Guangxi Key Laboratory of Low Carbon Energy Materials, School of Chemistry and Pharmaceutical Sciences, Guangxi Normal University, Guilin 541004, China
b.
Hebei Center for Industrial Energy-saving and Pollution Control Research, Hebei Vocational University of Technology and Engineering, Xingtai 054000, China
c.
Saudi Arabia Basic Industries Corporation (SABIC) at King Abdullah University of Science and Technology (KAUST), Thuwal 23955-6900, Saudi Arabia
Received Date:
11 January 2025 Accepted Date:
26 March 2025 Revised Date:
23 March 2025 Available Online:
15 September 2025
Abstract:
Amorphous bimetallic borides, as a new generation of catalytic nanomaterials with modifiable electronic properties, are of great importance in the design of high-efficiency catalysts for NaBH4 hydrolysis. This study synthesizes an amorphous Co3B-Mo2B5 catalyst using a self-sacrificial template strategy and NaBH4 reduction for both NaBH4 hydrolysis and the reduction of 4-nitrophenol. The catalyst delivers an impressive hydrogen generation rate of 7690.5 mL min−1 g−1 at 25 ℃, coupled with a rapid reaction rate of 0.701 min−1 in the reduction of 4-nitrophenol. The enhanced catalytic performance is attributed to the unique amorphous structure and the electron rearrangement between Co3B and Mo2B5. Experimental and theoretical analyses suggest electron transfer from Co3B to the Mo2B5, with the electron-deficient Co3B site favoring BH4− adsorption, while the electron-rich Mo2B5 site favoring H2O adsorption. Furthermore, Co3B-Mo2B5 demonstrated potential for energy applications, delivering a power output of 0.3 V in a hydrogen-air fuel cell.
The rapid depletion of fossil fuels has accelerated the advancement of clean energy conversion and hydrogen storage technologies [1]. Hydrogen is widely regarded as an efficient and environmentally friendly energy carrier, with a remarkable gravimetric energy density of 120 kJ/g, significantly surpassing that of petroleum (44 kJ/g) [2]. One of the key applications of hydrogen is in fuel cells, which serve as an effective medium for converting renewable energy into electricity [3]. However, hydrogen storage remains a significant bottleneck in the hydrogen energy value chain [4]. Among various storage methods, solid-state media, such as adsorbents and chemical hydrides, are considered the safest and most effective options [5]. In particular, sodium borohydride (NaBH4) has garnered attention as a promising hydrogen storage material, boasting a hydrogen content of 10.6 wt% [6]. Its potential is further enhanced by possibly regenerating the hydrolysis by-product, NaBO2, into NaBH4 through a rapid ball milling process [7]. While NaBH4 aqueous solutions can release substantial amounts of hydrogen, the self-hydrolysis reaction is relatively slow, which limits its practical applications. Therefore, there is an urgent need for highly efficient catalysts to accelerate the hydrolysis of NaBH4 and facilitate rapid hydrogen release [8,9].
Traditional catalysts for borohydride hydrolysis predominantly rely on precious metals, such as palladium (Pd) [10], platinum (Pt) [11] and ruthenium (Ru) [12]. However, these metals' high cost and limited availability have driven research into non-precious metal alternatives [13]. Cobalt-molybdenum (Co-Mo) bimetallic borides have emerged as promising candidates due to their excellent catalytic performance. Notable examples include Co-Mo-B@TiO2 [14], g-C3N4/Co-Mo-B/Ni [15], Co-Mo-B/CC-F [16], and Co-Mo-B/NF [17]. Despite their good catalytic activity for sodium borohydride hydrolysis, several challenges persist. Conventional hard-template carriers, such as TiO2, nickel foams, and carbon cloth, often suffer from low active site dispersion due to structural limitations, which restrict their catalytic efficiency. To overcome these challenges, the self-sacrificial template strategy has emerged as a practical approach [18]. This strategy enhances the active site density and induces electronic rearrangement in Co-Mo bimetallic boride catalysts, thereby improving their catalytic performance. Recent studies have also highlighted the advantages of amorphous catalysts, which feature flexible structures and abundant structural defects [19]. These characteristics facilitate efficient electron transfer between active sites and improve the catalysts' stability and resistance to deactivation. When combined with Co-Mo bimetallic boride catalysts, amorphous structures can significantly enhance the efficiency of sodium borohydride hydrolysis.
In addition to hydrogen storage applications, Co-Mo bimetallic boride catalysts have shown significant potential in reducing organic pollutants, such as 4-nitrophenol (4-NP), a common industrial contaminant. The reduction of 4-NP to 4-aminophenol (4-AP) is significant due to the widespread use of 4-NP in industrial processes, particularly in dye synthesis and as a precursor in pharmaceutical production [20]. This catalytic reduction involves hydrogen transfer, making it a promising reaction for hydrogenation. As a hydrogen donor, NaBH4 aligns well with this application [21]. The efficiency of this process heavily depends on the catalyst used, and Co-Mo bimetallic borides exhibit outstanding catalytic properties. These catalysts facilitate the hydrogenation of 4-NP and enhance overall reaction kinetics, making them attractive candidates for large-scale applications in both clean energy and environmental remediation.
In this study, we designed an amorphous Co3B-Mo2B5 catalyst using a Mo-MOF self-sacrificial template to improve the efficiency of NaBH4 hydrolysis and the catalytic reduction of 4-NP to 4-AP. Employing a comprehensive suite of characterization techniques, including XRD, SEM, BET, zeta potential analysis, and XPS, we examined the catalyst components' crystallinity, microstructure, porosity, and chemical states. As anticipated, the amorphous Co3B-Mo2B5 catalyst outperformed many noble metal catalysts, achieving highly efficient NaBH4 hydrolysis and reducing 4-NP to 4-AP in 420 s. Furthermore, as reported in previous literature, we proposed a plausible reaction mechanism based on the Michaelis-Menten model.
We synthesized Co3B-Mo2B5 catalysts using a self-sacrificial template strategy combined with a simple NaBH4 reduction technique, as illustrated in Fig. 1a. Initially, Mo-MOF was prepared via a hydrothermal method. This Mo-MOF was subsequently used as a substrate, onto which CoCl2·6H2O has uniformly loaded; subsequently, this Mo-MOF was the Co2+/Mo-MOF composite. Finally, the Co2+/Mo-MOF precursor reacted with NaBH4 in the presence of urea, resulting in boride. Co3B and Mo2B5 form a stable composite catalyst through intermetallic chemical bonds in this synthesis process. Scanning electron microscopy (SEM) analysis revealed that the Mo-MOF sample exhibited a smooth, rod-like morphology (Fig. 1b). After impregnation with a cobalt chloride solution, cobalt salt was visible on the surface of the Mo-MOF (Fig. 1c). Following treatment with NaBH4, the Co2+/Mo-MOF precursor transformed into a 3D porous nanofoam structure characterized by interconnected microchannels (Fig. 1d). Figs. S1a and b (Supporting information) show that Mo2B5 and Co3B exhibit irregular nano-aggregates. Transmission electron microscopy (TEM) analysis further confirmed a comparable nanoparticle-like morphology (Fig. 1e). High-resolution TEM (HR-TEM) images revealed a disordered atomic arrangement with no distinct lattice fringes (Fig. 1f), verifying the amorphous nature of Co3B-Mo2B5. Additionally, the selected area electron diffraction (SAED) pattern displayed a diffuse, continuous halo ring (inset in Fig. 1f), further supporting the amorphous structure [22]. Energy-dispersive X-ray spectroscopy (EDS) and elemental mapping demonstrated the homogeneous distribution of Co, Mo, and B elements throughout the Co3B-Mo2B5 catalyst (Fig. 1g and Fig. S2 in Supporting information).
Figure 1
Figure 1.
(a) Schematic protocol of the synthesis strategy for Co3B-Mo2B5. SEM images of (b) Mo-MOF, (c) Co2+/Mo-MOF, and (d) Co3B-Mo2B5. (e) TEM and (f) HR-TEM of Co3B-Mo2B5. (g) HAADF-TEM image and elemental mappings of Co3B-Mo2B5 (Co, Mo and B).
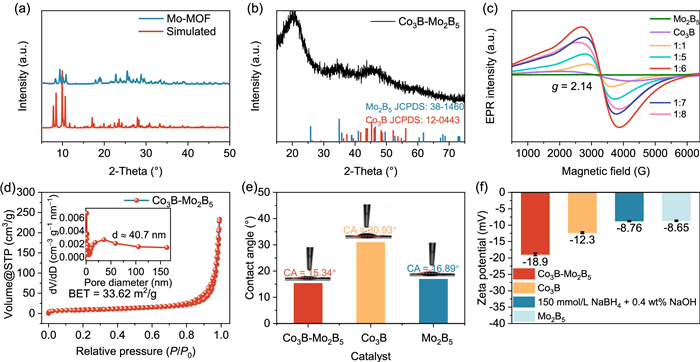
The structure of the catalyst was analyzed using X-ray powder diffraction (XRD). As shown in Fig. 2a, the pristine Mo-MOF exhibits four primary diffraction peaks at approximately 8.39°, 9.32°, 10.70°, and 19.06°, which correspond closely to the theoretical diffraction patterns predicted for Mo-MOF [23]. In contrast, Fig. 2b displays faint peaks corresponding to Co3B (JCPDS No. 12–0443) and Mo2B5 (JCPDS No. 38–1460), confirming the amorphous or low-crystalline nature of the Co3B-Mo2B5 compound. This observation is consistent with the HR-TEM results [24]. Furthermore, Figs. S3a and b (Supporting information) show that the prepared Co3B and Mo2B5 catalysts do not perfectly align with the standard reference spectra. This discrepancy is attributed to the significant expansion of the material during the sodium borohydride reduction reaction, leading to low crystallinity and the absence of distinct diffraction peaks in the XRD patterns [25]. Additionally, the strong absorption peak near 26° in Fig. S3a corresponds to amorphous carbon [26]. Cobalt defects in the samples were further analyzed using electron paramagnetic resonance (EPR), as shown in Fig. 2c. Co3B and Co3B-Mo2B5 exhibit nearly symmetric EPR signals centered around g = 2.14, while Mo2B5 shows no such signal. This indicates the presence of cobalt defects, which trap unpaired electrons [27]. The increased defect density is believed to enhance the synergistic interaction between the components, thereby improving catalytic performance [28]. The N2 adsorption-desorption isotherms in Fig. 2d reveal that Co3B-Mo2B5 has a Brunauer–Emmett–Teller (BET) surface area of 33.62 m2/g, which facilitates the exposure of active sites. The corresponding pore size distribution indicates that Co3B-Mo2B5 is a mesoporous material. This large specific surface area and hierarchical mesoporous structure provide numerous active adsorption sites, promoting catalytic efficiency [29]. To evaluate the influence of hydroxyl groups on the surface of Co3B-Mo2B5, contact angle (CA) measurements were performed. The results show contact angles of 15.34°, 30.93°, and 16.8° for Co3B-Mo2B5, Co3B, and Mo2B5, respectively (Fig. 2e). The increased hydrophilicity of Co3B-Mo2B5 is attributed to the incorporation of molybdenum, which enhances reactivity by improving interactions between the catalyst and hydroxyl groups [30,31]. Finally, zeta potential measurements were conducted to examine the effects of a mixed solution (150 mmol/L NaBH4 + 0.4 wt% NaOH) on the surface potentials of the catalysts shown in Fig. 2f, Co3B-Mo2B5 exhibited optimized adsorption of OH−/ BH4−, with a higher negative potential of −18.9 mV than Co3B and Mo2B5. This indicates more significant negative charge adsorption, contributing to its enhanced catalytic activity [32].
Figure 2
Figure 2.
XRD patterns of (a) Mo-MOF and (b) Co3B-Mo2B5. (c) EPR spectra of Co3B-Mo2B5, Co3B, and Mo2B5. (d) N2 adsorption-desorption isotherm with the inset showing the corresponding pore size distribution of Co3B-Mo2B5. (e) Bubble contact angle images of different catalysts. (f) Zeta potentials of the Co3B-Mo2B5, Co3B, Mo2B5 and a 150 mmol/L NaBH4 + 0.4 wt% NaOH solution.
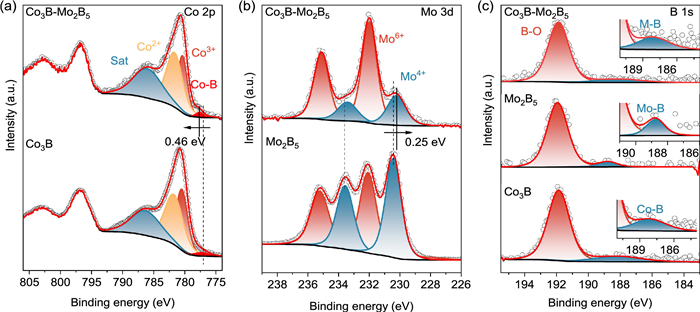
X-ray photoelectron spectroscopy (XPS) was utilized to analyze the elemental composition and chemical valence states on the surface of the catalysts. The survey spectrum (Fig. S4 in Supporting information) confirmed the presence of Co, Mo, and B elements, consistent with the results from elemental mapping. In the high-resolution C 1s spectra of Co3B-Mo2B5, Co3B, and Mo2B5, three distinct peaks were observed: C–C/C═C at 284.80 eV, C–O/C–N at 286.0 eV, and C═O at 288.88 eV. These peaks were the calibration standard for other XPS spectra (Figs. S5a–c in Supporting information) [33]. The high-resolution Co 2p XPS spectra of Co3B-Mo2B5 and Co3B, shown in Fig. 3a, were deconvoluted into Co–B (777.59 eV), Co3+ (780.37 eV), Co2+ (781.64 eV), and satellite peaks [34]. In the Mo 3d spectra of Co3B-Mo2B5 and Mo2B5 (Fig. 3b), two Mo 3d5/2−3d3/2 spin-orbit doublets were observed at 230.23/233.43 eV and 231.98/235.12 eV, corresponding to Mo4+ and Mo6+, respectively [35,36]. Notably, the binding energies of Co–B and Mo4+ in Co3B-Mo2B5 shifted positively by 0.46 eV and negatively by 0.25 eV, respectively, compared to Co3B and Mo2B5. This suggests electron transfer from the Co3B to the Mo2B5, with the electron-lack Co3B site favoring BH4− adsorption [37]. The B 1s XPS spectra exhibited B–O bonds at 191.89 eV for all three materials. Additionally, distinct peaks at 188.07, 188.74, and 188.29 eV corresponded to the metal−B (M-B) bonds in Co3B-Mo2B5, Mo2B5, and Co3B, respectively (Fig. 3c) [38]. In Co3B-Mo2B5, the peak at 188.07 eV in the B 1s spectrum, attributed to M–B (M = Co, Mo), was shifted positively by approximately 1.0 eV compared to elemental B (187.0 eV). This shift suggests electron transfer from alloyed B to the vacant d-orbitals of metallic Co and Mo, resulting in electron deficiency at B and electron enrichment at Co/Mo [39]. These findings highlight the strong interactions between the Co3B and Mo2B5 components within the catalyst. These interactions lead to variations in electron cloud densities, which enhance charge carrier transport and significantly improve the overall catalytic performance.
Figure 3
Figure 3.
High-resolution XPS spectra of (a) Co 2p, (b) Mo 3d and (c) B 1s of the Co3B-Mo2B5, Co3B and Mo2B5.
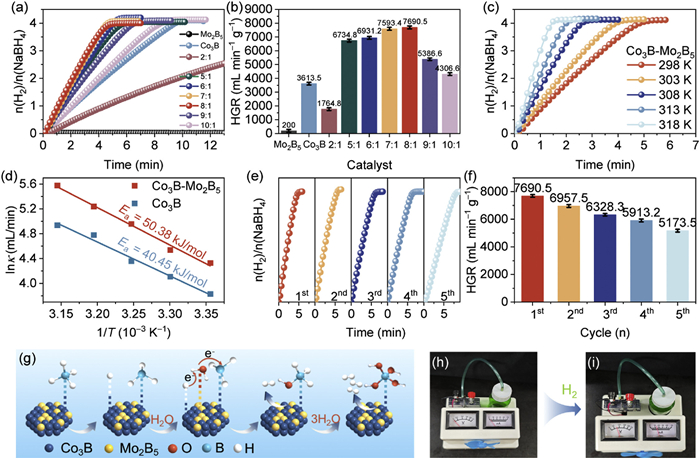
The dehydrogenation of NaBH4 hydrolysis by the catalysts is evaluated using drainage methods in a batch reactor (Fig. S6 in Supporting information). Figs. 4a and b show that Mo2B5 does not catalyze hydrogen release from NaBH4 hydrolysis, while Co3B takes over 9 min to complete the reaction. In contrast, the amorphous Co3B-Mo2B5 catalyst, optimized at a Co/Mo molar ratio of 8:1, achieves complete NaBH4 hydrolysis in just 5 min, with an impressive hydrogen generation rate (HGR) of 7690.5 mL min−1 g−1, surpassing most recently reported catalysts (Table S1 in Supporting information). The catalytic activity improves as the Co/Mo ratio increases to 8:1, after which performance declines. The lower activity observed at a Co/Mo ratio of 2:1 is likely due to the excessive presence of Mo2B5, which covers Co3B's active sites, thereby reducing the available surface-active sites and overall catalytic efficiency [40]. The Co3B-Mo2B5 significantly reduces the dehydrogenation time compared to Mo2B5 and Co3B. This is primarily due to the amorphous nature and electron rearrangement within the Co3B-Mo2B5 structure, which facilitates the hydrogen release process more efficiently. The actual metal content of the catalysts was determined through ICP-MS analysis (Table S2 in Supporting information). However, excessive Co content leads to a decline in HGR, likely due to nanoparticle agglomeration, which reduces the availability of active sites (Fig. S7 in Supporting information). Figs. S8a and b (Supporting information) illustrate the variation in HGR with different masses of Co3B-Mo2B5 catalyst (ranging from 5 mg to 20 mg). Among the tested masses, 10 mg of Co3B-Mo2B5 exhibited the highest hydrogen generation rate. At higher catalyst masses, the solution viscosity increases, inhibiting the efficient diffusion of the byproduct NaBO2. This accumulation of NaBO2 on the catalyst surface blocks active sites and restricts access to reactants, ultimately reducing catalytic efficiency [9]. The influence of NaOH concentration on the hydrolysis of NaBH4 is depicted in Fig. S9a (Supporting information). The catalytic activity of Co3B-Mo2B5 decreases in the absence of NaOH. However, increasing the NaOH concentration from 0.4 wt% to 0.8 wt% has no significant impact on dehydrogenation activity. These observations suggest that OH− ions have a dual function in catalyzing the hydrolysis reaction: at lower concentrations, they stabilize the NaBH4 solution, whereas, at higher concentrations, they hinder the response by limiting the availability of active hydrogen sites [41]. Additionally, the effect of NaBH4 concentration on the hydrolysis reaction was examined. As shown in Fig. S9b (Supporting information), similar hydrogen generation rates were observed across different NaBH4 concentrations. The slope of ln(rate) versus ln(concentration of NaBH4) was determined to be −0.086 (inset in Fig. S9b), indicating that the hydrolysis reaction catalyzed by Co3B-Mo2B5 follows a quasi-zero-order kinetic model [42].
Figure 4
Figure 4.
(a) The equivalent H2 per mole of sodium borohydride versus time with different catalysts and (b) the corresponding HGR values. (c) Relationship between the H2 generation rates and reaction time of different reaction temperatures (298−318 K). (d) Summarized Arrhenius diagram from (c). (e) Reusability test of Co3B-Mo2B5 catalyst at 25 ℃. (f) Summarized HGR from (e). (g) Catalytic mechanism diagram of H2 production by hydrolysis of NaBH4 based on Co3B-Mo2B5 catalyst. (h, i) Photographs of a working electric fan powered by a customized H2–air fuel cell. All tests were performed in 150 mmol/L NaBH4 + 0.4 wt% NaOH solution.
Experiments were conducted on Co3B and Co3B-Mo2B5 catalysts over a temperature range of 298–318 K to investigate the reaction kinetics further. As expected, the dehydrogenation kinetics improved with increasing temperature (Fig. 4c). The Arrhenius plot (Fig. 4d) revealed that the apparent activation energies (Ea) for NaBH4 hydrolysis catalyzed by Co3B-Mo2B5 and Co3B were 50.38 and 40.45 kJ/mol, respectively. Although Co3B-Mo2B5 has higher activation energy than Co3B, its superior performance can be attributed to multiple factors, including a higher density of surface-active sites, a synergistic effect optimizing the reaction mechanism, and a more optimized electronic structure that facilitates electron transfer and reactant activation. In addition to catalytic performance, the reusability of the catalysts is a critical factor for practical applications in large-scale hydrogen production. Figs. 4e and f show that the hydrogen generation rate (HGR) gradually decreased with successive cycles, with the HGR in the fifth cycle being 66% of the initial value. To determine the cause of this decline in activity, XRD, XPS, and ICP analyses were performed on the Co3B-Mo2B5 catalyst after five cycles. The XRD pattern (Fig. S10 in Supporting information) indicated that the structural composition of Co3B-Mo2B5 remained largely unchanged after five cycles. Similarly, XPS analysis (Fig. S11 in Supporting information) confirmed the continued presence of Co, Mo, and B elements, consistent with the pre-reaction composition. Furthermore, high-resolution XPS spectra of Co 2p, Mo 3d, and B 1s (Fig. S12 in Supporting information) showed no significant changes after the five cycles, suggesting that the catalyst's chemical states were largely preserved [3]. However, ICP analysis further confirmed that the slight decrease in the Co/Mo ratio after five cycles is one of the factors contributing to the performance decline (Table S2).
A reaction mechanism based on the Michaelis-Menten model is proposed for the catalytic hydrolysis of NaBH4 by the Co3B-Mo2B5 catalyst (Fig. 4g) [42]. The mechanism begins with the cleavage of the B–H bond, resulting in the formation of adsorbed hydrogen (Hads). This Hads subsequently reacts with a hydrogen atom from a water molecule, generating hydrogen gas (H2) and forming borohydride (BH3OH). In the following steps, the remaining hydrogen atoms in borohydride are replaced by hydroxide ions (OH−), ultimately leading to the dissociation of tetrahydroxoborate (B(OH)4−). Additionally, experiments demonstrate that the Co3B-Mo2B5 catalyst delivers a maximum current of 15 mA in a hydrogen-air fuel cell, achieving a power output of 0.3 V (Figs. 4h and i). These findings underscore the catalyst's capability to enhance hydrogen generation efficiency and fuel cell performance.
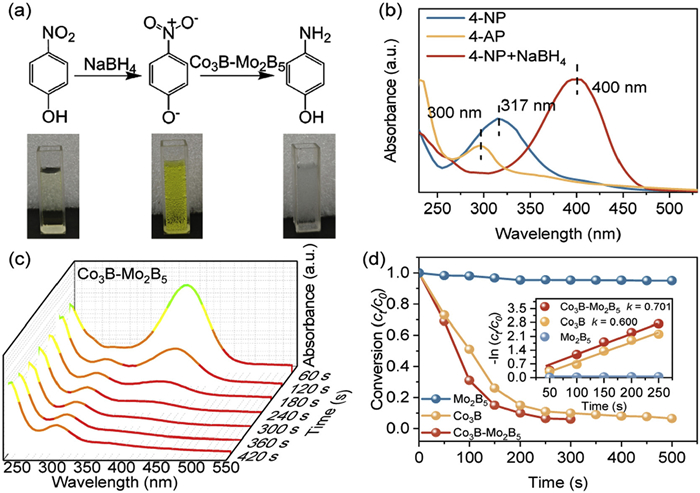
To evaluate the catalytic performance of Co3B-Mo2B5, the reduction of 4-NP using an excess of NaBH4 as the reductant was selected as a model reaction. The reduction process was monitored using UV–vis spectroscopy. As shown in Figs. 5a and b, the aqueous solution of 4-NP appeared light yellow and exhibited an absorption peak at 317 nm. In the presence of excess NaBH4, 4-NP underwent deprotonation to form the 4-nitrophenyl anion, causing the absorption peak to shift from 317 nm to 400 nm and resulting in a yellow color change in the solution [43]. Upon adding Co3B-Mo2B5, the solution turned colorless, and a new absorption peak at 300 nm appeared, indicating the formation of 4-AP [44]. As depicted in Fig. 5c, the 2 mg Co3B-Mo2B5 catalyst wholly and rapidly converted 4-NP to 4-AP within 460 s, whereas pure Co3B required 600 s, and Mo2B5 showed negligible conversion (Figs. S13a and b in Supporting information). A pseudo-first-order kinetics model was applied to investigate the reaction kinetics of the three catalysts, as the significant excess of NaBH4 ensured a constant NaBH4 concentration throughout the reaction. The reaction rate constant (k) for 4-NP degradation by Co3B-Mo2B5 was determined to be 0.701 min−1, with a corresponding removal rate of 94% (Fig. 5d and inset). This rate is significantly higher than the control catalysts, and many previously reported catalysts (Table S3 in Supporting information), demonstrating that Co3B-Mo2B5 exhibits faster catalytic kinetics and holds great potential for practical applications [45].
Figure 5
Figure 5.
(a) Photographs of 4-NP, NaBH4 + 4-NP and 4-AP solution, respectively. (b) UV–vis spectra of corresponding solutions. (c) Time-dependence of UV–vis absorption spectra for reducing 4-NP with NaBH4 catalyzed by Co3B-Mo2B5. (d) Removal efficiencies of 4-NP over different materials with the inset showing the corresponding fitting curves of kinetic data.
In summary, we successfully synthesized the amorphous bifunctional catalyst Co3B-Mo2B5 using a self-sacrificial template. This catalyst exhibits exceptional performance at 25 ℃, achieving a remarkable hydrogen generation rate of 7690.5 mL min−1 g−1 during NaBH4 hydrolysis and a reaction rate of 0.701 min−1 in the reduction of 4-NP. The enhanced catalytic activity is attributed to the unique amorphous structure, which provides abundant reactive sites, along with electron rearrangement between Co3B and Mo2B5 that modifies the electron cloud density. The possible mechanistic insights gained from these studies reveal that the Co3B facilitates the activation of BH4−, while the Mo2B5 preferentially activates water, thereby enabling efficient hydrogen production. In summary, this study not only provides new insights into the mechanism of action of bimetallic borides in the hydrolysis of NaBH4 and the reduction reaction of 4-NP, but also significantly advances the understanding and development of amorphous materials in catalysis and energy storage/conversion technology applications.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Chenxi Shang: Writing – original draft, Conceptualization. Boxuan Lu: Methodology. Chongbei Wu: Data curation. Shuqing Zhou: Investigation. Luyan Shi: Data curation. Tayirjan Taylor Isimjan: Writing – review & editing. Xiulin Yang: Writing – review & editing, Supervision, Funding acquisition.
Acknowledgments
This work has been supported by the National Natural Science Foundation of China (Nos. 52363028, 21965005), Natural Science Foundation of Guangxi Province (Nos. 2021GXNSFAA076001, 2018GXNSFAA294077), Guangxi Technology Base and Talent Subject (Nos. GUIKE AD23023004, GUIKE AD20297039).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111152.
[1]
Z. Li, S. Ji, C. Wang, et al., Adv. Mater. 35 (2023) 2300905. doi: 10.1002/adma.202300905
Y. Wang, K. Zou, D. Wang, et al., Renew. Energy 154 (2020) 453–460.
[18]
X. Xiao, H. Zhao, L.F. Li, et al., Rare Met. 42 (2023) 1186–1194.
[19]
M. Tan, B. Huang, L. Su, et al., Adv. Energy Mater. 14 (2024) 2402424.
[20]
X. Huang, D. Wu, D. Cheng, J. Colloid Interface Sci. 507 (2017) 429–436.
[21]
S.S. Muir, X. Yao, Int. J. Hydrog. Energy 36 (2011) 5983–5997.
[22]
X. Wang, B. Liu, S. Ma, et al., Nat. Commun. 15 (2024) 2600.
[23]
X. Cao, B. Zheng, W. Shi, et al., Adv. Mater. 27 (2015) 4695–4701.
[24]
Y. Wang, J. Liu, H. Yuan, et al., Adv. Funct. Mater. 33 (2023) 2211909.
[25]
S. Wintersteller, O. Yarema, D. Kumaar, et al., Nat. Commun. 15 (2024) 1011.
[26]
L. Wang, J. Huang, Z. Huang, et al., Chem. Eng. J. 472 (2023) 144924.
[27]
Z. Deng, C. Ma, Z. Li, et al., ACS Appl. Mater. Interfaces 14 (2022) 46595–46602.
[28]
M. Sathiya, J.B. Leriche, E. Salager, et al., Nat. Commun. 6 (2015) 6276.
[29]
Y. Luo, J. Xu, P. Mou, et al., Small 20 (2024) 2304981.
[30]
D. Kim, X. Qin, B. Yan, Y. Piao, Chem. Eng. J. 408 (2021) 127331.
[31]
Z. Na, X. Liu, W. Li, et al., Chem. Eng. J. 439 (2022) 135513.
[32]
Z. Dai, Y. Zheng, Z. Guo, et al., J. Hazard. Mater. 473 (2024) 134689.
[33]
M. Guo, L. Wang, Z. Huang, et al., ACS Nano 18 (2024) 17901–17912.
[34]
K.M. Hung, J.J. Wu, Chem. Eng. J. 484 (2024) 149772.
[35]
J. Wang, M. Zhou, R. Fu, et al., Adv. Funct. Mater. 34 (2024) 2315326.
[36]
Z. Shi, X. Zhang, X. Lin, et al., Nature 621 (2023) 300–305.
[37]
X. Liu, Y. Yao, W. Li, et al., Small 20 (2024) 2308549.
[38]
Z. Tian, K. Zhou, M. Xie, et al., Chem. Eng. J. 447 (2022) 137495.
[39]
Q. Hu, G. Li, Z. Han, et al., Adv. Energy Mater. 9 (2019) 1970109.
[40]
S. Xiao, Y. Xiao, S. Hu, et al., Energy Storage Mater. 75 (2025) 104047.
[41]
L. Shi, K. Zhu, Y. Yang, et al., Chin. Chem. Lett. 35 (2024) 109222.
[42]
H. Li, X. Hu, L. Wang, et al., Chem. Eng. J. 481 (2024) 148547.
[43]
W. Wang, W. Xue, Z. Huang, et al., Chem. Eng. J. 499 (2024) 156667.
[44]
B. Jacob, M. Mohan, D. K C, H. Thomas, Environ. Res. 251 (2024) 118567.
[45]
S. Dou, S. Zhou, H. Huang, et al., Chem. Eur. J. 26 (2020) 16923–16931.
Figure 1
(a) Schematic protocol of the synthesis strategy for Co3B-Mo2B5. SEM images of (b) Mo-MOF, (c) Co2+/Mo-MOF, and (d) Co3B-Mo2B5. (e) TEM and (f) HR-TEM of Co3B-Mo2B5. (g) HAADF-TEM image and elemental mappings of Co3B-Mo2B5 (Co, Mo and B).
Figure 2
XRD patterns of (a) Mo-MOF and (b) Co3B-Mo2B5. (c) EPR spectra of Co3B-Mo2B5, Co3B, and Mo2B5. (d) N2 adsorption-desorption isotherm with the inset showing the corresponding pore size distribution of Co3B-Mo2B5. (e) Bubble contact angle images of different catalysts. (f) Zeta potentials of the Co3B-Mo2B5, Co3B, Mo2B5 and a 150 mmol/L NaBH4 + 0.4 wt% NaOH solution.
Figure 4
(a) The equivalent H2 per mole of sodium borohydride versus time with different catalysts and (b) the corresponding HGR values. (c) Relationship between the H2 generation rates and reaction time of different reaction temperatures (298−318 K). (d) Summarized Arrhenius diagram from (c). (e) Reusability test of Co3B-Mo2B5 catalyst at 25 ℃. (f) Summarized HGR from (e). (g) Catalytic mechanism diagram of H2 production by hydrolysis of NaBH4 based on Co3B-Mo2B5 catalyst. (h, i) Photographs of a working electric fan powered by a customized H2–air fuel cell. All tests were performed in 150 mmol/L NaBH4 + 0.4 wt% NaOH solution.
Figure 5
(a) Photographs of 4-NP, NaBH4 + 4-NP and 4-AP solution, respectively. (b) UV–vis spectra of corresponding solutions. (c) Time-dependence of UV–vis absorption spectra for reducing 4-NP with NaBH4 catalyzed by Co3B-Mo2B5. (d) Removal efficiencies of 4-NP over different materials with the inset showing the corresponding fitting curves of kinetic data.


 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
