. Xu, S. He, H. Zhang, et al., J. Mater. Chem. A 3 (2015) 24261–24271. doi: 10.1039/C5TA06838J
D.Y. Osadchii, A.I. Olivos-Suarez, A.V. Bavykina, J. Gascon, Langmuir 33 (2017) 14278–14285. doi: 10.1021/acs.langmuir.7b02929
X. Liang, S. Wang, J. Feng, et al., Inorg. Chem. Front. 10 (2023) 2961–2977. doi: 10.1039/d2qi02436e
M. Zhang, K. Yang, J. Cui, et al., Chem. Eng. J. 386 (2020) 124023. doi: 10.1016/j.cej.2020.124023
H. Li, Y. Chen, H. Zhao, et al., J. Mater. Chem. C 12 (2024) 3141–3153. doi: 10.1039/d3tc03775d
H. Zhang, C. Zhou, H. Zeng, et al., Process Saf. Environ. Prot. 166 (2022) 11–22.
H. Zhang, A. Chen, Z. Bi, et al., ACS Nano 17 (2023) 24070–24079. doi: 10.1021/acsnano.3c09020
J. Nie, J. Shi, T. Huang, et al., Adv. Funct. Mater. 34 (2024) 2314172. doi: 10.1002/adfm.202314172
E. Zhu, C. Shi, J. Yu, et al., Appl. Catal. B: Environ. Energy 347 (2024) 123796. doi: 10.1016/j.apcatb.2024.123796
L. Long, X. Wang, H. Fu, et al., ACS Appl. Mater. Interfaces 16 (2024) 21838–21848. doi: 10.1021/acsami.4c00911
C. de Los Rios, J. Egea, J. Marco-Contelles, et al., J. Med. Chem. 53 (2010) 5129–5143. doi: 10.1021/jm901902w
R. Menicagli, S. Samaritani, G. Signore, F. Vaglini, L.D. Via, J. Med. Chem. 47 (2004) 4649–4652. doi: 10.1021/jm0495374
A.L. Isfahani, I. Mohammadpoor-Baltork, V. Mirkhani, et al., Adv. Synth. Catal. 355 (2013) 957–972. doi: 10.1002/adsc.201200707
S.S. Poly, Y. Hashiguchi, I. Nakamura, T. Fujitani, S.M.A.H. Siddiki, Catal. Sci. Technol. 12 (2022) 4679–4687. doi: 10.1039/d2cy00426g
F. Wang, Z. Deng, Y. Wang, et al., Tetrahedron 123 (2022) 132985. doi: 10.1016/j.tet.2022.132985
Q. You, F. Wang, C. Wu, et al., Org. Biomol. Chem. 13 (2015) 6723–6727. doi: 10.1039/C5OB00724K
Z.W. Liu, F. Peng, H.J. Wang, et al., Angew. Chem. Int. Ed. 50 (2011) 3257–3261. doi: 10.1002/anie.201006768
L. Peng, S.S.A. Shah, Z. Wei, Chin. J. Catal. 39 (2018) 1575–1593. doi: 10.1016/S1872-2067(18)63130-4
W. Gong, H. Zhang, L. Yang, et al., J. Ind. Eng. Chem. 106 (2022) 492–502. doi: 10.1016/j.jiec.2021.11.032
X. Yu, Z.Y. Yu, X.L. Zhang, et al., J. Am. Chem. Soc. 141 (2019) 7537–7543. doi: 10.1021/jacs.9b02527
M. Bae, Y. Kang, D.W. Lee, D. Jeon, J. Ryu, Adv. Energy Mater. 12 (2022) 2201452. doi: 10.1002/aenm.202201452
O. Robles, D. Romo, Nat. Prod. Rep. 31 (2014) 318–334. doi: 10.1039/C3NP70087A
C.P. Li, J. Chen, C.S. Liu, M. Du, Chem. Commun. 51 (2015) 2768–2781. doi: 10.1039/C4CC06263A

Figure 1 (a) 1-D chain-like architecture of Cd-CPs along the a-axis. (b) 1-D chain-like architecture of Cd-CPs along the c-axis (C: gray, N: blue, O: red, Cd: cyan, and all H atoms are omitted for clarity).

Figure 3 (a, b) SEM images of the Cd-CPs. (c, d) SEM images of the Fe-CPs. (e, f) SEM images of the Fe-P. (g, h) TEM images of the Cd-CPs. (i, j) TEM images of the Fe-CPs. (k, l) TEM images of the Fe-P. (m) HAADF-STEM image and EDS elemental mappings of Cd-CPs. (n) HAADF-STEM image and EDS elemental mappings of Fe-P. (o) HRTEM images of Fe-P.

Scheme 4 Dehydrogenation coupling of 2-amino-3-pyridinemethanol (2a) with various aryl ketones (3). Reagents and conditions: 2a (0.5 mmol), 3 (0.75 mmol), Na2CO3 (1.0 equiv.), Cd-CPs (2 mol% Cd), H2O (2 mL), reflux, 12 h. Yields of isolated product.

Scheme 5 Dehydrogenation coupling of 2-amino-3-pyridinemethanol (2a) with various ketones (5). Reagents and conditions: 2a (0.5 mmol), 5 (0.75 mmol), Na2CO3 (1.0 equiv.), Cd-CPs (2 mol% Cd), H2O (2 mL), reflux, 12 h. Yields of isolated product.

Scheme 6 Substrate scope for various 1,3,5-triazines. Reagents and conditions: 6a (0.5 mmol), 7 (1.0 mmol), Na2CO3 (0.75 equiv.), Cd-CPs (2 mol% Cd), 80 ℃, 10 h. Yields of isolated product.

Figure 4 The OER catalytic performance of Fe-P, Fe-400, Fe-CPs, Cd-P, Cd-400, Cd-CPs and IrO2 in 1.0 mol/L KOH. (a) LSV curves. (b) Tafel plots fitted from the corresponding LSV curves. (c) Comparison of the overpotential (η) and the Tafel slopes at 10 mA/cm2 of Fe-P (Ⅰ), Fe-400 (Ⅱ), Fe-CPs (Ⅲ), Cd-P (Ⅳ), Cd-400 (Ⅴ), Cd-CPs (Ⅵ), and IrO2 (Ⅶ) respectively. (d) Cdl values of different samples. (e) The Nyquist diagram for Fe-P, Fe-400, and Fe-CPs respectively. (f) LSV curve of Fe-P after 1000 cycles.
Table 1. Optimization of reaction conditions.a
|
|||||
| Entry | Catalyst | Additive | Temp (℃) | Time (h) | Yield (%)b |
| 1 | ̶ | Na2CO3 | Reflux | 12 | NR |
| 2 | Cd-CPs | – | Reflux | 12 | NR |
| 3 | Cd-CPs | Et3N | Reflux | 12 | Trace |
| 4 | Cd-CPs | DBU | Reflux | 12 | Trace |
| 5 | Cd-CPs | NaOMe | Reflux | 12 | 61 |
| 6 | Cd-CPs | NaHCO3 | Reflux | 12 | 58 |
| 7 | Cd-CPs | Na2CO3 | Reflux | 12 | 76 |
| 8 | Cd-CPs | K2CO3 | Reflux | 12 | 49 |
| 9c | Cd-CPs | Na2CO3 | Reflux | 12 | 91 |
| 10d | Cd-CPs | Na2CO3 | Reflux | 12 | 85 |
| 11e | Cd-CPs | Na2CO3 | Reflux | 12 | 81 |
| 12c, f | Cd-CPs | Na2CO3 | Reflux | 12 | 83 |
| 13c, g | Cd-CPs | Na2CO3 | Reflux | 12 | 42 |
| 14c | Fe-CPs | Na2CO3 | Reflux | 12 | 76 |
| 15c | Fe-P | Na2CO3 | Reflux | 12 | NR |
| 16 c | Cd-CPs | Na2CO3 | 80 | 12 | 69 |
| 17 c | Cd-CPs | Na2CO3 | 120 | 12 | 88 |
| 18 c | Cd-CPs | Na2CO3 | Reflux | 8 | 76 |
| 19 c | Cd-CPs | Na2CO3 | Reflux | 14 | 90 |
| a Reagents and conditions: 2a (0.5 mmol), 3a (0.75 mmol), additive (1.0 equiv.), catalyst (2 mol% Cd or Fe), H2O (2 mL). b Yields of isolated product. c TBAB (0.5 equiv.). d TBAI (0.5 equiv.). e TEAB (0.5 equiv.). f Oxygen atmosphere. g Nitrogen atmosphere. |
|||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们









 下载:
下载: