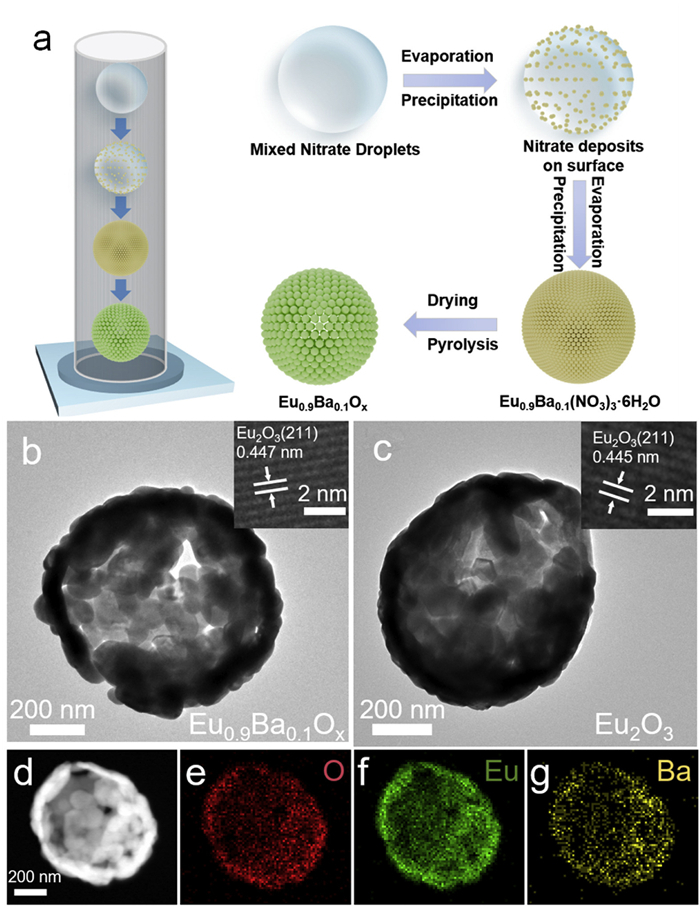
Figure 1.
Structure of the Eu0.9Ba0.1Ox catalyst. (a) Schematic diagram of catalyst synthesis (b, c) TEM image and HRTEM image of Eu0.9Ba0.1Ox and Eu2O3 (d-g) Corresponding EDX mapping images of Eu0.9Ba0.1Ox.
Ba-doping to modulate surface electronic states in Eu0.9Ba0.1Ox catalysts for oxidation coupling of methane
Shibo Zhang , Xiang Chu , Meng Zhao , Baokang Geng , Xiao Wang , Shuyan Song , Hongjie Zhang

Methane, the main component of shale gas and natural gas, holds immense potential for the single step production of value added hydrocarbons and oxygenated compounds owing to its abundant availability [1–4]. However, it is hard to directly activate or convert methane due to its chemical internes including the low polarizability, negligible electron affinity and high C–H bond dissociation energy [5,6]. Therefore, high temperature is usually required to overcome the thermodynamic barriers and initiate reaction [7–9]. Among various direct transformation methods, the OCM reaction has garnered research interest in the past decades because it can produce valuable C2+ hydrocarbons [10]. The OCM is believed to require the following processes: (1) The C–H bond in methane dissociates to give methyl radicals, catalyzed by the surface active oxygen; (2) The coupling reaction involving the obtained methyl radical result in the formation of ethane; (3) The dehydrogenation process converts ethane into ethylene [11]. Simultaneously, methane may also undergo deep dehydrogenation and oxidation, resulting in the formation of carbon dioxide and carbon monoxide, which poses a major obstacle to achieving high yields of C2 hydrocarbons during the OCM process, rendering it a formidable challenge [12]. Therefore, it is imperative to search for well-crafted catalysts that can optimize the adsorption/desorption behavior of intermediates and effectively attain high yields of the desired products.
Rare-earth (RE) oxide with unfilled 4f orbitals and the high thermal stability, have been a hot research topic in OCM reactions [13–16]. However, despite their potential, the selectivity and yield of C2 product over RE oxide catalysts remain suboptimal. To address this issue, surface modification and regulation of the catalysts represent a promising strategy [17]. In the OCM reaction, oxygen vacancies play a crucial role in activating oxygen to produce reactive oxygen species. These species, in turn, facilitate the subsequent dehydrogenation steps of methane and ethane [18]. Many efforts have been made to increase the amount of oxygen vacancies on the catalysts surface [19]. For instance, Sim et al. successfully doped Al into La2O3 lattice, which promoted the formation of oxygen vacancies and consequently increased the C2 selectivity from 35% to 42% [20]. Zhang et al. combined La and Ce to form a stable La2Ce2O7 compound with a disordered defective cubic fluorite structure and increased C2 yield from 12% to 15% [21]. It is therefore clearly evident that there is still a great deal of room for improvement in terms of C2 selectivity and conversion rates in OCM. Hence, the strategic design of the material surface, coupled with the augmentation of oxygen vacancies, emerges as promising strategies for optimizing the performance of OCM reactions.
In this study, Ba-doped Eu2O3 catalysts were prepared using ultrasonic spray pyrolysis, exhibiting high C2 selectivity and high stability for OCM reactions. A variety of characterization techniques were employed to gain insight into the structural and physicochemical properties of the Ba-doped catalysts, including transmission electron microscopy (TEM), X-ray diffraction (XRD) analysis, X-ray photoelectron spectroscopy (XPS), electron paramagnetic resonance (EPR) and programmed temperature rise desorption (TPD) analysis. The findings demonstrate that Ba doping in Eu2O3 can markedly enhance the number of basic sites and the concentration of oxygen vacancies on the catalyst surface. This results in the generation of reactive oxygen species, which in turn improves the C2 selectivity and thus enhances the catalytic performance.
As illustrated in Fig. 1a, Ba-doped Eu2O3 catalyst was prepared via ultrasonic spray pyrolysis. The spray solution was initially underwent ultrasonic treatment within a spray generator, and followed by calcining in a tube furnace, wherein the aerosol precursor decomposed into powder [22]. The SEM and TEM images displays that the obtained products are regular spherical polyhedron with a diameter of 0.5–3 μm (Fig. S1 in Supporting information and Figs. 1b and c). The sharp contrast observed between the center and the edges allows for the inference that the primary grains possess a hollow structure. The creation of the unique hollow structure can be attributed to the rapid evaporation of water from the droplet surface within the tube furnace, which is accompanied by the swift deposition of nitrate onto the particle surfaces, leading to the formation of M(NO3)3–6H2O shells. Once the solvent evaporation is complete, the nitrate undergoes dehydration and ultimately decomposes, yielding single shell hollow oxide particles [23]. Throughout the entire process, the spray droplets can serve as individual reactors. The high-resolution TEM image in the inset demonstrates that the crystal region of pure Eu2O3 exhibits a lattice stripe spacing of 0.445 nm, which corresponds to the (211) planar spacing of cubic Eu2O3. In contrast, the lattice fringe spacing of the Ba-doped Eu2O3 crystal region is 0.447 nm, which can be attributed to the substitution of Ba2+ for Eu3+ along with a bigger ionic radius of Ba (135 pm) than that of Eu (95 pm). Further characterization by the corresponding EDX elemental mapping reveals the uniform distribution of Eu, Ba, and O elements (Figs. 1d-g). The X-ray diffraction (XRD) pattern of Eu2O3 (Fig. S2 in Supporting information) is in accordance with the standard card (PDF #34–0392) and exhibits a space group of la
As shown in Fig. 2, we evaluated the OCM catalytic performance of various catalysts. As shown in Fig. 2a, while the reaction temperature rises from 550 ℃ to 800 ℃, both the methane conversion and C2+ selectivity exhibit a steady augmentation. That is account for the fact that higher temperatures facilitate the cleavage of the C–H bond within the methane molecule. Specifically, the methane conversion of Eu0.9Ba0.1Ox catalyst was 30.9% as the temperature reached 800 ℃. Among all the products, the selectivity for ethane and ethylene were 19.2% and 30.9% respectively, while there was a minor proportion of C3 products and a significant majority of by-products consisting of CO2. As a contrast, the methane conversion over bare Eu2O3 was 30.1%, but the C2+ selectivity was relatively low at 41.6%. The C2+ selectivity of bare BaO was comparable to that of Eu0.9Ba0.1Ox, however, the methane conversion was significantly lower, standing at just 8.7% (Fig. 2b, Figs. S3 and S4 in Supporting information). The results underscore the superiority of Ba-doping in terms of enhancing the selectivity of C2+. To ascertain the role of oxygen, we conduct further investigation into the performance of OCM reaction under O2-lean and O2-rich conditions. As shown in Fig. 2c, as the feeding ratio of CH4 to O2 was increased from 2:1 to 3:1, the methane conversion continued to decrease from 37.8% to 30.9%, but the C2+ selectivity continued to increase from 42.9% to 50.2%. The reason for this phenomenon is that an O2-rich environment favors not only the activation of methane but also the undesirable overoxidation of methane, leading to a simultaneous increase in methane conversion and a decrease in C2+ selectivity, and a similar situation occurs with the reduction of GHSV (Fig. S5 in Supporting information) [15]. Subsequently, an investigation was extended to encompass other rare earth elements beyond Eu, as exemplified in Fig. 2d. In a comparative analysis, the catalytic activity exhibited by Eu0.9Ba0.1Ox surpasses that of Ce0.9Ba0.1Ox, Pr0.9Ba0.1Ox and Er0.9Ba0.1Ox catalysts (Figs. S6-S10 in Supporting information). The C2+ yields of the Eu0.9Ba0.1Ox catalysts were superior to most of the catalysts reported in the literature (Fig. S11 in Supporting information).
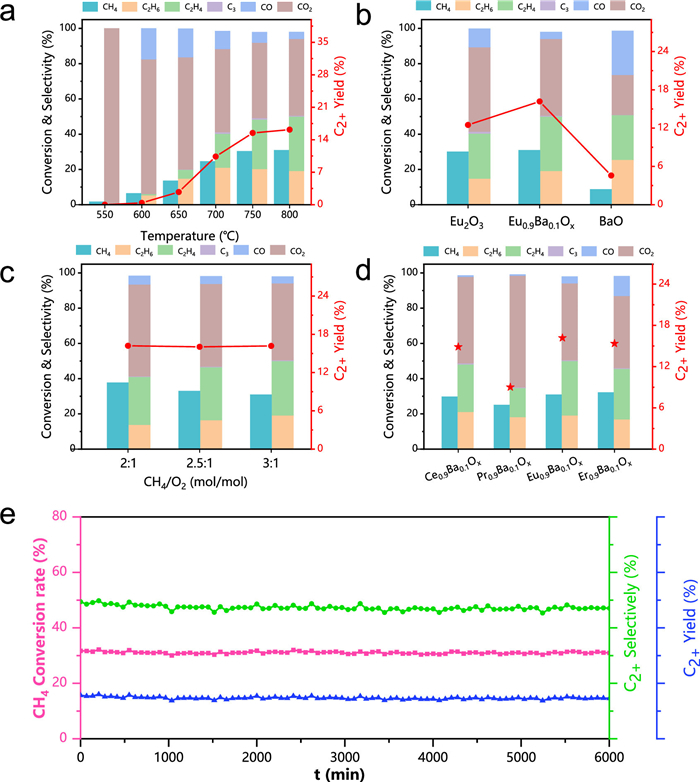
Finally, the findings depicted in Fig. 2e illustrate that the Eu0.9Ba0.1Ox catalyst maintains a methane conversion rate within a narrow band of 31%−32% over a prolonged reaction period of 100 h. The catalyst exhibits high thermal stability and no carbon buildup (Fig. S12 in Supporting information). Concurrently, the yield of C2 products remains consistently stable at approximately 15%−16%, exhibiting extremely stable OCM reaction activity. After undergoing rigorous durability testing, the samples were thoroughly characterized. As shown in Fig. S13 (Supporting information), the XRD pattern remained nearly indistinguishable from their initial state, with no impurity diffraction peaks detected. TEM analysis provided additional confirmation that the morphology and structural integrity of the catalysts sustained unchanged throughout the entire testing period (Fig. S14 in Supporting information).
Fig. 3a and Fig. S15 (Supporting information) illustrate the Raman spectra of pure Eu2O3 and Eu0.9Ba0.1Ox, which were acquired under ambient conditions utilizing a 633 nm excitation laser. Both samples exhibit a prominent band at 339.5 cm−1, along with two distinct peaks at 285.8 and 432.2 cm−1. The band centered at 339.5 and 432.2 cm−1 are assigned to the Fg mode of Eu2O3 crystal, whereas the band at 285.8 cm−1 is ascribed to the combined Fg + Eg mode [26]. In the Eu0.9Ba0.1Ox sample, the aforementioned peaks exhibit a slight shift towards lower wavelengths, which can be attributed to the phonon confinement effects arising from an increased Ov concentration [27]. To gain better qualitative and quantitative insights into the unpaired electrons in substances, EPR measurement was constructed. As shown in Fig. 3b, the g-value signal associated with unpaired electrons captured by oxygen vacancies (2.003) displays a noticeable augmentation in the Eu0.9Ba0.1Ox catalyst [28]. The presence of Ov facilitates the dissociative adsorption of O2 to generate surface-active electrophilic oxygen, which is a pivotal active site for OCM reactions [29]. All of the aforementioned results powerful confirm that the incorporation of Ba atoms into Eu2O3 lattice plays an instrumental role in enhancing the concentration of oxygen vacancies and enabling efficient oxygen activation.
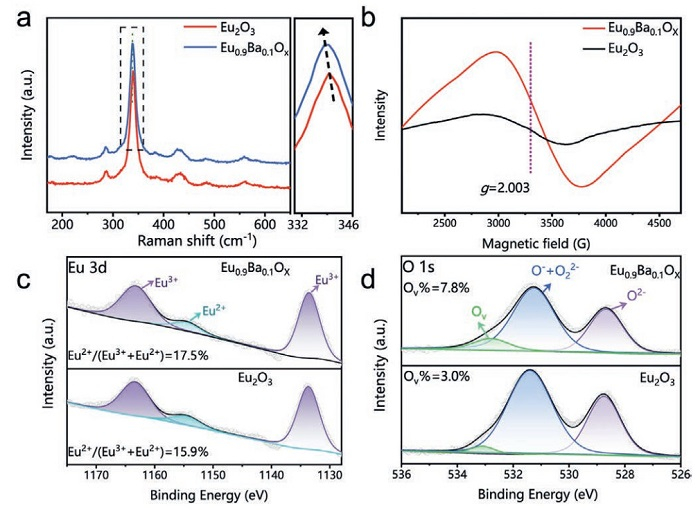
We further investigate the surface elemental composition and fine chemical state of the Eu2O3 catalyst both before and after doping Ba by XPS analysis [30]. In the Eu 3d spectra (Fig. 3c), it can be found that the peak at 1133.6 and 1163.3 eV corresponds to Eu3+ and the peak at 1154.6 eV is attributed to Eu2+ [31]. By calculating the areas under the Eu3+ and Eu2+ peaks, it was determined that the percentage of the Eu2+ state in the Eu0.9Ba0.1Ox catalyst increased from 15.9% to 17.5% after Ba doping. The concentration of oxygen vacancies in the catalyst is closely related to the percentage of the Eu2+ state. In order to maintain charge conservation, a decrease in the Eu valence state leads to the appearance of oxygen vacancies. As shown in Fig. 3d, the O 1s spectrum in Eu0.9Ba0.1Ox catalyst can be deconvoluted into three secondary peaks, which have been assigned to the oxygen vacancies Ov (532.8 eV), surface-adsorbed oxygen O22− and O− (529.8 eV) and lattice oxygen O2− (528.8 eV) [32]. It is noteworthy that the substantial rise in Ov content, from 3.0% in Eu2O3 to 7.8% in Eu0.9Ba0.1Ox, coincides with the increased in Eu2+ content on the catalyst surface. The proportion of Eu2+ and Ov in the Eu0.9Ba0.1Ox catalyst is almost constant before and after the reaction (Figs. S16 and S17 in Supporting information). The above results further confirm that the incorporation of Ba atoms into Eu2O3 lattice can effectively promote the formation of oxygen vacancies, as evidenced by both Raman and EPR analyses. It is also demonstrated that the quantity of Ov is the key to promote the OCM catalytic performance.
For better understanding the structure-activity relationship of catalysts, the O2-TPD characterization was utilized to get a deeper insight into the intrinsic correlation between surface oxygen species and catalytic performance. As shown in Fig. 4a, the O2-TPD profile was divided into three sections: the desorption peaks of surface physically adsorbed oxygen (50–300 ℃), surface chemisorbed oxygen (300–550 ℃), and surface lattice oxygen species (550–800 ℃) [33]. It has been proven that the presence of surface chemisorbed oxygen facilitates the activation of methane and enhance the subsequent dehydrogenation of ethane whereas the surface lattice oxygen (O2−) promotes the complete oxidation of hydrocarbons to COx. As expected, it can be observed that the two catalysts exhibit distinct desorption profiles for oxygen species. Specifically, the amount of desorbed chemisorbed oxygen was higher in the Eu0.9Ba0.1Ox catalyst compared to Eu2O3, aligning with our findings regarding their respective catalytic performances. In H2-TPR curve (Fig. S18 in Supporting information), the Eu0.9Ba0.1Ox catalysts have stronger and more pronounced reduction peaks compared to the Eu2O3 catalysts, indicating that the oxygen on the surface of the Eu0.9Ba0.1Ox catalysts is more likely to bind with hydrogen and form oxygen vacancies.

It is well known that the acidity and alkalinity of the catalyst surface are significant variables which significantly influence the efficacy of the OCM process. On the one hand, the conversion of oxygen into reactive oxygen species on the catalyst surface necessitates a specific alkalinity. On the other hand, the generated reactive oxygen species also requires a certain alkalinity to be stabilized [34]. Accordingly, the alkaline sites on the catalyst surface were investigated through CO2-TPD (Fig. 4b). The alkaline sites were classified into three categories based on the CO2 desorption temperature, namely weakly alkaline sites at 50–250 ℃, moderately strong alkaline sites at 250–600 ℃, and strongly alkaline sites at 600–800 ℃ [35]. However, weakly basic sites are prone to be inactive at elevated temperatures, while strongly basic sites are susceptible to inactivation due to the formation of carbonates upon combining with CO2; thus, medium-strength basic sites are optimal for achieving high C2 selectivity. It can be found that the number of medium-strength basic sites of the Ba-doped Eu0.9Ba0.1Ox catalyst increased significantly in comparison with that of Eu2O3, which is consistent with the observed catalytic activity.
In order to identify whether CH4 is activated to methyl radicals by lattice oxygen of metal oxides or surface reactive oxygen species in this catalytic system, we performed CH4-TPSR experiments. Prior to test, the Eu0.9Ba0.1Ox catalyst was first purged with He at 700 ℃ to remove reactive oxygen species (O2−, O−, etc.) on the catalyst surface. The results are shown in Fig. 4c, where the coupling product of the OCM reaction (C3H6), was rapidly formed at 580 ℃, and the deep oxidation product (COx), appeared at 750 ℃. This indicates that the coupling reaction starts to occur above 580 ℃. To verify the possibility of the reactive oxygen species mechanism, the Eu0.9Ba0.1Ox catalyst was directly subjected to CH4-TPSR experiments. As shown in Fig. 4d, the product peaks of C3H6 and CO2 can be detected at a low temperature of 360 ℃. The above comparative results intuitively indicate that reactive oxygen species play an important role in the activity of Eu0.9Ba0.1Ox catalysts, which is because the coupling reaction reacts violently at 360 ℃ in the presence of reactive oxygen species, and the rapid coupling reaction temperature rises to 580 ℃ after removal of the reactive oxygen species.
Based on the comprehensive analysis of the above characterization results, the catalytic pathway for OCM reaction over the Eu0.9Ba0.1Ox catalyst is clearly illustrated in Fig. 4e. The doping of Ba ions significantly increased the number of oxygen vacancies on the catalyst surface, thereby effectively facilitated the adsorption and activation of O2. This, in turn, fostered the evolution of O2− species and further promoted the dissociative coupling of methane to form ethane. Additionally, the presence of O2− species also facilitated the dehydrogenation of ethane to ethylene, ultimately enhancing the overall performance of the oxidative coupling of methane (OCM) reaction.
In conclusion, a monolayer hollow Eu0.9Ba0.1Ox catalyst was successfully prepared and utilized for the OCM reaction. The doping of Ba ions in cubic Eu2O3 promoted the formation of Ov and increased the number of active oxygen sites. The Eu0.9Ba0.1Ox catalyst exhibited satisfactory performance and stability in the OCM reaction, achieving a CH4 conversion of 30.9%, a selectivity of 52.4% and a yield of 16.2% for C2+ product, and maintained its catalytic activity for up to 100 h. The Ba doping of Eu2O3 increased the number of surface oxygen vacancies, which is conducive to the activation of oxygen into active O2−, and increased the number of medium-strength basic sites on the surface, which is conducive to the stabilization of the generated reactive oxygen species. This strategy holds great promise for advancing the design of highly active and stable rare earth oxide-based materials for catalytic applications.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Shibo Zhang: Writing – original draft, Investigation, Formal analysis. Xiang Chu: Investigation. Meng Zhao: Writing – review & editing, Formal analysis. Baokang Geng: Investigation. Xiao Wang: Writing – review & editing, Supervision, Project administration. Shuyan Song: Writing – review & editing, Validation, Supervision, Project administration. Hongjie Zhang: Supervision.
This work was supported by the financial aid from National Science and Technology Major Project of China (No. 2023YFB3508000), National Natural Science Foundation of China (Nos. 22020102003, 22025506, 22271274 and U23A20140), Jilin Province Science and Technology Development Plan Project (Nos. 20230101035JC, 20230101022JC and 20240402056GH), Jilin Province Innovation and Entrepreneurship Talent Project (No. E31S6105) and Yunan Province Science and Technology Development Plan Project (No. 202301BC070001–003). X. Wang acknowledges funding from National Natural Science Foundation of China Outstanding Youth Science Foundation of China (Overseas).
Supplementary material associated with this article can be found, in the online version, at doi:
X. Sun, G. Liu, T. Shen, et al., Small 20 (2024) e2310857. doi: 10.1002/smll.202310857
M. Li, A.C. van Veen, Appl. Catal. B: Environ. 237 (2018) 641–648. doi: 10.1016/j.apcatb.2018.06.032
S.R. Patlolla, K. Katsu, A. Sharafian, et al., Renew. Sust. Energ. Rev. 181 (2023) 113323. doi: 10.1016/j.rser.2023.113323
J. Kim, M. Ferree, S. Gunduz, et al., ChemCatChem 15 (2023) 122026.
P. Wang, R. Shi, Y. Zhao, et al., Angew. Chem. Int. Ed. 62 (2023) e202304301. doi: 10.1002/anie.202304301
Y. Wang, G. Hong, Y. Zhang, et al., Angew. Chem. Int. Ed. 62 (2023) e202310525. doi: 10.1002/anie.202310525
A.L.A. Marinho, F.S. Toniolo, F.B. Noronha, et al., Appl. Catal. B: Environ. 281 (2021) 119459. doi: 10.1016/j.apcatb.2020.119459
A.L.A. Marinho, R.C. Rabelo-Neto, F. Epron, et al., Appl. Catal. B: Environ. 268 (2020) 118387. doi: 10.1016/j.apcatb.2019.118387
N. Sun, J. Zhang, L. Ling, et al., Fuel 354 (2023) 129398. doi: 10.1016/j.fuel.2023.129398
J. Kim, Y.J. Kim, M. Ferree, et al., Appl. Catal. B: Environ. 321 (2023) 122026. doi: 10.1016/j.apcatb.2022.122026
A. Zanina, V.A. Kondratenko, H. Lund, et al., ACS Catal. 12 (2022) 15361–15372. doi: 10.1021/acscatal.2c04916
C. Karakaya, R.J. Kee, Prog. Energy Combust. Sci. 55 (2016) 60–97. doi: 10.1016/j.pecs.2016.04.003
Y. Jiang, H. Fu, Z. Liang, et al., Chem. Soc. Rev. 53 (2024) 714–763. doi: 10.1039/d3cs00708a
X. Wu, X. Wang, L. Zhang, et al., Angew. Chem. Int. Ed. 63 (2024) e202317594. doi: 10.1002/anie.202317594
K. Zhao, Y. Gao, X. Wang, et al., Nat. Commun. 14 (2023) 7749. doi: 10.1038/s41467-023-43682-5
J. Xu, Y. Zhang, X. Xu, et al., ACS Catal. 9 (2019) 4030–4045. doi: 10.1021/acscatal.9b00022
K. Wang, M. Lv, T. Si, et al., J. Hazard. Mater. 461 (2024) 132479. doi: 10.1016/j.jhazmat.2023.132479
Y. Gambo, A.A. Jalil, S. Triwahyono, et al., J. Ind. Eng. Chem. 59 (2018) 218–229. doi: 10.1016/j.jiec.2017.10.027
Y. Ivanova, R. Petrov, S. Reshetnikov, et al., Mater. Today Chem. 26 (2022) 101124. doi: 10.1016/j.mtchem.2022.101124
Y. Sim, J. Yoo, J.M. Ha, et al., J. Energy Chem. 35 (2019) 1–8.
Z. Zhang, Y. Gong, J. Xu, et al., Catal. Today 400-401 (2022) 73–81. doi: 10.1016/j.cattod.2021.11.012
H. Du, K. Huang, M. Li, et al., Nano Res. 11 (2018) 1490–1499. doi: 10.1007/s12274-017-1766-1
J. Leng, Z. Wang, J. Wang, et al., Chem. Soc. Rev. 48 (2019) 3015–3072. doi: 10.1039/c8cs00904j
K. Yuan, Y.W. Zhang, Inorg. Chem. Front. 7 (2020) 4256–4280. doi: 10.1039/d0qi00750a
M. Luo, M. Wang, W. Niu, et al., Chem. Eng. J. 412 (2021) 128471. doi: 10.1016/j.cej.2021.128471
M.V. Abrashev, N.D. Todorov, J. Appl. Phys. 116 (2014) 103508. doi: 10.1063/1.4894775
J. Ibáñez, O. Blázquez, S. Hernández, et al., J. Raman Spectrosc. 49 (2018) 2021–2027. doi: 10.1002/jrs.5488
S.M. Wu, X.L. Liu, X.L. Lian, et al., Adv. Mater. 30 (2018) e1802173. doi: 10.1002/adma.201802173
R. Feng, P. Niu, B. Hou, et al., J. Energy Chem. 67 (2022) 342–353. doi: 10.1016/j.jechem.2021.10.018
C. Zheng, J. Feng, Z. Wei, et al., Chem. Eng. J. 467 (2023) 143390. doi: 10.1016/j.cej.2023.143390
M. Wang, P. Guo, Y. Zhang, et al., J. Hazard. Mater. 349 (2018) 224–233. doi: 10.1016/j.jhazmat.2018.01.058
Y. Zhu, X. Wu, Z. Wu, et al., Adv. Funct. Mater. 34 (2024) 2409324. doi: 10.1002/adfm.202409324
T. Wu, Y. Wei, J. Xiong, et al., J. Energy Chem. 91 (2024) 331–344. doi: 10.1016/j.jechem.2023.12.025
T. Wu, P. Zhang, Y. Wei, et al., ACS Catal. 14 (2024) 1882–1902. doi: 10.1021/acscatal.3c05094
R.S. Pal, S. Rana, S.K. Sharma, et al., Chem. Eng. J. 458 (2023) 141379. doi: 10.1016/j.cej.2023.141379

Figure 1 Structure of the Eu0.9Ba0.1Ox catalyst. (a) Schematic diagram of catalyst synthesis (b, c) TEM image and HRTEM image of Eu0.9Ba0.1Ox and Eu2O3 (d-g) Corresponding EDX mapping images of Eu0.9Ba0.1Ox.

Figure 2 Catalytic performance investigations. (a) Temperature effect for Eu0.9Ba0.1Ox. (b) Catalytic performances of Eu2O3, Eu0.9Ba0.1Ox and BaO. (c) CH4/O2 (mol/mol) ratios of the Eu0.9Ba0.1Ox catalyst. (d) Redox OCM performance comparison of Ce0.9Ba0.1Ox, Pr0.9Ba0.1Ox, Eu0.9Ba0.1Ox and Er0.9Ba0.1Ox. (e) Long-term stability test of the Eu0.9Ba0.1Ox catalyst at 800 ℃, 3:1 CH4/O2 ratio, and 2000 mL h−1 gcat−1 GHSV for 6000 min.

Figure 3 Characterizations of catalyst structure. (a) Raman spectra of Eu0.9Ba0.1Ox and Eu2O3. (b) EPR spectra of Eu0.9Ba0.1Ox and Eu2O3. (c) Eu 3d and (d) O 1s XPS spectra of Eu0.9Ba0.1Ox and Eu2O3.

Figure 4 Mechanism investigation. (a) CO2-TPD of Eu0.9Ba0.1Ox and Eu2O3. (b) O2-TPD of Eu0.9Ba0.1Ox and Eu2O3. (c) CH4-TPSR spectra of Eu0.9Ba0.1Ox catalysts pretreated with He at 700 ℃. (d) CH4-TPSR spectra of Eu0.9Ba0.1Ox catalysts without He pretreatment. (e) Structural schematic diagram of Eu0.9Ba0.1Ox.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: