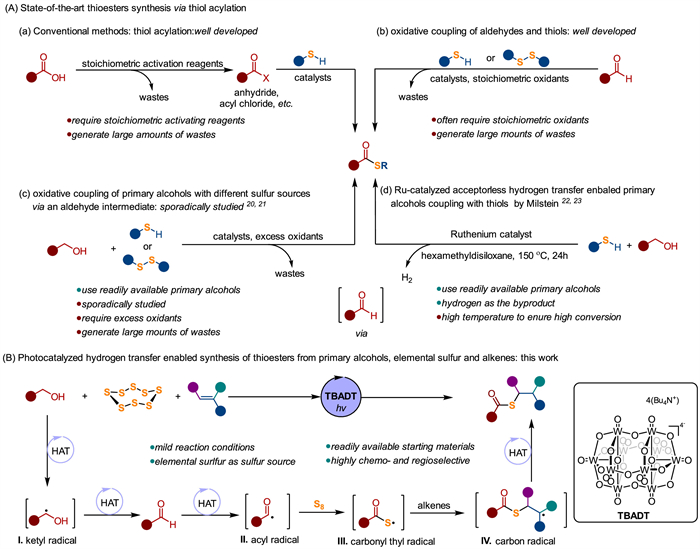
Figure 1.
(A) State-of-the-art thioester synthesis via thiol acylation. (B) This work: Photocatalysed hydrogen transfer enabled synthesis of thioester from primary alcohols, elemental sulfur and alkenes.

The thioester functional group is prevalent in natural products, pharmaceuticals, polymers, and food products [1-6]. Thioesters are integral to various biological processes, notably, the role of acetyl coenzyme A in metabolism [7,8]. Additionally, thioester serve as a versatile platform in synthetic chemistry, facilitating structural diversification through acyl transfer and coupling reactions [9,10]. Consequently, the synthesis of thioesters has intrigued significant interest in both organic synthesis and biosynthetic chemistry.
Traditional methods for thioester synthesis typically involve acylating thiols with carboxylic acid derivatives in the presence of activating reagents or catalysts, which often generate significant waste (Fig. 1A, a) [11-14]. Alternatively, oxidative coupling of aldehydes with thiols offers a more efficient route, through it generally requires excess oxidants (Fig. 1A, b) [15,16]. While primary alcohols are abundant in natural products and commodity chemicals, making them ideal starting materials for constructing complex molecular scaffolds [17-19], their use in thioester synthesis has been sporadically explored, and generally demands excess oxidants (Fig. 1A, c) [20,21]. A recent advancement by the Milstein group introduced a more sustainable approach using a ruthenium-catalyzed acceptorless hydrogen transfer to form thioester from primary alcohols and thiols (Fig. 1A, d). However, this method requires a high temperature (150 ℃) probably due to the reversible nature of the reaction [22,23]. Additionally, thiols are malodorous, unstable, and have a strong affinity to transition metal catalysts, making them less ideal as sulfur sources.
We propose a photo-mediated hydrogen transfer strategy for the synthesis of thioesters from primary alcohols, elemental sulfur and alkenes (Fig. 1B). This method offers a greener and more sustainable approach by using readily available and non-toxic sulfur source, which is abundance, non-toxicity, high-stability, and odourless, especially given the global growing surplus of elemental sulfur [24-33]. Recent studies have shown that elemental sulfur can be activated and participate in radical reactions under photocatalytic conditions for the synthesis of sulfur-containing molecules [28-33]. Notably, thioesters can be obtained from the coupling of alkenes with carbonyl thiyl radicals, which are generated by reacting elemental sulfur with acyl radicals derived from α-ketoacids or aldehydes [32,33]. We envisioned that primary alcohols could generate acyl radical via consecutive photo-mediated hydrogen atom transfer (HAT), proceeding through an aldehyde intermediate. Photo-HAT has proven effective for selectively activating α-hydroxy C—H bonds in alcohols, forming ketyl radical (Ⅰ) that can undergo various radical transformations [34-36]. Primary alcohols can thereof undergo hydrogen transfer to form aldehydes [37-39], which are more accessible for photo-HAT, producing acyl radical (Ⅱ) [40-43]. Subsequently, acyl radical (Ⅱ) can react with elemental sulfur, generating carbonyl thiyl radical (Ⅲ), which then add to alkenes, yielding thioesters. Herein, we describe the photocatalyzed hydrogen transfer enabled synthesis of thioesters from primary alcohols, elemental sulfur and alkenes. This method, employing robust tetrabutylammonium decatungstate (TBADT) as the photocatalyst, operates under mild conditions, accommodates a wide range of primary alcohols and alkenes, and demonstrates high functional group tolerance. However, the method still has room for improvement, particularly regarding the use of the high molecular weight photocatalyst and the excess amounts of alcohol and elemental sulfur.
We began our investigation of the three-component thioester synthesis by optimizing reaction conditions using elemental sulfur as the sulfur source, along with 3-phenylpropanol (1a) and alkene (2a) as model substrates (Tables S1–S5 in Supporting information). After thorough exploration, we achieved an 84% yield of thioester 3a using TBADT as the photocatalyst in acetonitrile at ambient temperature, under violet light irradiation for 4 h (Table 1, entry 1). The yield of 3a is sensitive to the amount of sulfur used, with deviations from the optimal quantity resulting in diminished yields (entries 2 and 3). While varying the amount of 1a had a relatively minor effect on the yield, but 3.5 equiv. of 1a provided optimal results across most reactions (entries 4 and 5, Fig. 2,Fig. 3). The formation of 3a occurred rapidly within the initial three hours, after which the reaction rate slowed (entry 6 and Fig. S5 in Supporting information), thereby 4 h was established as the ideal reaction time for most substrates. Control experiments validated the essential role of both the photocatalyst and light in this transformation (entries 7–9). Additionally, the reaction demonstrated good resistance to water and moderate tolerance of air, underscoring its robustness (entries 10 and 11).
 DownLoad:
CSV
DownLoad:
CSV
 |
||
| Entry | Deviations from the standard conditions | 3a (%) |
| 1 | None | 84 |
| 2 | S (2.0 equiv.) | 63 |
| 3 | S (5.2 equiv.) | 79 |
| 4 | 1a (4.5 equiv.) | 81 |
| 5 | 1a (2.5 equiv.) | 80 |
| 6 | Reaction for 3 h | 78 |
| 7 | w/o TBADT | n.d. |
| 8 | w/o light irradiation | n.d. |
| 9 | Using 2 W 455 nm blue LEDs | n.d. |
| 10 | With 2.0 equiv. of H2O | 77 |
| 11 | Under air atmosphere | 31 |
| a Standard reaction conditions: 1a (3.5 equiv.), S (3.6 equiv.), 2a (0.25 mmol), TBADT (5 mol%), CH3CN (0.5 mol/L), 2 W 385 nm LEDs, under argon atmosphere, at 35–40 ℃, 4 h, GC yields with n-hexadecane as the internal standard. n.d.: not detected. | ||
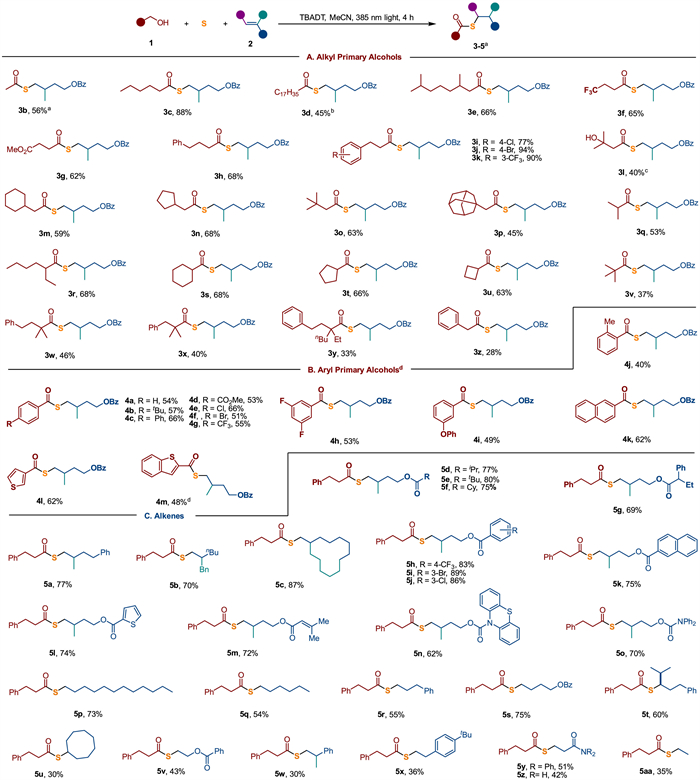

With the optimized reaction conditions in hand, we next explored the generality of this protocol with respect to the alcohol component (Fig. 2A). A wide variety of primary alcohols were successfully coupled with sulfur and 2a, affording the desired thioesters (3b-3z) in acceptable to high yields. Alkyl primary alcohols of varying chain lengths afforded thioester 3b-3d in moderate to high yields. This protocol also demonstrated good tolerance for a range of functional groups, including trifluoromethyl (3f and 3k), ester (3g), aryl halides such as chloride (3i) and bromide (3j), as well as a tertiary alcohol (3l). Additionally, primary alcohols with four-, five- and six-membered rings at the α- or β-carbon of the hydroxyl group were compatible with the reaction, giving thioesters in good yields (3m, 3n, 3s-3u). However, as steric bulk around the alcohol increased, yields tended to decrease (3w-3z), likely due to the formation of relatively stable tertiary carbon radicals that led to alcohol dehydrocarbonylation. A similar outcome was observed with 2-phenylethanol, where the reaction led to the formation of toluene from dehydrocarbonylation via a benzyl radical [44].
Aryl methanols are also competent substrates for this three-component transformation (Fig. 2B). Benzyl alcohols with a range of substituents, including both electron-donating (methyl, t-butyl, phenoxyl, and phenyl) and electron-withdrawing (ester, fluoro, chloro, bromo, and trifluoromethyl) functionalities, reacted smoothly with alkene 2a and sulfur, delivering thioesters (4a-4i) in good yields. Naphthyl methanol also participated successfully, yielding thioester 4k in 62%. Notably, heteroaryl methanols were compatible with the reaction, giving thioesters 4l and 4m in good yields.
We then turned our attention to exploring the scope of alkenes in this three-component reaction (Fig. 2C). Using 3-phenylpropanol (1a) as the primary alcohol component, a wide range of nucleophilic alkenes were successfully converted into thioesters (5a-5aa). 1,1-Disubstituted alkenes bearing various functionalities, such as ester (5a-5o), trifluoromethyl (5h), aryl halides (5i, 5j), and heterocycles (5l, 5n), were efficiently transformed into the corresponding thioesters, with yields ranging from 62% to 89%. Notably, the reaction selectively converted the 1,1-disubstituted nucleophilic alkene, while leaving the tri-substituted nucleophilic counterpart unaffected (5m). Mono-substituted, 1,2-disubstituted and 1,1,2-trisusbtituted alkenes also underwent effective conversion to thioesters (5p-5u). However, cyclooctene exhibited low reactivity under these conditions (5u). Remarkably, activated alkenes, including vinyl ester, aryl ethylenes, and acrylamides were also suitable substrates for this transformation, leading to diverse thioester products (5v-5z). Notably, gaseous ethylene could also participate in the reaction when introduced via an ethylene balloon, although the yield of the thioester (5aa) was low (35%).
To demonstrate the synthetic utility of this methodology, we applied it to the late-stage functionalization of complex natural products and pharmaceuticals (Fig. 3). Natural products containing olefin functionality, such as (-)-isopulegol, (+)-dihydrocarvone, (-)-β-pinene, camphene, nootkatone, and citronellol all reacted smoothly with 3-phenylproponol (1a) and sulfur, producing the desired thioester (6a-6f) with high chemo- and regioselectivity. In cases, where two alkene units were present, the reaction preferentially targeted the electron-rich and less sterically hindered alkene (6e). Notably, the tri-substituted alkene in cetronellol also underwent the transformation efficiently (6f). Similarly, alkene derived from pharmaceutical molecules, such as fenbufen, zaltoprofen, estrone, and indomethacin, were successfully incorporated into the reaction, demonstrating high chemo- and regioselectivity for thioester formation (6g-6j). These results highlight the versatility of this approach in functionalizing complex molecules, offering a valuable tool for the late-stage modification of bioactive compounds.
A series of mechanistic experiments were conducted to elucidate the reaction mechanism (Figs. 4A–C). The reaction involving 3-phenylpropanol (1a), elemental sulfur and alkene 2a was deliberately stopped at an early stage to identify potential intermediates (Fig. 4A, a). Analysis of the reaction mixture revealed that 15% of 1a was converted to 3-phenylpropanal (7, 11%) and thiocarboxylic acid (8, 4%), with t thioester 3a forming at 40% yield. When aldehyde 7 was subjected to the same reaction conditions with sulfur and 2a, it underwent complete conversion to thioester 3a (Fig. 4A, b), as did thiocarboxylic acid 8 (Fig. 4A, c). These results indicate that aldehyde and thiocarboxylic acid are key intermediates in the reaction.
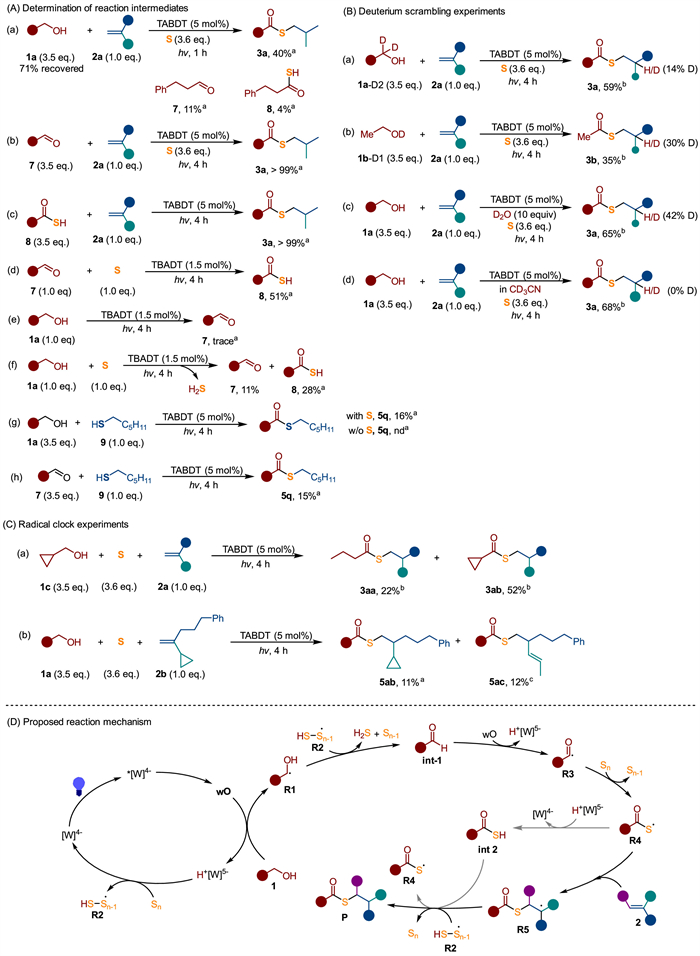
Further investigation revealed that aldehyde 7 was produced early in the reaction, while thiocarboxylic acid 8 accumulated as the formation of 3a slowed down (Fig. S5 in Supporting information). Notably, aldehyde 7 could react with sulfur to yield thiocarboxylic acid 8 under identical reaction conditions (Fig. 4A, d). This suggests that aldehyde is an intermediate that undergoes further transformation to thiocarboxylic acid. Direct conversion of 3-phenylpropanol (1a) to aldehyde 7 was not observed (Fig. 4A, e), but the presence of sulfur was essential for converting 1a to both aldehyde 7 (11%) and thiocarboxylic acid 8 (28%) (Fig. 4A, f), with release of H2S gas, as confirmed by lead acetate test paper (Fig. S8 in Supporting information). This suggests that sulfur as both a hydrogen acceptor and a sulfur source. While alkenes are known to react with H2S under photochemical conditions to form thiols [45,46], which can subsequently undergo coupling with alcohols or aldehydes to form thioesters [20,22], no thioester formation was observed when 1a and 1-hexanethiol (9) were reacted without sulfur (Fig. 4A, g). Even, in the presence of sulfur, the reaction of 1-hexanethiol 9 with 1a or aldehyde 7 did not efficiently afford the thioester (Fig. 4A, g and h), suggesting that a thiol-based pathway is less likely. Deuterium scrambling experiments further supported the involvement of hydrogen transfer in the reaction. Deuterium from labeled alcohols, 1a-D2 and 1b-D1, was incorporated into products 3a and 3b, albeit at low levels (Fig. 4B, a and b). Deuterium incorporation was not observed when using deuterated solvent (Fig. 4B, d), but the addition of D2O led to deuterium incorporation into 3a (Fig. 4B, c). These results suggest that H/D exchange occurs between the alcohol, D2O, and intermediate species during the reaction, likely facilitated by the acidic nature of protonated photocatalyst and reversible HAT [33,47-49].
Additionally, the reaction with cyclopropyl methanol (1c) produced 52% of the ring-intact thioester 3ab and 22% of the ring-opening product 3aa (Fig. 4C, a), indicating the involvement of a ketyl and/or acyl radical intermediate [33,50]. Similarly, the reaction with cyclopropyl-substituted alkene 2b yielded both the ring-opening product 5aa (12%) alongside the ring-remaining product 5z (11%), suggesting the formation of a carbon radical through the addition of a carbonyl thiyl radical to alkene 2b (Fig. 4C, b).
A plausible reaction mechanism based on the mechanistic investigations is proposed in Fig. 4D. The process begins with the excitation of the decatunstate photocatalyst ([W]4−) by violet light, generating a relaxed triplet excited state (wO) through intersystem crossing (ISC) [51]. The excited state wO (E(wO/[W]5−) = 2.44 V vs. SCE) then selectively activates the α-hydroxyl C—H bond of the primary alcohol (1) via HAT, affording the ketyl radical (R1) and reduced decatungstate (H+[W]5−) [37,52,53]. The reduced decatunstate (H+[W]5−) reacts with polysulfur to regenerate the ground state ([W]4−) and produce a hydropolysulfur radical (R2), which participates in the subsequent steps of the reaction. The ketyl radical R1 undergoes HAT with the hydropolysulfur radical (R2), forming an aldehyde intermediate (int-1), along with H2S and a polysulfur species (Sn−1) [54,55]. Alternatively, R1 may be oxidized by wO to directly form the aldehyde intermediate (int-1) after the release of a proton [50]. The aldehyde intermediate (int-1) then undergoes selective C—H bond activation by wO, generating an acyl radical (R3). This R3 reacts with elemental sulfur to generate a carbonyl thiyl radcial (R4) and polysulfur species (Sn−1). The electrophilic radical R4 adds to alkene 2, delivering a carbon radical (R5). However, the direct reduction of R5 by H+[W]5− is challenging due to their mismatched redox potentials [56]. Instead, the thioester product (P) is formed through HAT from R2 to R5. Alternatively, the carbonyl thiyl radical (R4) could react with H+[W]5− to generate a thiocarboxylic acid (int 2), which can then act as hydrogen atom donor to R5, leading to the formation of the thioester product.
In summary, we have developed a three-component reaction for the efficient synthesis of thioesters from readily available feedstock chemicals through photocatalyzed hydrogen transfer of primary alcohols under mild conditions. This approach enabled the rapid and efficient formation of a diverse array of thioesters, demonstrating excellent functional group compatibility. The synthetic utility of this method is examplified by its successful application to in-corporate the thioester functional group into complex pharmaceuticals and natural products. We anticipate that this work will advance the application of photocatalyzed hydrogen transfer of primary alcohols, offering streamlined and sustainable synthetic routes to access complex organic scaffolds.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Tingting Zhang: Methodology, Investigation, Formal analysis. Jing Zhang: Writing – review & editing, Writing – original draft, Visualization, Validation, Supervision, Resources, Project administration, Funding acquisition, Formal analysis, Data curation, Conceptualization.
We thank National Natural Science Foundation of China (Nos. 22071185 and 22271224), the Fundamental Research Funds for the Central Universities (No. 2042019kf0008), and Wuhan University startup funding for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
Y. Kanda, T. Ashizawa, S. Kakita, et al., J. Med. Chem. 42 (1999) 1330–1332. doi: 10.1021/jm9900366
N. Wang, P. Saidhareddy, X. Jiang, Nat. Prod. Rep. 37 (2020) 246–275. doi: 10.1039/c8np00093j
K. Taori, V.J. Paul, H. Luesch, J. Am. Chem. Soc. 130 (2008) 1806–1807. doi: 10.1021/ja7110064
X.L. Liu, Y. Shi, J.S. Kang, P. Oelschlaeger, K.W. Yang, ACS Med. Chem. Lett. 6 (2015) 660–664. doi: 10.1021/acsmedchemlett.5b00098
S. Aksakal, R. Aksakal, C.R. Becer, Poly. Chem. 9 (2018) 4507–4516. doi: 10.1039/c8py00872h
N. Martin, V. Neelz, H.E. Spinnler, Food Qual. Prefer. 15 (2004) 247–257. doi: 10.1016/S0950-3293(03)00065-X
F. Pietrocola, L. Galluzzi, J.M. Bravo-San Pedro, F. Madeo, G. Kroemer, Cell Metab. 21 (2015) 805–821. doi: 10.1016/j.cmet.2015.05.014
J. Franke, C. Hertweck, Cell Chem. Biol. 23 (2016) 1179–1192. doi: 10.1016/j.chembiol.2016.08.014
V. Agouridas, O.E. Mahdi, V. Diemer, et al., Chem. Rev. 119 (2019) 7328–7443. doi: 10.1021/acs.chemrev.8b00712
S. Sikandar, A.F. Zahoor, S. Naheed, et al., Mol. Divers. 26 (2022) 589–628. doi: 10.1007/s11030-021-10194-7
S. Iimura, K. Manabe, S. Kobayashi, Chem. Commun. 38 (2002) 94–95.
H. Wang, Z. Liu, A. Das, et al., Nat. Synth. 2 (2023) 1116–1126. doi: 10.1038/s44160-023-00353-z
C. Santi, B. Battistelli, L. Testaferri, M. Tiecco, Green Chem. 14 (2012) 1277–1280. doi: 10.1039/c2gc16541d
M. Kazemi, L. Shiri, J. Sulfur Chem. 36 (2015) 613–623. doi: 10.1080/17415993.2015.1075023
T. Uno, T. Inokuma, Y. Takemoto, Chem. Commun. 48 (2012) 1901–1903. doi: 10.1039/c2cc17183j
C.L. Yi, Y.T. Huang, C.F. Lee, Green Chem. 15 (2013) 2476–2484. doi: 10.1039/c3gc40946e
V. Zoufal, S. Mairinger, M. Brackhan, et al., J. Nucl. Med. 61 (2020) 1050–1057. doi: 10.2967/jnumed.119.237198
N. Kosaric, Z. Duvnjak, A. Farkas, et al., Ethanol, in: C. Ley, B. Elvers, et al. (Eds.), Ullmann’s Encyclopedia of Industrial Chemistry, seventh ed., Wiley-VCH, 2011, pp. 333–403.
J. Ott, V. Gronemann, F. Pontzen, et al., Methanol, in: C. Ley, B. Elvers, et al. (Eds.), Ullmann’s Encyclopedia of Industrial Chemistry, 7th ed., Wiley-VCH, 2011, pp. 1–27.
V.J. Roy, P.P. Sen, S.R. Roy, J. Org. Chem. 86 (2021) 16965–16976. doi: 10.1021/acs.joc.1c02111
X. Zhu, Y. Shi, H. Mao, Y. Cheng, C. Zhu, Adv. Synth. Catal. 355 (2013) 3558–3562. doi: 10.1002/adsc.201300584
J. Luo, M. Rauch, L. Avram, et al., Nat. Catal. 3 (2020) 887–892. doi: 10.1038/s41929-020-00514-9
M. Rauch, J. Luo, L. Avram, Y. Ben-David, D. Milstein, ACS Catal. 11 (2021) 2795–2807. doi: 10.1021/acscatal.1c00418
T.V. Choudhary, J. Malandra, J. Green, S. Parrott, B. Johnson, Angew. Chem. Int. Ed. 45 (2006) 3299–3303. doi: 10.1002/anie.200503660
H. Liu, X. Jiang, Chem. Asian J. 8 (2013) 2546–2563. doi: 10.1002/asia.201300636
T.B. Nguyen, Adv. Synth. Catal. 362 (2020) 3448–3484. doi: 10.1002/adsc.202000535
H. Huang, G.-J. Deng, S. Liu, Synlett 32 (2021) 142–158. doi: 10.1364/ol.409729
M. Wang, Z. Dai, X. Jiang, Nat. Commun. 10 (2019) 2661. doi: 10.1038/s41467-019-10651-w
A. Porey, S.O. Fremin, S. Nand, et al., ACS Catal. 14 (2024) 6973–6980. doi: 10.1021/acscatal.4c01289
T.B. Nguyen, Asian J. Org. Chem. 6 (6) (2017) 477–491. doi: 10.1002/ajoc.201700011
T.B. Nguyen, Adv. Synth. Catal. 359 (2017) 1066–1130. doi: 10.1002/adsc.201601329
S. Murakami, T. Nanjo, Y. Takemoto, Org. Lett. 23 (2021) 7650–7655 2021. doi: 10.1021/acs.orglett.1c02904
H. Tang, M. Zhang, Y. Zhang, et al., J. Am. Chem. Soc. 145 (2023) 5846–5854. doi: 10.1021/jacs.2c13157
S. Protti, M. Fagnoni, D. Ravelli, ChemCatChem 7 (2015) 1516–1523. doi: 10.1002/cctc.201500125
D. Ravelli, M. Fagnoni, T. Fukuyama, T. Nishikawa, I. Ryu, ACS Catal. 8 (2018) 701–713. doi: 10.1021/acscatal.7b03354
L. Capaldo, D. Ravelli, M. Fagnoni, Chem. Rev. 122 (2022) 1875–1924. doi: 10.1021/acs.chemrev.1c00263
T. Yamase, N. Takabayashi, M. Kaji, J. Chem. Soc., Dalton Trans. 1984 (1984) 793–799.
J.J. Zhong, W.P. To, Y. Liu, W. Lu, C.M. Che, Chem. Sci. 10 (2019) 4883–4889. doi: 10.1039/c8sc05600e
X.J. Yang, Y.W. Zheng, L.Q. Zheng, et al., Green Chem. 21 (2019) 1401–1405. doi: 10.1039/c8gc03828g
Y.L. Liu, Y.J. Ouyang, H. Zheng, H. Liu, W.T. Wei, Chem. Commun. 57 (2021) 6111–6120. doi: 10.1039/d1cc02112e
C. Raviola, S. Protti, D. Ravelli, M. Fagnoni, Green Chem. 21 (2019) 748–764. doi: 10.1039/c8gc03810d
M.A. Theodoropoulou, N.F. Nikitas, C.G. Kokotos, Bellstein J. Org. Chem. 16 (2020) 833–857. doi: 10.3762/bjoc.16.76
A. Banerjee, Z. Lei, M.Y. Ngai, Synthesis 51 (2019) 303–333. doi: 10.1055/s-0037-1610329
D.J. Abrams, J.G. West, E.J. Sorensen, Chem. Sci. 8 (2017) 1954–1959. doi: 10.1039/C6SC04607J
W.E. Vaughan, F.F. Rust, J. Org. Chem. 7 (1942) 472–476. doi: 10.1021/jo01200a004
K.I. Sugimoto, W. Ando, S. Oae, Bull. Chem. Soc. Jpn. 37 (1964) 365–368. doi: 10.1246/bcsj.37.365
T. Yamase, T. Usami, J. Chem. Soc., Dalton Trans. 1988 (1988) 183–190.
I. Ryu, A. Tani, T. Fukuyama, et al., Angew. Chem. Int. Ed. 50 (2011) 1869–1872. doi: 10.1002/anie.201004854
Y. Kuang, H. Cao, H. Tang, et al., Chem. Sci. 11 (2020) 8912–8918. doi: 10.1039/d0sc02661a
D. Dondi, M. Fagnoni, A. Albini, Chem. Eur. J. 12 (2006) 4153–4163. doi: 10.1002/chem.200501216
V.D. Waele, O. Poizat, M. Fagnoni, A. Bagno, D. Ravelli, ACS Catal. 6 (2016) 7174–7182. doi: 10.1021/acscatal.6b01984
T. Fukuyama, K. Yamada, T. Nishikawa, et al., Chem. Lett. 47 (2018) 207–209. doi: 10.1246/cl.171068
K. Yamada, T. Fukuyama, S. Fujii, et al., Chem. Eur. J. 23 (2017) 8615–8618. doi: 10.1002/chem.201701865
A.W. Horton, Dehydrogenation of alcohols with sulfur, Patent, US2522676, 1950.
B. Zheng, L. Zhong, X. Wang, et al., Nat. Commun. 15 (2024) 5507. doi: 10.1038/s41467-024-49374-y
A. Prieto, M. Taillefer, Org. Lett. 23 (2021) 1484–1488. doi: 10.1021/acs.orglett.1c00189

Figure 1 (A) State-of-the-art thioester synthesis via thiol acylation. (B) This work: Photocatalysed hydrogen transfer enabled synthesis of thioester from primary alcohols, elemental sulfur and alkenes.

Figure 2 Substrate scope of alcohols. Conditions: 1 (3.5 equiv.), S (3.6 equiv.), 2 (0.25 mmol, 1.0 equiv.), TBADT (5 mol%), CH3CN (0.5 mL), LEDs (2 W, 385 nm) irradiation for 4 h at 35–40 ℃, isolated yields. a 1 (5.0 equiv.). b CH3CN (1.0 mL). c 1 (1.5 equiv.). d 8 h reaction.

Figure 4 Mechanistic investigations and proposed reaction mechanism. See Supporting information for details. a GC yields with n-hexadecane as the internal standard. b Isolated yield, D% determined by 1H NMR; c NMR yield determined using mesitylene as the internal standard.
Table 1. Evaluation of the reaction parameters.a
|
||
| Entry | Deviations from the standard conditions | 3a (%) |
| 1 | None | 84 |
| 2 | S (2.0 equiv.) | 63 |
| 3 | S (5.2 equiv.) | 79 |
| 4 | 1a (4.5 equiv.) | 81 |
| 5 | 1a (2.5 equiv.) | 80 |
| 6 | Reaction for 3 h | 78 |
| 7 | w/o TBADT | n.d. |
| 8 | w/o light irradiation | n.d. |
| 9 | Using 2 W 455 nm blue LEDs | n.d. |
| 10 | With 2.0 equiv. of H2O | 77 |
| 11 | Under air atmosphere | 31 |
| a Standard reaction conditions: 1a (3.5 equiv.), S (3.6 equiv.), 2a (0.25 mmol), TBADT (5 mol%), CH3CN (0.5 mol/L), 2 W 385 nm LEDs, under argon atmosphere, at 35–40 ℃, 4 h, GC yields with n-hexadecane as the internal standard. n.d.: not detected. | ||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: