Department of Gynecological Oncology, Zhongnan Hospital of Wuhan University, School of Pharmaceutical Sciences, Wuhan University, Wuhan 430071, China
b.
State Key Laboratory of Virology and Biosafety, Frontier Science Center for Immunology and Metabolism, Hubei Province Engineering and Technology Research Center for Fluorinated Pharmaceuticals, Key Laboratory of Combinatorial Biosynthesis and Drug Discovery (Wuhan University), Ministry of Education, Wuhan University, Wuhan 430071, China
c.
Hubei Key Laboratory of Tumor Biological Behaviors, Hubei Cancer Clinical Study Center, Wuhan 430071, China
cdong@whu.edu.cn (C. Dong). 1 These authors contributed equally to this work.
Received Date:
11 December 2024 Accepted Date:
26 March 2025 Revised Date:
25 March 2025 Available Online:
15 April 2026
Abstract:
Sirtuin 2 (SIRT2) is one of the key members of sirtuins family that plays important role in regulating many physiological processes. Recent evidences have revealed that SIRT2 is associated with the development, progression and metastasis of ovarian cancer. In this study, guided by an in-depth analysis of the clinical characteristics of the expression pattern of SIRT2 in ovarian cancer patients, the first SIRT2-targeted hydrophobic tagging (HyT) degraders have been developed. These acyl thiourea degraders exhibited remarkable anti-proliferative activity in several ovarian cancer cells. Among them, the most effective compound Ⅱ-6 exhibited excellent anti-tumor activity both in vitro and in vivo (half maximal inhibitory concentration (IC50) = 0.002 ± 0.001 µmol/L). In addition, Ⅱ-6 was found to effectively suppress cancer cell proliferation and migration, as well as cell cycle arrest and apoptosis. Moreover, further investigation revealed that compound Ⅱ-6 indirectly induced DNA damage through the H4K20me2/53BP1 pathway by degradation of SIRT2. The study not only exemplifies the advantage of the novel HyT degradation strategy but also prove the great potential of SIRT2 as a promising target for drug development of ovarian cancer.
Ovarian cancer (OC) is one of the three major gynecological malignancies, accounting for 4% of all cancer cases in women. The non-specific symptoms and the lack of effective screening strategies result in 70%–75% of OC patients being diagnosed at stage Ⅲ or Ⅳ, making OC one of the most fatal gynecological cancers globally [1]. According to the data released by the World Health Organization (WHO), an estimated 12,740 women in United States are threatened with death from OC in 2024. The 5-year relative survival rate is still < 47% among these patients even though the screening, diagnosis, and therapy have made progress in recent years [2,3].
OC is currently treated in a mainstream way with primary debulking surgery followed by radiotherapy and chemotherapy [4]. The first-line treatment of advanced epithelial OC include an adjuvant combination of carboplatin/cisplatin, paclitaxel, and bevacizumab, as well as maintenance therapies such as poly(ADP-ribose) polymerase inhibitors (PARPis) [5]. Platinum compounds cause antineoplastic activity, when they bind to DNA, creating intrastrand adducts and interstrand cross-link, that damage the genetic material [6]. PARP inhibitors can inhibit key enzyme (PARP1, PARP2) activities in DNA-damage response (DDR) [7]. Although 50%–81% of advanced OC patients initially show sensitivity to chemotherapy, 70%–85% of OC patients experience recurrence after first-line treatment and gradually become increasingly resistant to various treatment options [8]. Moreover, cisplatin and PARP inhibitors show significant toxicity to normal tissues, such as the kidneys, heart, and liver [9,10]. Thus, the existing treatment is not able to meet clinical needs because of low survival rates, inevitable drug resistance, delayed recurrence, and toxic side effects.
The sirtuin (SIRT) protein family is a group of nicotinamide-adenine dinucleotide (NAD)-dependent histone deacetylases that are widely distributed and have been preserved through evolution [11,12]. The sequence and length of the N- and C-terminal domains of the SIRTs differ, which partly accounts for their distinct localization and functionality [13]. According to the evidence, SIRTs are responsible for regulating gene expression, energy metabolism, DDR, cellular stress response in almost all cancer types [13,14]. Despite the important roles played by sirtuins in caloric restriction, aging, apoptosis, transcription, and inflammation, our understanding of their functions remains limited. The mainstream view is currently that sirtuin is as a double-edged sword in tumorigenesis. According to the genetic background, tumor type and stage, sirtuin can be defined as either tumor suppressor or oncogene, and the function of sirtuin subtypes is also significantly different even in the same tissue [15]. In recent years, with the increasing understanding of sirtuin, it has become a very potential target in cancer chemotherapy. Seven mammalian homologs of the yeast Sir2 are referred to as SIRT1–7 in the sirtuin family [11,12]. SIRT1 functions in both the nucleus and the cytoplasm; SIRT3–5 are mitochondrial proteins; SIRT6 and SIRT7 are nuclear proteins; SIRT2 is the only subtype that is predominantly present in the cytoplasm but shuttles between the nucleus and cytoplasm during the G2 to mitosis phase [16]. The effects of SIRT2 on tumor growth are complex, with multiple competing mechanisms involved. As a tumor-promoting factor, SIRT2 can induce regulatory T cells, inhibit tumor-associated macrophages, myeloid-derived cells, and tumor-associated neutrophils in the tumor microenvironment, help tumor cells evade immune surveillance, and limit the tumor immune response, thereby promoting tumor development [17]. In addition, SIRT2 has the potential to promote deacetylation-dependent lactate dehydrogenase A (LDH-A) excitation, speed up glycolytic reactions, and promote tumor cell proliferation and migration [18]. Furthermore, SIRT2 interacts with various factors within the tumor microenvironment to inhibit the growth and development of tumor cells [15]. So far, several structurally distinct selective small-molecule SIRT2 inhibitors have been developed, crystallographic analyses showed that they bind to the unique hydrophobic site of SIRT2, providing important clues for future application (Fig. S1 in Supporting information). Nicotinamide acts as a non-competitive inhibitor of SIRT diacylation, exhibiting the capability to reduce the proliferation of human prostate cancer cells and leukemia cells [19,20]. Sirtinol and cambinol have similar β-naphthol structures [21,22]. GW5074 is an indole-based SIRT inhibitor [23]. Compounds 2–5 showed significant inhibitory activity towards the proliferation of breast cancer cells, non-small cell lung cancer cells, and so on. Compound 6 exhibited moderate inhibitory effects specifically against the PANC-1 cells [24]. Meanwhile, TM, tenovin-1, tenovin-6, SirReal2, and AGK2 possessed broad-spectrum antitumor properties, displaying varying degrees of cytotoxicity against multiple tumor cell lines [25-28].
Interestingly, a detailed mechanism study reveals that the SIRT2-targeted inhibitors such as AGK2 (11), SirReal2 (8), and TM (7) specifically hinder deacetylation activity, but not the defattyacylation activity [25-30]. Surprisingly, TM-P4-Thal, the first SIRT2-targeted proteolysis-targeting chimera (PROTAC) specifically inhibits both the deacetylase and defattyacylase activities of SIRT2 in living cells, providing more options for the research of SIRT2-targeted cancer treatment [29-32]. Despite the lack of clinically useful SIRT2-targeted degraders at present, the potential of selective SIRT2 degradation has been validated in both in vitro and in vivo. Since SIRT2 plays an important role in regulating OC progression, therefore, in order to correct the one-sided understanding of SIRT2, an in-depth analysis of the clinical characteristics of the OC patients bearing SIRT2, and development of a diversity-oriented novel SIRT2-targeting small molecule degraders may provide useful guidance for SIRT2-targeted OC therapy.
In recent years, targeted protein degradation (TPD) has emerged from the chemical biology toolbox as one of the most exciting areas for development of new therapeutics in the pharmaceutical industry [30-32]. In contrast to small molecule inhibitors, TPDs are able to catalyze the degradation of target proteins in an "event-driven" manner, rather than the continuous occupation of the binding pocket relying on high affinity [33]. Thus, TPD is capable of causing a complete function loss of protein of interest (POI), even traditional "undruggable" targets at low concentrations. Hydrophobic tagging (HyT) degradation, as another extension of TPD, harnesses the fact that exposure of hydrophobic residues on the protein surface marks it as an unfolded protein, ultimately eliminating the protein through ubiquitin-proteasome system (UPS) or autophagy [34]. HyT degrader usually consists of a hydrophobic tag (such as adamantane) linked with the target binding ligand. The binding of HyT to a POI, increases the hydrophobicity of the target protein, which results in the recruitment of heat shock proteins, ultimately targeting it for proteasomal degradation. Just like a precision-guided missile, hydrophobic tagging-protein degraders (HyT-PDs) can adhere to target proteins, and swiftly activate the protein quality control (PQC) machinery to eliminate these "damaged" or "misfolded" proteins after being sensed to maintain cellular homeostasis [35-38]. So far, adamantane, fluorene, pyrene, norbornene, etc. have been used as the hydrophobic group in HyT-PDs and successfully induced the efficient degradation of POI targets, such as human epidermal growth factor receptor 3 (HER3), enhancer of zeste homolog 2 (EZH2), and PAPR1 [39,40], and demonstrated favorable anti-proliferative effects in various types of tumors. Therefore, the HyT-PD technology offers a new way to target and degrade POI. Herein, guided by an in-depth analysis of the clinical characteristics of the patients, we disclosed the design, synthesis, and biological evaluation of a novel series of SIRT2 HyT degraders based on the tenovin-6 for treating OC. The optimization of the hydrophobic groups, linker and functional groups resulted in series of SIRT2-targeted HyT degraders. The preferred degrader Ⅱ-6 was highly effective in inhibiting the growth of various OC cells at low concentrations in vitro. The biosafety of compound Ⅱ-6 was superior to tenovin-6 and cisplatin in normal ovarian cells. More importantly, compound Ⅱ-6 effectively suppressed cancer cell proliferation by selectively inducing SIRT2 degradation and significantly inhibiting downstream protein kinase B-mammalian target of rapamycin (AKT-mTOR) signaling pathways, which result in DNA damage and ultimately causes cell apoptosis through the down regulation of SET domain-containing protein 8 (SET8) and methylated histone H4 lysine 20 (H4K20). To the best of our knowledge, compound Ⅱ-6 is the first HyT degrader targeting SIRT2. This study highlights the potential for understanding of the underlying role of SIRT2 in OC, as well as paving the way for the development of anti-OC drugs.
Considering that OC is a highly aggressive form of cancer that primarily affects women and has a poor prognosis, and studies have shown that the expression of various SIRTs differs depending on the specific subtype, grade and stage of the tumor [41]. In order to investigate the expression pattern of SIRT2 in OC tissues, we conducted the study with 55 patients who had been diagnosed with OC and were at various stages (Ⅰ–Ⅳ) according to the staging criteria by the International Federation of Gynecology and Obstetrics (FIGO 2014). The characteristics of the patients were detailed in Table S1 (Supporting information). After conduction an in-depth analysis of the comprehensive clinical data from 55 OC patients, it was found that there was a correlation between SIRT2 mRNA levels and certain clinical characteristics. The study utilized quantitative real-time polymerase chain reaction (PCR) on 14 normal ovarian epithelial tissue specimens and 55 OC tissue samples from Zhongnan Hospital of Wuhan University. As shown in Fig. S2 (Supporting information), the mRNA expression of SIRT2 in OC tissues was significantly higher than that in normal ovarian epithelial tissues (Fig. S2A). In addition, compared with normal ovarian epithelium tissues, the brown staining areas in OC tissues significantly increased in the immunohistochemical staining results of pathological sections (Fig. S2B). At the same time, the results from Western blot also verified the high expression of SIRT2 protein in OC tissues (Fig. S2C). In view of the above results, subsequent research was carried out at the cellular level. The expression of SIRT2 protein in different OC cells (SKOV3, A2780, and OVCAR3) and normal ovarian epithelial cell (IOSE80) was investigated. As shown in Fig. S2D, compared to normal ovarian epithelial cell, the expression of SIRT2 was increased in most OC cells.
The Gene Expression Omnibus Series (GSE) database was used to analyze the expression of SIRT2 in OC. The results showed the mRNA expression of SIRT2 is markedly up-regulated in OC tissues compared to normal tissues (Fig. S2E). Furthermore, the Gene Expression Profiling Interactive Analysis (GEPIA) online database was applied to analyze the SIRT2 expression and the prognostic significance of patients diagnosed with OC. According to our data, there was a correlation between SIRT2 expression and overall survival (OS) in OC (Fig. S2F). The data clearly showed that the group with high SIRT2 expression (n = 211) had significantly lower OS rate compared to the group with low-expression (n = 212), with a hazard ratio (HR) of 1.4 for OS between the two groups (P = 0.011). While, significant difference was observed in disease-free survival (DFS) in the short term (< 110 months). Surprisingly, no statistically difference in DFS was observed after 110 months (Fig. S2G). This observation would be due to the effects of specific stages of OC on the expression of SIRT2. In the early or intermediate stages of the disease, high SIRT2 expression may accelerate disease progression by regulating relevant signaling pathways, thereby leading to a shortened DFS. Nevertheless, multiple therapeutic interventions (such as surgery, chemotherapy, and radiation therapy) might impact disease progression and prognosis, leading to changes in the trend of DFS [42].
Taken together, in most cases, a high expression of SIRT2 is obviously correlated with an unfavorable prognosis for patients. Therefore, the research may provide a potential approach on the development of SIRT2-targeted drugs for treatment of OC. In this study, novel small molecule degraders targeting SIRT2 were designed and synthesized by means of HyT degradation technology (Fig. 1A). The potential application of these degraders in OC was evaluated. Tenovin-6, a SIRT2 inhibitor, was selected as the core structure to construct the goal SIRT2-targeted degraders. Subsequently, series acyl thiourea-based HyT degraders containing different linker and hydrophobic groups, such as amantadine, diphenylmethanamine, 9H-fluoren-9-amine, Boc1-Trp, Boc2-Lys, and Boc3-Arg. were designed and synthesized accordingly (Fig. 1B). The detailed synthetic procedures of all the desired acyl thiourea HyT degraders were outlined (Schemes S1–S3 in Supporting information).
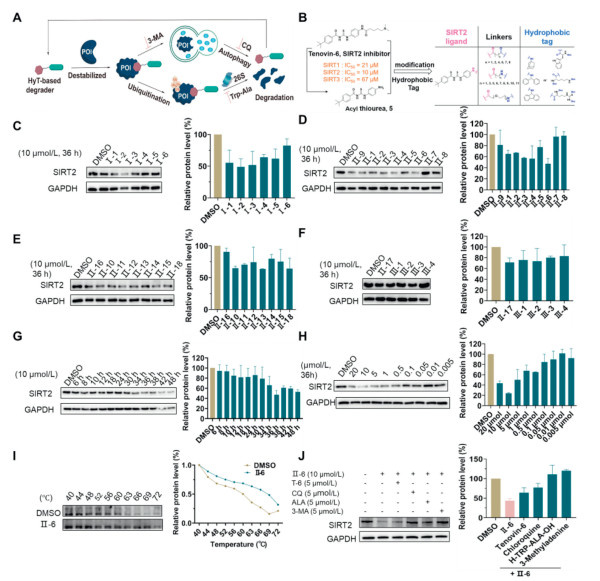
Figure 1
Figure 1.
Degradation activity of these HyT degraders evaluated. A2780 cells were individually pretreated with 10 µmol/L degraders for 36 h before protein level analysis. (A) The mechanism of SIRT2 degradation induced by HyT degraders. (B) Design of ultimate degraders based on acyl thiourea skeletons. (C) Western blot assay was performed to examine the degradation activity of amantadine series degraders Ⅰ-1–6. (D) Degradation activity of Boc1-Trp series degraders Ⅱ-1–9. (E) Degradation activity of Boc2-Lys series degraders Ⅱ-10–16 and Ⅱ-18. (F) Degradation activity of Boc3-Arg series degrader Ⅱ-17 and Ⅲ-1–4. (G) Western blot analysis showed that Ⅱ-6 degraded SIRT2 protein in a time-dependent manner. (H) SIRT2 protein was degraded in a dose-dependent manner. (I) The CETSA assay of Ⅱ-6. (J) Ⅱ-6 induced SIRT2 degradation through proteasome-mediated proteolysis and the autophagy lysosome pathway. Data are presented as mean ± standard deviation (SD) (n = 3). GAPDH, glyceraldehyde-3-phosphate dehydrogenase; CQ, chloroquine.
The degraders were firstly evaluated for their anti-proliferative activity against OC cells, including SKOV3, OVCAR3, A2780 and ovarian epithelial cell (IOSE80 cell) as well using an in vitro cell counting kit-8 (CCK-8) assay. As shown in Table S2 (Supporting information), most of the compounds exhibited inhibitory activity in the nanomolar to submicromolar range against these OC cells. However, there was lower inhibition observed in SKOV3 and OVCAR3 cells. In particular, most of the compounds exhibit superior inhibitory effects in the A2780 cells compared to the OVCAR3 and SKOV3 cells. Particularly, the compounds Ⅰ-1–Ⅰ-6, tagged with amantadine, were more effective to inhibit proliferation of cancer cells. Among them, compound Ⅰ-6 containing ten carbon-atom linker achieved an half maximal inhibitory concentration (IC50) value of 0.008 µmol/L which was much lower than that of cisplatin (IC50 = 1.28 µmol/L). However, length of linker had no obvious effect on the anti-tumor activity of compounds Ⅰin vitro. We found that the replacement of amantadine with Boc1-Trp such as Ⅱ-6 increased potency accordingly (IC50 = 0.002 ± 0.001 µmol/L). Moreover, in case of compounds Ⅱ, the longer linker weakened the anti-tumor activity in vitro (Ⅱ-8, Ⅱ-15). For example, when the compounds Ⅱ-7 and Ⅱ-8 with twelve carbon atom alkyl linker was used in this assay, the IC50 values went up to 0.13 and 0.04 µmol/L, respectively. In addition, among compounds Ⅱ-10–16 comprising Boc2-Lys as the hydrophobic group also displayed excellent inhibitory activity with an IC50 value up to 10 nmol/L (Ⅱ-16). It was notable that compounds Ⅲ-1–4 derived from the aromatic diphenylmethanamine or 9H-fluoren-9-amine hydrophobic groups displayed inferior anti-proliferation activity in OVCAR3 cells, while no big difference was observed in A2780 cells. Altogether, Ⅱ-6 exhibited the best inhibitory activity and the lowest cytotoxicity, therefore, it was selected as a potential candidate for further studies.
To explore whether the synthesized degraders could degrade the SIRT2 protein, we investigated the degradability of these acyl thiourea in A2780 cells. As shown in Figs. 1C–F, we found that some degraders, such as Ⅰ-3, Ⅱ-4, Ⅱ-6, Ⅱ-12, would able to significantly decrease the protein level of SIRT2 when cancer cells were treated with a concentration of 10 µmol/L for 36 h. However, degraders Ⅱ-7–8 and Ⅲ-1–4 showed little to no effect on degrading SIRT2 protein in A2780 OC cell. Of all the degraders tested, the Ⅱ-6 exhibited the most promising degradation activity and was chosen for further investigation.
In order to systematically evaluate the SIRT2 selective degradation efficiency of the HyT degrader Ⅱ-6, we checked the SIRT2 protein lever in A2780 OC cells after certain hours at various concentrations. The results were presented in Fig. 1. At first, we examined the time course of Ⅱ-6-induced SIRT2 degradation in A2780 cells. As the incubation time increased, 10 µmol/L of Ⅱ-6 lowered SIRT2 lever even further. With the treatment of 10 µmol/L of Ⅱ-6, the SIRT2 level decreased obviously at 36 h (Fig. 1G). Thus, SIRT2 was degraded by Ⅱ-6 in a time-dependent manner. Next, we further tested the SIRT2 degradation at different concentrations. Even at 0.1 µmol/L, the SIRT2 level was significantly decreased compared to that of a vehicle-treated sample (Fig. 1H). Furthermore, the high efficiency of Ⅱ-6 induced SIRT2 degradation was achieved at 10 µmol/L for 36 h. While, at 20 µmol/L Ⅱ-6, the SIRT2 level slightly changed likely due to the hook effect. Moreover, to demonstrate the selectivity of Ⅱ-6 towards the localization of SIRT2 protein (nuclear and cytosolic), both confocal laser scanning microscope (CLSM) and Western blot analyses were employed (Figs. S3A and B in Supporting information). The experimental results indicated that SIRT2 protein was distributed in the cytoplasm and the nucleus of OC. Notably, the expression level of SIRT2 protein in the nucleus was significantly reduced after treatment with 10 µmol/L of Ⅱ-6 for 36 h.
To confirm the direct interaction between Ⅱ-6 and SIRT2, the cellular thermal shift assay (CETSA) was applied for further validation of their specific interactions, as displayed in Fig. 1I. Taking DMSO as the blank control, the thermal stability of the SIRT2 protein decreased with a serial increase in temperatures ranging from 44 ℃ to 72 ℃. However, with the addition of Ⅱ-6, the stability of SIRT2 improved significantly, indicating the possible formation of an Ⅱ-6/SIRT2 complex. Compared with the significant degradation of SIRT2 protein observed at 63 ℃ in the control group, the remarkable degradation of SIRT2 protein in the Ⅱ-6 treatment group occurred at an increased temperature of 72 ℃, and the thermal dissolution curve shifted significantly to the right, indicating that its thermal stability was significantly improved in the presence of Ⅱ-6. These findings demonstrated that Ⅱ-6 could selectively target SIRT2 protein.
According to literature reports, HyT degradation agents can directly recruit heat shock protein 70 (Hsp70) or 20S proteasomes through hydrophobic fragments, mediating POI ubiquitination or degradation; or hydrophobic group can disrupt POI stability or mimic misfolded proteins to recruit endogenous partner proteins, thereby mediating proteasome degradation (Fig. 1A) [43]. To further explore the intricate mechanism of Ⅱ-6 induced SIRT2 degradation, the SIRT2 degradation was examined by blocking the ubiquitin proteasome pathway and autophagolysosomal pathway with L-tryptophanyl-L-alanine (H-TRP-ALA-OH), chloroquine, and 3-methyladenine (3-MA), respectively. The results showed that the intervention of tenovin-6 can partly prevent the degradation of SIRT2 (Fig. 1J), which suggests that the affinity with SIRT2 is essential for the degradation. As expected, SIRT2 protein degradation induced by Ⅱ-6 could be effectively blocked and reversed with proteasome inhibitors (H-TRP-ALA-OH) and autophagy inhibitors (3-MA and chloroquine). The results indicated that Ⅱ-6-induced SIRT2 protein degradation was not only UPS dependent, but autophagy-lysosome pathway as well.
To investigate the effect of Ⅱ-6 on SIRT2, Schrödinger Maestro was used to model the conformation of the 16-SIRT2 complex and Ⅱ-6-SIRT2 complex [44]. The docking models with the lowest binding energy of 16 (green) and Ⅱ-6 (yellow) for the SIRT2 complex were shown in Fig. S4A (Supporting information). The molecular docking results showed that the HyT degrader Ⅱ-6 was able to bind to the binding site of SIRT2 in the similar binding mode as the parent compound 16. Based on the docking results, we found that the aniline group of compound 16 could form a strong hydrogen-bond with amino residue of Asp170. Compared with the 16-SIRT2 complex, the Ⅱ-6-SIRT2 complex was slightly different, the phenyl ring of the acyl thiourea moiety could occupy the so-called "EC-Site" of SIRT2 [45], and bind with amino residues of Phe96 and Phe119 via hydrogen bond interaction. More importantly, the HyT group of Ⅱ-6 was preferred to be extended to the outer side of "EC-Site", and pointed towards the ADP ribose (ADPR) binding sites of SIRT2. The Boc1-Trp group in Ⅱ-6 formed strong hydrogen bonds with amino residues Asn286 and Glu242, respectively.
To systematically and comprehensively verify the inhibitory effect of Ⅱ-6 at the cellular level, 72-h CCK-8 proliferation assay and colony formation assay were performed. OC A2780 cells were pretreated with the degrader Ⅱ-6, together with cisplatin as the positive control. In agreement with its potency in vitro, Ⅱ-6 reduced cell viability in a dose-dependent and time-dependent manner, whereas cisplatin showed only weak effects (Figs. S4B and C in Supporting information). Additionally, Ⅱ-6 exhibited slightly toxic effect on normal ovarian cells (IOSE80) (Fig. S4D in Supporting information), which indicated its potential as a promising agent for OC therapy. Furthermore, we found that Ⅱ-6 specifically inhibited the colony formation in A2780 cell (Fig. S5A in Supporting information) at lower concentration of 0.5 µmol/L, suggesting that Ⅱ-6 was an anti-tumor compound for OC cells. Surprisingly, cisplatin effectively inhibited colony production at 5 and 1 µmol/L, respectively. The primary cause of death among OC patients is distant metastasis [46]. In the complex process of tumor cell migration, epithelial cells gradually undergo a notable change. They not only lose their inherent epithelial characteristics, such as tight inter cellular connections and polarity, but also gradually acquire the properties of mesenchymal cells, which include an enhanced migratory potential and invasiveness [47]. This process, namely epithelial-mesenchymal transition (EMT), indicates that tumor epithelial cells acquire high mobility, which is the key to the tumor metastasis. EMT-targeted therapy can inhibit the migration and invasion of tumor cells in primary tumors. Meanwhile, EMT can reduce the proportion of cancer stem cells in tumors, prevent the colonization of circulating tumor cells in primary tumors, and reduce the ability of circulating tumor cells to produce secondary tumors when inoculated at a distance [48]. The migration of tumor cells is an important factor of tumor metastasis and one of the main factors for the failure of cancer treatment in the late stage. Degrader Ⅱ-6 has been found to inhibit the migration of A2780 at a concentration of 0.05 µmol/L (Fig. S5B in Supporting information). Furthermore, the migration of tumor cells was almost completely blocked within 48 h after treated with 0.5 µmol/L of Ⅱ-6. These results substantiate the potent anti-proliferative and anti-metastatic efficacy of Ⅱ-6.
The results of wound healing assay revealed that compared to the control group, the migration distance in the Ⅱ-6-treated group was significantly shortened, and the migration inhibition effect was inversely correlated in the presence of Ⅱ-6, further illuminating its anti-migratory potential. Within the tumor micro environment, cancer cells and immune cells emit cytokines, growth factors, and chemokines that surround the epithelial cells. This process triggers the activation of EMT transcription factors (Snail, Slug, Twist, zinc finger E-box binding homeobox 1 (ZEB1), and ZEB2), ultimately initiating the EMT in those epithelial cells [49]. Snail, a crucial transcription factor involved in driving EMT, exhibits its activation is related to tumor pathological grade, lymphatic metastasis, and poor prognosis [50]. The level of snail was significantly inhibited after treatment with Ⅱ-6, leading to the blocking of EMT (Fig. S5C in Supporting information), which manifested that the epithelial marker E-cadherin was up-regulated while mesenchymal markers N-cadherin, ZEB1, matrix metalloproteinase-9 (MMP9), and vimentin were down-regulated. All of the results robustly demonstrating that Ⅱ-6 inhibits the EMT process, thereby weakening the migration and invasion capabilities of OC cells.
After thoroughly exploring the effectiveness of Ⅱ-6 in inhibiting the proliferation and migration of tumor cells, we further focused on its regulation of critical life activities within tumor cells. The disordered proliferation and migration of tumor cells are the two core features of their malignancy, and the abnormal regulation of the cell cycle and the evasion of apoptosis mechanisms are the key molecular mechanisms behind these behaviors [51]. To comprehensively understand the anti-tumor mechanism of Ⅱ-6, we further explore whether Ⅱ-6 can intervene in the cell cycle progression of tumor cells and induce cell apoptosis. Flow cytometry analysis (Fig. S5D in Supporting information) unveiled the unique mechanism of Ⅱ-6 inducing cell cycle arrest at the S phase in A2780 cells. Compared to the control, Ⅱ-6 inhibited cell proliferation by disrupting the critical stage of DNA replication, the proportion of cells in the S phase significantly increased, whereas that in the G2/M phase decreased. Subsequently, to validate the pro-apoptotic effect of Ⅱ-6, we conducted the Annexin V-fluorescein isothiocyanate/propidium iodide (FITC/PI) staining which revealed a distinct dose-dependent manner of apoptosis induction in A2780 cells (Fig. S5E in Supporting information). When tumor cells receive stimulation, some triggers in the mitochondrial pathway are activated, subsequently activating the pro-apoptotic proteins (BAX, Bak) and the anti-apoptotic proteins (Bcl-2, Bcl-XL and Mcl-1), leading to the disruption of mitochondrial outer membrane permeability (MOMP). As a result, proteins usually confined in the intermembrane space diffuse into the cytosol, bind to apoptosis protease activating factor-1 (Apaf-1), and then trigger the formation of a complex named apoptosome. This complex recruits the initiator pro-caspase-9 to its caspase recruitment domain (CARD), which activates caspase cascade reaction followed by proteolysis [52]. The balance between the regulatory factors of pro-apoptotic and anti-apoptotic proteins is the critical point to determine whether a cell undergoes apoptosis. The imbalance of cell apoptosis is considered to be one of the hallmarks of cancer. In this study, compound Ⅱ-6 exerted its apoptotic effect by significantly induced up-regulation of the pro-apoptotic protein BAX and down-regulation of the anti-apoptotic proteins Bcl-2 and Bcl-XL (Fig. S5F in Supporting information), thereby providing definitive evidence for the underlying mechanism of Ⅱ-6-induced apoptosis. Up to now, this study has elucidated the anti-proliferation, migration and pro-apoptotic effects of compound Ⅱ-6 in OC cells from multiple dimensions, laying a solid cellular and molecular foundation for its development as a potential therapeutic drug for OC.
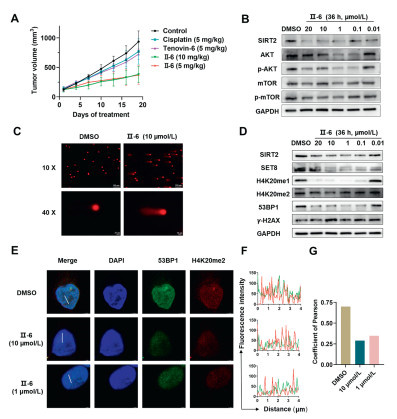
Given the remarkable anti-tumor activity and SIRT2 degradation capability of compound Ⅱ-6 observed in vitro, subsequently, we implemented pharmacodynamic explorations to thoroughly evaluate the anti-tumor efficacy in xenograft mouse model. Pharmacodynamics was explored based on the experimental flowchart shown in Fig. S6A (Supporting information). When administered at a dosage of 5 mg/kg, the degrader Ⅱ-6 significantly delayed the tumor progression than the conventional chemotherapeutic agent (cisplatin) and the small molecule SIRT2 inhibitor (tenovin-6) in the ovarian tumor xenograft mouse model (Fig. 2A and Fig. S6B in Supporting information). Additionally, compared to the control group, there was no animal mortality and significant change in mice body weight observed during the study period (Fig. S6C in Supporting information). This result emphasized the exceptional safety of Ⅱ-6 at the tested dosage.
Figure 2
Figure 2.
Antitumor effect in xenograft model and mechanism of Ⅱ-6 in A2780. (A) Tumor volume change of mice during the antitumor treatment. Data are presented as mean ± SD (n = 5). (B) Ⅱ-6 was associated with the SIRT2/AKT/mTOR signaling pathway. (C) The alkaline comet assay image, which showed Ⅱ-6 induced DNA damage in A2780 cells exposed to 10 µmol/L concentration. Scale bar: 100 µm for 10× image and 20 µm for 40× image. (D) Western blot was used to analyze the levels of SET8 and H4K20me1/H4K20me2 after SIRT2 degradation, as well as the levels of DNA damage markers 53BP1 and γ-H2AX. (E) CLSM analysis for H4K20me2 and 53BP1. Scale bar: 3 µm. (F) The plot profiles show fluorescence intensity variations along an oblique line, quantified by ImageJ. (G) Co-localization between two entities is assessed using Pearson coefficients, calculated by JACoP in ImageJ.
To further analyze whether Ⅱ-6 affects the organs of mice while inhibiting tumor growth, we conducted histopathological analysis on the important organs (heart, liver, spleen, lung, and kidney) of nude mice through hematoxylin and eosin (H & E) staining (Fig. S7 in Supporting information). The staining results in the heart, liver, spleen, lung, and kidneys showed no significant difference among all groups, indicating that Ⅱ-6 treatment had no obvious adverse effects on the organs of nude mice. In addition, the presence of Ki-67 within the nucleus serves as a proliferation indicator; as its expression level increases, it signifies an elevated number of cells undergoing the cell division phase, thereby accelerating tumor growth [53]. Moreover, the Ⅱ-6 induced tumor growth reduction in mouse xenograft tumor models was also confirmed by the results of Ki67 antibody staining (Fig. S7).
To assess the stability and safety of compound Ⅱ-6, a preliminary evaluation of metabolism and toxicity was investigated by utilizing the online ADMET software (https://admetmesh.scbdd.com). As shown in Table S3 (Supporting information), Ⅱ-6 was a potential inhibitor or substrate for cytochrome P450 family 2 subfamily C member 9 (CYP2C9), CYP2D6 and CYP3A4. In addition, it revealed that Ⅱ-6 was safe with no potential toxic side effects against multiple targets. Furthermore, the acute toxicity and the plasma stability of compound Ⅱ-6 were conducted accordingly. Ⅱ-6 was quite stable in the complex metabolic factors of plasma, and slight acute toxicity (Figs. S8 and S9 in Supporting information). Taken together, these findings suggested that Ⅱ-6 was a good candidate with low toxicity and could serve as a potential lead for an anti-tumor drug to treat OC.
It was reported that SIRT2 promoted the migration and invasion of gastric cancer through Rat sarcoma/extracellular signal-regulated Kinase/c-Jun N-terminal kinase/MMP-9 (RAS/ERK/JNK/MMP-9) pathway [54], the SIRT2/myelocytomatosis viral oncogene homolog (cMYC) pathway in cholangiocarcinoma [55], and the SIRT2/AKT/p21 pathway in colon cancer [56]. So far, there is no report on the signaling pathway mediated by SIRT2 in OC. Since, the phosphatidylinositol 3-kinase (PI3K)/AKT/mTOR signaling pathway plays an important role in the regulation of OC cell survival, growth, proliferation, angiogenesis, transcription, translation, and metabolism [57]. AKT, a serine/threonine kinase, holds a pivotal position in the PI3K signaling pathway, upregulation AKT activates mTOR, which is contributed to tumorigenesis. As we know, AKT’s activity and phosphorylation state are regulated by SIRT2. Thus, we proposed that SIRT2 might involve in the AKT signaling pathway in OC. Therefore, the pathway enrichment analysis was performed accordingly, the experimental result was shown in Fig. 2. We observed significant inhibition of AKT and p-AKT under the action of 100 nmol/L Ⅱ-6, which in turn leading to the down-regulation of its downstream signaling molecule mTOR (Fig. 2B). This results clearly demonstrated that the overall activity of the SIRT2/AKT/mTOR signaling pathway was effectively inhibited by degrader Ⅱ-6.
Previous reports have demonstrated that SIRT2′s deacetylation activity triggers the activation of DNA-dependent protein kinase (DNA-PK), which subsequently enhances the efficiency of repairing DNA double-strand breaks (DSBs) via the non-homologous end joining (NHEJ) [58]. To further verify whether the degrader Ⅱ-6 can induce DNA damage, we performed the comet assay. In the comet assay, damaged cellular DNA (comprising fragments and broken bonds) was segregated from intact DNA, forming a comet tail shape visible under fluorescence microscopy, while undamaged DNA appears as a round shape without a tail. Compared with control group, Ⅱ-6 elicited a discernible level of DNA damage upon interaction with A2780 cells (Fig. 2C). To further validate this conclusion, we systematically assessed the impact of Ⅱ-6 on downstream DNA damage markers, γH2AX and 53BP1, using a combination of Western blot and fluorescence confocal microscopy. The protein level of 53BP1 gradually decreased as the concentration of Ⅱ-6 increased, while significantly elevating γH2AX levels (Fig. 2D and Fig. S6D in Supporting information), which indicated that more DSBs were produced after being treated with Ⅱ-6.
To investigate the mechanism for the degradation of Ⅱ-6 on DSBs related protein, we focused on SET8 protein, another acetylation substrate of SIRT2 [59]. The absence of SET8 triggers a range of pathological changes, mainly because of the down-regulation of essential factors involved in homologous recombination (HR), including RADiation sensitive 51 (RAD51) and Breast Cancer 2 (BRCA2). This reduction affects the stability and repair proficiency of DNA replication processes, ultimately leading to the accumulation of DNA damage [60]. As expected, SET8 expression lever decreased significantly as SIRT2 degradation (Fig. 2D). As the sole methyltransferase in mammals that targets H4K20, the down-regulation of SET8 directly impacted the methylation status of H4K20, which not only led to a decrease in the expression of H4K20me1, but also the expression of H4K20me2 as well (Figs. 2D and E, Fig. S6E in Supporting information). 53BP1 is a key medium protein that guides DSBs repair through NHEJ, studies have shown that 53BP1 can specifically bind to nucleosomes containing H2AK15ub through ubiquitin-dependent recruitment motifs (UDR). This binding depends on the simultaneous involvement of histone H4K20 dimethylation (H4K20me2) via its tandem Tudor domains, which ensures the precise localization of 53BP1 at DNA damage sites [61]. In other words, the presence of H4K20me2 is essential for the recruitment of 53BP1 to DSBs. From this, we hypothesize that the down-regulation of H4K20me2 would disrupt the normal function of 53BP1.
CLSM analysis was performed to precisely validate the spatial colocalization between H4K20me2 foci and 53BP1 foci. It was worth mentioning that our fluorescence confocal analysis showed a significant degree of colocalization between 53BP1 and H4K20me2 when normal conditions were present (colocalization coefficient = 0.699). The treatment of Ⅱ-6 led to significant reduction in H4K20me2 protein levels (Fig. 2E), which resulted in a weakening of their colocalization efficiency (Figs. 2F and G), with the colocalization coefficient decreasing to 0.29 at Ⅱ-6, 10 µmol/L). This modification had a direct impact on the interaction between H4K20me2 and 53BP1, which ultimately hindered the efficient progression of DDR processes. The results provided a solid theoretical foundation and experimental evidence for the potential of Ⅱ-6 as a potential drug for OC at the molecular level.
To acquire a thorough understanding of how acyl thiourea hydrophobic tag degrader Ⅱ-6 affected the gene transcriptional activities of A2780 cells, we conducted a comprehensive gene transcriptomics analysis using RNA sequencing technology. The analysis demonstrated from a transcriptomic perspective that Ⅱ-6 inhibited the HR mechanism, thus preventing the effective repair process of DSB and ultimately leading to the accumulation of DNA damage. Kyoto encyclopedia of genes and genomes (KEGG) analysis revealed significant enrichment in the AKT-mTOR signaling pathway and the DNA damage signaling pathway (Fig. S10 in Supporting information). In addition, a detailed mechanistic study revealed that the SIRT2 degradation caused by Ⅱ-6 could down-regulate the expression of SET8, its deacetylation substrate, which, in turn, led to a significant reduction in the expression levels of H4K20me1 and H4K20me2 at the histone H4K20 site, a modification that plays a crucial role in maintaining chromatin structure and regulating gene expression [62]. Furthermore, the intricate biochemical regulation directly hindered the recruitment function of the DDR factor 53BP1, resulting in the accumulation of DNA damage (Fig. S11 in Supporting information). Therefore, SIRT2 degradation could impact SET8-mediated epigenetic modifications, and consequently regulate the expression of downstream genes and influence biological behaviors of OC cells. The detailed transcriptomic validation information was outlined in the Supporting information.
In summary, we firstly demonstrated that SIRT2 was highly expressed in OC and correlated with poor prognosis of patients using an in-depth analysis of the clinical characteristics of the expression pattern of SIRT2 in OC patients. Based on the HyT degradation strategy, series of novel SIRT2-targeted acyl thiourea-based HyT degraders were designed and synthesized; their anti- OC activity and detailed mechanism were evaluated accordingly. Gratefully, most compounds exhibited excellent antiproliferative activity against various OC cell lines. Also, excellent degradation activities were observed in A2780 cells. Among them, compound Ⅱ-6 featuring Boc1-Trp exhibited superior degradation, antiproliferative abilities (IC50 = 0.002 ± 0.001 µmol/L), and higher security (selectivity index (SI) = 405.50) as well. Furthermore, it significantly inhibited the metastasis of OC cells and induced cell cycle arrest and apoptosis in vitro. Concurrently, it also showed promising anti-tumor growth effects in vivo. The study establishes a solid foundation for HyT degradation as an effective strategy for treating OC.
Ethical statement
The clinical OC specimens involved in this study were all obtained from participants who voluntarily consented to take part. All research procedures strictly adhered to the ethical principles of the Declaration of Helsinki and were approved by the Ethics Committee of Zhongnan Hospital of Wuhan University (approval number: 2021087). In addition, the animal experiments conducted in this study were carried out in accordance with animal welfare ethical requirements and were reviewed and approved by the Experimental Animal Ethics Committee of Wuhan University (approval number: ZN2024146).
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The work was supported by National Natural Science Foundation of China (Nos. 82173676, 82473775, 82273774, 82073690), Basic and Clinical Medical Research Joint Fund of Zhongnan Hospital Wuhan University (No. ZNLH202201) and the Fundamental Research Funds for the Central Universities of China (No. 2042022dx0003). We gratefully acknowledge the computational support from Xiaogan 3D Scientific Computing Center.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111143.
[1]
R.L. Pullen Jr, Nursing 54 (2024) 17–28.
[2]
R.L. Siegel, A.N. Giaquinto, A. Jemal, CA Cancer J. Clin. 74 (2024) 12–49. doi: 10.3322/caac.21820
[3]
S. Lheureux, M. Braunstein, A.M. Oza, CA Cancer J. Clin. 69 (2019) 280–304. doi: 10.3322/caac.21559
[4]
J.S. Laurent, J.F. Liu, J. Clin. Oncol. 42 (2024) 127–133.
[5]
NCCN Clinical Practice Guidelines in Ovarian Cancer, 2024, https://www.nccnchina.org.cn/.
[6]
N.K. Karanam, L. Ding, A. Aroumougame, M.D. Story, Transl. Res. 217 (2020) 33–46. doi: 10.1016/j.trsl.2019.10.003
[7]
H. Onji, J. Murai, Cancer Sci. 113 (2022) 2943–2951. doi: 10.1111/cas.15477
[8]
A. Pawłowska, A. Rekowska, W. Kuryło, et al., Int. J. Mol. Sci. 24 (2023) 10859. doi: 10.3390/ijms241310859
Figure 1
Degradation activity of these HyT degraders evaluated. A2780 cells were individually pretreated with 10 µmol/L degraders for 36 h before protein level analysis. (A) The mechanism of SIRT2 degradation induced by HyT degraders. (B) Design of ultimate degraders based on acyl thiourea skeletons. (C) Western blot assay was performed to examine the degradation activity of amantadine series degraders Ⅰ-1–6. (D) Degradation activity of Boc1-Trp series degraders Ⅱ-1–9. (E) Degradation activity of Boc2-Lys series degraders Ⅱ-10–16 and Ⅱ-18. (F) Degradation activity of Boc3-Arg series degrader Ⅱ-17 and Ⅲ-1–4. (G) Western blot analysis showed that Ⅱ-6 degraded SIRT2 protein in a time-dependent manner. (H) SIRT2 protein was degraded in a dose-dependent manner. (I) The CETSA assay of Ⅱ-6. (J) Ⅱ-6 induced SIRT2 degradation through proteasome-mediated proteolysis and the autophagy lysosome pathway. Data are presented as mean ± standard deviation (SD) (n = 3). GAPDH, glyceraldehyde-3-phosphate dehydrogenase; CQ, chloroquine.
Figure 2
Antitumor effect in xenograft model and mechanism of Ⅱ-6 in A2780. (A) Tumor volume change of mice during the antitumor treatment. Data are presented as mean ± SD (n = 5). (B) Ⅱ-6 was associated with the SIRT2/AKT/mTOR signaling pathway. (C) The alkaline comet assay image, which showed Ⅱ-6 induced DNA damage in A2780 cells exposed to 10 µmol/L concentration. Scale bar: 100 µm for 10× image and 20 µm for 40× image. (D) Western blot was used to analyze the levels of SET8 and H4K20me1/H4K20me2 after SIRT2 degradation, as well as the levels of DNA damage markers 53BP1 and γ-H2AX. (E) CLSM analysis for H4K20me2 and 53BP1. Scale bar: 3 µm. (F) The plot profiles show fluorescence intensity variations along an oblique line, quantified by ImageJ. (G) Co-localization between two entities is assessed using Pearson coefficients, calculated by JACoP in ImageJ.

 DownLoad:
DownLoad:

 下载:
下载:



 下载:
下载:
