School of Environment, School of Chemistry and Chemical Engineering, State Key Laboratory of Water Pollution Control and Green Resource Recycling, State Key Laboratory of Analytical Chemistry for Life Science, the Frontiers Science Center for Critical Earth Material Cycling, Nanjing University, Nanjing 210023, China
b.
Key Laboratory of Energy Thermal Conversion and Control of Ministry of Education, School of Energy and Environment, Southeast University, Nanjing 210096, China
c.
Key Laboratory for Organic Electronics & Information Displays and Institute of Advanced Materials, Nanjing University of Posts & Telecommunications, Nanjing 210096, China
d.
School of Energy and Environment, City University of Hong Kong, Hong Kong SAR 999077, China
wenleizhu@nju.edu.cn (W. Zhu). 1 These authors contributed equally to this work.
Received Date:
26 December 2024 Accepted Date:
18 March 2025 Revised Date:
14 March 2025 Available Online:
01 July 2026
Abstract:
The development of photosynthetic biological systems (PBSs) presents a promising approach to mitigating global climate change. However, the practical application of PBSs remains hindered by their low product yields. Key determinants of production efficiency include light utilization, electron transfer efficiency, and catalyst stability. To address these challenges, we developed a high-performance Cupriavidus necator/CdS@Au@Poly dimethyl diallyl ammonium chloride (C. necator/CdS@Au@PDDA) biohybrid system for the photocatalytic conversion of CO2 into bioplastic poly(3-hydroxybutyrate) (PHB). The incorporation of Au nanoclusters extends the visible light absorption range and alleviates photocorrosion of CdS, while the PDDA modification enhances electron transfer rates and enables the material to firmly adhere to the bacterial surface. In situ H2 production by CdS@Au@PDDA drives CO2 fixation through bacterial metabolic pathways, achieving a quantum efficiency of 2.76% ± 0.22% and a maximum PHB yield of 53.6 ± 5.2 mg/L, representing the highest yield reported for C. necator-based artificial PBSs. This biohybrid system demonstrates the effective integration of advanced nanomaterials with microbial processes, offering a robust platform for sustainable bioplastic production through carbon-neutral artificial photosynthesis technology and providing a novel perspective for addressing the global challenge of microplastic pollution.
Global warming and climate change represent one of the most pressing challenges faced by humanity in the 21st century. Photosynthetic biological systems (PBSs), which could utilize complexes of inorganic nanomaterials and non-photosynthetic bacteria as catalysts, offer a promising solution to these environmental crises [1-3]. By leveraging solar energy, PBSs could convert CO2 into high-value chemicals or fuels [4]. In these systems, artificial photosensitizers (e.g., semiconductors) capture light to generate electrons or catalyze water splitting to produce hydrogen [5-8]. These electrons or hydrogen are subsequently transferred to bacteria through electron transfer proteins, mediators, or hydrogenases, where bacterial metabolic pathways drive the conversion of CO2 into high-value chemicals or fuels [9-11]. PBSs combine the high product selectivity of bacteria, and the superior light-harvesting capabilities of semiconductors, achieving significant progress in CO2 reduction and solar-to-chemical energy conversion [12]. However, the primary barrier to the practical application of PBSs lies in their low product yields [13]. Extensive studies have investigated key factors such as light utilization efficiency, mass transfer efficiency of electron donors, and catalyst stability [1]. Research efforts have primarily focused on modifying specific bacteria with functional nanomaterials, including semiconductors, polymers, and metal-based nanomaterials, to develop efficient artificial photosynthetic hybrids for high-performance CO2 photocatalytic conversion [2,14].
Moreover, microplastic and nanoplastic pollution has emerged as another critical challenge in the field of environmental science [15,16]. These pollutants are widely distributed in water, soil, and air, posing potential threats to ecosystems and human health [17,18]. By designing efficient PBSs to convert CO2 into biodegradable plastics, such as poly(3-hydroxybutyrate) (PHB), it is possible to not only mitigate CO2 emissions but also significantly reduce the use of conventional plastics, offering a novel strategy for addressing plastic pollution and promoting sustainable development [19-21]. PHB is a biodegradable polymer with properties comparable to conventional plastics, making it a promising alternative in sustainable material applications. It exhibits high biocompatibility, non-toxicity, and excellent mechanical properties, making it suitable for biomedical applications such as sutures, tissue engineering, and drug delivery systems. Additionally, its biodegradability under natural conditions positions it as a competitive alternative to petroleum-based plastics in packaging and disposable product industries. PHB synthesized via microbial biosynthesis exhibits high crystallinity (60%−80%), providing excellent mechanical strength but inherent brittleness, with a molecular weight (0.5–1.2 × 10⁶ Da) comparable to industrial PHB. Cupriavidus necator (formerly known as Ralstonia eutropha) could utilize hydrogen as an energy source and CO2 as a carbon source, producing biodegradable PHB under carbon-rich, nitrogen- or phosphorus-limited conditions [22-28]. Given the low solubility of H2 in water and its tendency to escape, we designed a system where hydrogen-producing photocatalysts were coated on the surface of C. necator, enabling in situ cascade catalysis on the inorganic and biological interface. Inorganic semiconductors typically possess suitable band structures for photocatalytic hydrogen production [7,29]. However, these materials often suffer from high electron-hole recombination rates, susceptibility to oxidation in the presence of oxygen and water, and photodegradation under light irradiation, resulting in a significant reduction in catalytic activity [30-34]. To address these issues, further modifications of such inorganic semiconductors are necessary, including the construction of composite catalysts to enhance their stability and performance.
In this study, we constructed a solar-driven C. necator/CdS@Au@PDDA hybrid system for CO2 reduction into bioplastics (Fig. 1). The Au nanoclusters (Au NCs) on the CdS surface could significantly enhance the light absorption range, utilization efficiency of photogenerated charge carriers, electron transfer efficiency, and photostability, enabling efficient photocatalytic hydrogen production through water splitting [35]. As a functional biocompatible polymer, the coating of PDDA on the CdS@Au surface not only enhances the conductivity of CdS but also enables the material to firmly adhere to the bacterial surface. Hydrogen generated by CdS@Au@PDDA was transferred into bacterial cells via hydrogenase on the bacterial surface, serving as an energy source for CO2 reduction into PHB. The biohybrid system achieved a notable PHB production of 53.6 ± 5.2 mg/L with a total quantum efficiency of 2.76% ± 0.22%, surpassing the yield of natural photosynthetic processes and representing the highest reported production rate among artificial photosynthetic systems utilizing C. necator. This work highlights the potential of engineered hybrid biocatalytic systems in promoting sustainable CO2 utilization and mitigating plastic pollution, while providing a pathway for the development of environmentally friendly and efficient photocatalytic materials.
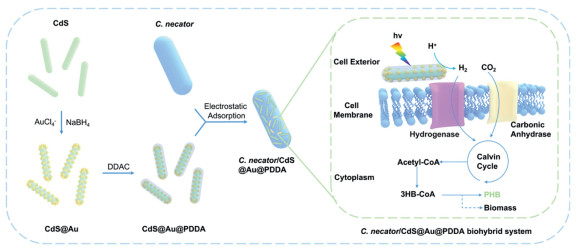
Figure 1
Figure 1.
Preparation and photocatalytic mechanism of the C. necator/CdS@Au@PDDA biohybrid system. Au NCs are synthesized by the reduction of AuCl₄⁻ with NaBH₄ and loaded onto CdS nanorods to form CdS@Au, followed by surface modification with PDDA. Subsequently, CdS@Au@PDDA adheres to the surface of C. necator via electrostatic adsorption. Under light irradiation, it generates photogenerated electrons, catalyzing the reduction of H⁺ to H2 and facilitating CO2 fixation. Through the synergistic action of carbonic anhydrase and hydrogenase, H2 and CO2 are converted into 3HB-CoA, which is further utilized for the biosynthesis of PHB and biomass.
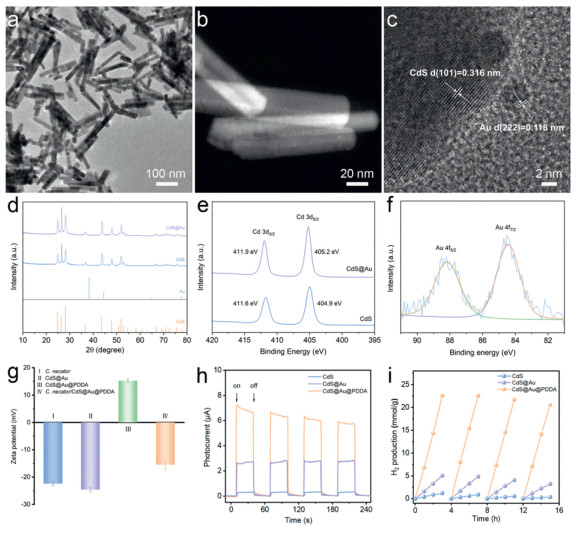
CdS nanorods were successfully synthesized via a typical hydrothermal method at 180 ℃ for 24 h. The resulting CdS nanorods exhibit a uniform rod-like morphology with lengths ranging from 100 nm to 200 nm (Fig. 2a). This regular and highly ordered structure provides a large specific surface area and excellent dispersibility for subsequent metal nanoparticle loading, which could effectively reduce particle aggregation. A gold loading of 1 wt% was subsequently deposited onto the surface of CdS nanorods via NaBH₄ reduction (Fig. S1 in Supporting information). The results demonstrate that Au NCs are uniformly distributed on the CdS nanorods without aggregation (Fig. 2b). HRTEM further revealed the lattice spacing of CdS and Au, with the CdS lattice spacing measured as 3.16 Å, corresponding to the (101) plane, and the Au lattice spacing as 1.18 Å, corresponding to the (222) plane (Fig. 2c). XRD patterns indicate that the primary diffraction peaks of CdS remain unchanged after Au modification, confirming that the hexagonal crystalline structure of CdS is preserved (Fig. 2d). Moreover, no significant diffraction peaks for Au are observed, suggesting the presence of Au in the form of nanoclusters rather than nanoparticles. To further investigate the influence of Au modification on the chemical environment of CdS, XPS analysis was performed (Fig. 2e and Fig. S2 in Supporting information). Compared with pristine CdS, the binding energies of Cd 3d and S 2p in CdS@Au are shifted by 0.3 and 0.1 eV toward higher energies, respectively. This shift indicates that the introduction of Au alters the electronic distribution on the CdS surface, reducing the electron density of Cd due to electron transfer from Cd to Au, which reflects strong interfacial interactions between Au and CdS (Fig. 2e). Additionally, the Au 4f spectrum exhibits binding energies of 88.45 eV (Au 4f7/2) and 88.15 eV (Au 4f5/2), indicating that Au exists in a zero-valence state and is primarily distributed as nanoclusters on the CdS surface (Fig. 2f).
Figure 2
Figure 2.
Characterization and performance analysis of CdS@Au@PDDA nanocomposite. (a) TEM image of CdS nanorods. (b) High-angle annular dark field (HAADF) STEM image of CdS@Au, showing Au clusters distributed uniformly across the surface of CdS nanorods. (c) High-resolution transmission electron microscopy (HRTEM) image of CdS@Au, illustrating the lattice spacings of CdS and Au crystals, measured as 0.316 nm for CdS and 0.118 nm for Au. (d) XRD patterns of CdS and CdS@Au. (e) XPS spectra of Cd 3d, demonstrating the influence of Au modification on the chemical state of Cd atoms. (f) XPS spectra of Au 4f, indicating the chemical state of Au on the CdS surface. (g) Zeta potential comparison showing the surface charge characteristics of various samples. (h) Photocurrent response under light on-off cycles. (i) Time-dependent H2 production curves from cycling photocatalytic tests under light irradiation (AM 1.5 G).
Previous studies have demonstrated that conductive polymers can act as charge transfer mediators, effectively improving the separation and migration efficiency of photogenerated charge carriers, thereby enhancing the photocatalytic performance of semiconductor materials [36-38]. Herein, PDDA was introduced onto the surface of CdS@Au based on our previously established method [39]. This modification not only significantly enhanced the photocatalytic performance of the material but also imparted a positive surface charge, facilitating subsequent interactions with bacteria. The zeta potential of unmodified CdS@Au was measured to be −22.5 mV, which is similar to that of C. necator (−22 mV) (Fig. 2g). After PDDA modification, the zeta potential of CdS@Au@PDDA increased to 15.2 mV, and this positive charge promoted the electrostatic adsorption of bacteria onto the material, forming a stable biohybrid system. Furthermore, UV–vis spectroscopy revealed that PDDA modification did not affect the visible light absorption of CdS (Fig. S3 in Supporting information).
The optical properties of the materials were subsequently characterized. UV–vis DRS results revealed that the absorption edge of pure CdS was at approximately 500 nm, corresponding to its typical bandgap of 2.4 eV (Fig. S4 in Supporting information). In contrast, the CdS@Au@PDDA sample exhibited significantly enhanced absorption in the range of 500–700 nm, primarily attributed to the surface plasmon resonance effect of Au, which markedly improved light absorption in the visible and near-infrared regions. Tauc plots showed that the bandgap of pure CdS was 2.40 eV, while the bandgap of CdS@Au@PDDA decreased slightly to 2.37 eV (Fig. S5 in Supporting information). This minor reduction in bandgap suggests that although Au NCs did not substantially alter the electronic structure of CdS, it effectively extended the light response range and enhanced optical performance, particularly in the visible region. Photocurrent response measurements indicated that CdS@Au (~2.7 μA) and CdS@Au@PDDA (~7.1 μA) exhibited significantly higher photocurrents than pure CdS (~0.3 μA) under light on-off cycling conditions (Fig. 2h). These results demonstrate that the synergistic effect of Au modification and PDDA functionalization significantly improved the separation efficiency of photogenerated charge carriers and reduced the probability of charge recombination, thereby enhancing the photoelectric performance of the material. To evaluate the stability of CdS@Au@PDDA, a four-cycle (16 h) stability test was conducted (Fig. 2i). The performance of pristine CdS decreased gradually after the second cycle, with the hydrogen production in the fourth cycle reduced to one-fourth of the first cycle (357.72 μmol/g). In comparison, CdS@Au exhibited improved cyclic stability, with the hydrogen production in the fourth cycle reaching approximately 60% of the first cycle (3247.29 μmol/g). Further PDDA modification resulted in a significant enhancement in both hydrogen production and stability, with the fourth-cycle hydrogen production reaching 20, 531.09 μmol/g. These findings highlight the effective synergistic interaction between the surface plasmon resonance effect of Au and the interfacial optimization provided by PDDA, which collectively enhanced the photocatalytic efficiency and cyclic stability of CdS.
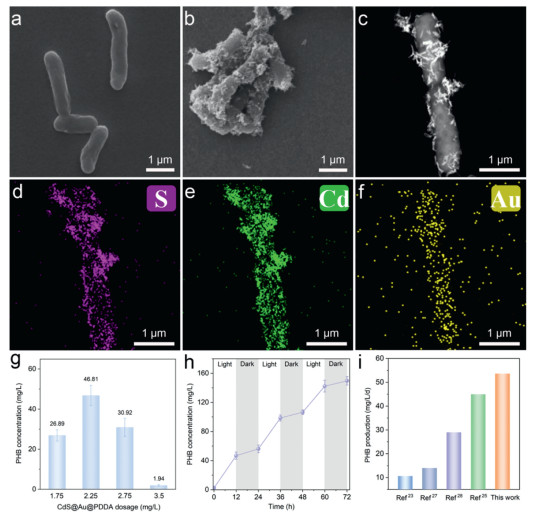
The composite of C. necator/CdS@Au@PDDA was successfully achieved through electrostatic interactions. The pristine C. necator cells exhibited a typical rod-shaped morphology, with smooth surfaces and lengths of approximately 1.5–4 μm (Fig. 3a). After composite formation, the bacterial surfaces were uniformly decorated with CdS@Au@PDDA nanorods (Fig. 3b). High-angle annular dark field (HAADF) STEM image further revealed that CdS@Au@PDDA nanorods were evenly distributed on the surface of dividing C. necator cells, demonstrating excellent stability of the composite material and robust interaction between the bacteria and the nanorods (Fig. 3c). To further investigate the elemental composition and distribution of CdS@Au@PDDA, EDS mapping was performed (Fig. 3d-f). The results showed that S, Cd, and Au were uniformly distributed across the bacterial surface, confirming the successful formation of C. necator/CdS@Au@PDDA. Such uniform distribution is crucial for enhancing photocatalytic performance, as it effectively minimizes charge recombination, thereby improving photocatalytic reaction efficiency.
Figure 3
Figure 3.
Morphology, elemental distribution, and photocatalytic performance for PHB synthesis of C. necator/CdS@Au@PDDA. (a) SEM image of C. necator. (b) SEM image of the C. necator/CdS@Au@PDDA. (c) HAADF-STEM image of dividing C. necator/CdS@Au@PDDA. (d-f) Energy-dispersive X-ray spectroscopy (EDS) elemental mapping images showing the distribution of S, Cd, and Au, indicating that CdS@Au@PDDA are uniformly distributed on the bacterial surface. (g) PHB concentration changes under different CdS@Au@PDDA dosages for the photocatalytic synthesis of PHB using C. necator/CdS@Au@PDDA. (h) The effect of light-dark cycles on PHB production. (i) Comparison of PHB production performance in this work with previously reported studies.
To evaluate the CO2 reduction capabilities of the C. necator/CdS@Au@PDDA, a series of 12-h photocatalytic experiments were conducted under simulated sunlight with l-cysteine as the sacrificial agent. The extracted photosynthetic PHB and its corresponding ¹H—NMR spectrum are shown in Fig. S6 (Supporting information). The results indicated that PHB production was not affected when the l-cysteine concentration was within the range of 0.3–0.5 wt% (Fig. S7 in Supporting information). Additionally, the amount of CdS@Au@PDDA nanorods played a decisive role in the energy supply to the system and significantly influenced photocatalytic efficiency. Excessive material loading inhibited bacterial activity and caused aggregation, while insufficient loading failed to meet energy demands, thus limiting CO2 fixation efficiency. At an optimal CdS@Au@PDDA loading of 2.25 mg/L, the PHB concentration reached a maximum of 46.81 mg/L; however, increasing the loading to 2.75 mg/L led to a decline in PHB production (Fig. 3g). These findings suggest that an appropriate amount of CdS@Au@PDDA enhances photocatalytic performance, while excessive loading may result in light-blocking effects or particle aggregation, thereby reducing the performance. Light intensity was also identified as a critical factor influencing CO2 fixation efficiency. Under simulated sunlight at 30 mW/cm2, the C. necator/CdS@Au@PDDA system exhibited optimal CO2 reduction performance, producing 46.81 ± 5.0 mg/L of PHB within 12 h (Fig. S8 in Supporting information). In contrast, a simple mixture of C. necator and CdS@Au generated only 7.18 ± 0.85 mg/L of PHB, corresponding to 15.4% of the composite system. This significant difference is attributed to the tight integration between the CdS@Au@PDDA and the bacteria, enabling the rapid in situ utilization of hydrogen produced by CdS@Au nanorods. While Au exhibits excellent conductivity, stability, and catalytic activity, its high price may hinder industrial applications. However, our study demonstrates that only a minimal amount of Au is required to achieve significant performance enhancements. The optimized Au loading effectively improves electron transfer efficiency and photocorrosion resistance without excessive material costs. One possible alternative to Au is the use of other transition metals such as silver (Ag), copper (Cu), or nickel (Ni), which possess similar charge transfer capabilities at a lower cost. However, these materials often exhibit inferior stability compared to Au, particularly under long-term photocatalytic conditions, where oxidation and agglomeration can lead to reduced performance. Another promising approach is the use of conductive polymers as substitutes or co-modifiers, which can enhance charge transport while reducing reliance on precious metals. While Au-based systems may incur higher initial costs, their superior durability and efficiency could reduce long-term operational expenses by minimizing catalyst degradation and replacement frequency. Future research will focus on optimizing material composition and exploring cost-effective alternatives to balance performance and economic viability.
We then conducted a 72-h day-night experiment to assess the stability of C. necator/CdS@Au@PDDA (Fig. 3h). During the initial light phase (0–12 h), PHB concentration increased rapidly, which is due to the efficient light harvesting and hydrogen generation by CdS@Au@PDDA under illumination, therefore directly driving bacterial metabolism. During the subsequent dark phase (12–24 h), PHB concentration stabilized, suggesting that bacterial metabolism relied on residual hydrogen and accumulated reducing equivalents to continue CO2 reduction, ultimately achieving a maximum PHB yield of 53.6 ± 5.2 mg/L, reaching the highest PHB yield of C. necator in the PBS system (Fig. 3i and Table S1 in Supporting information) [23,25,27,28]. In contrast, the system produced only 2.49 ± 0.63 mg/L PHB after 12 h under dark conditions. Additionally, the quantum efficiency of 2.76% ± 0.22% surpasses that of traditional CdS-based biohybrid systems, which often exhibit values below 2%. In summary, the integration of bacteria with CdS@Au@PDDA nanocomposites effectively leveraged photocatalytically generated in situ hydrogen to enhance PHB synthesis. This synergistic strategy offers valuable insights into the application of photocatalysis in microbial metabolic regulation and high-value bioproduct synthesis, while providing a novel pathway for the development of environmentally friendly photocatalytic materials.
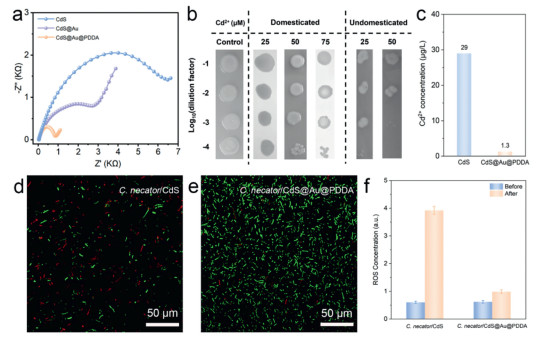
To elucidate the superior PHB production achieved by C. necator/CdS@Au@PDDA, we investigated the charge transfer performance of the nanomaterial interface using EIS [40]. The Nyquist plot shows that CdS exhibits the largest charge transfer resistance (Rct), indicating its poor charge transfer capability (Fig. 4a). After modification with Au NCs, the Rct of CdS@Au decreased to 4 kΩ, attributed to the enhanced conductivity imparted by Au NCs. Further incorporation of C. necator/CdS@Au@PDDA reduced Rct to 820 Ω, demonstrating that the PDDA layer not only effectively protected the CdS substrate but also enhanced the separation and migration efficiency of photogenerated charges, as well as electron transfer capabilities. These enhancements collectively contributed to the improved H2 production rate of the C. necator/CdS@Au@PDDA system. It is noteworthy that photocorrosion is an unavoidable challenge in photocatalytic processes, as CdS releases Cd2+ under light irradiation, negatively impacting C. necator activity. To improve the Cd2+ tolerance of C. necator, the bacteria were acclimated through 20 generations before photosynthetic experiments. The growth ability of acclimated and non-acclimated C. necator strains was evaluated under different Cd2+ concentrations using dilution spot assays (Fig. 4b). The results showed that acclimated strains exhibited higher tolerance at 25 and 50 μmol/L Cd2+ concentrations, while non-acclimated strains displayed reduced growth under the same conditions. The acclimation process effectively improved the bacterial adaptation to heavy metal stress, particularly at high Cd2+ concentrations [41]. Furthermore, bacterial growth curves confirmed that the presence of 25 µmol/L Cd2+ significantly affected the growth dynamics of non-domesticated C. necator, particularly by prolonging the early logarithmic phase and reducing overall growth rates (Fig. S9 in Supporting information). This delay is attributed to Cd2+ toxicity, which disrupts essential cellular functions, including membrane integrity and enzymatic activity. In contrast, domesticated C. necator strains exhibited improved tolerance, with growth curves showing a recovery in doubling time and final cell density similar to Cd2+-free conditions. This suggests that prolonged exposure led to adaptive mechanisms, such as enhanced expression of metal efflux pumps or intracellular Cd2+ sequestration proteins [41]. In contrast, the PHB production of non-acclimated C. necator in the photocatalytic CO2 fixation experiment was only 19.67 ± 2.21 mg/L, accounting for only 43% of the PHB production achieved by the acclimated strain. To further evaluate material stability, the Cd2+ release concentrations of CdS and CdS@Au@PDDA were compared under identical photocatalytic conditions. As shown in Fig. 4c, after 12 h of photocatalytic reaction, the Cd2+ release concentration from CdS reached as high as 29 μg/L, whereas CdS@Au@PDDA released only 1.3 μg/L of Cd2+. This significant difference indicates that Au NCs modification and PDDA coating not only enhanced the chemical stability of the material but also effectively reduced Cd2+ leaching, thereby maintaining high bacterial activity during the reaction and significantly reducing potential environmental toxicity. Moreover, continuous operation could lead to progressive Cd2+ accumulation, necessitating mitigation strategies such as periodic media replacement or chelation-based Cd2+ removal to further suppress leaching.
Figure 4
Figure 4.
Electrochemical properties, biocompatibility, and ROS quenching capacity of C. necator/CdS@Au@PDDA. (a) EIS spectra illustrating the interfacial charge transfer resistance of CdS, CdS@Au, and CdS@Au@PDDA composites. (b) Cd2+ tolerance assays comparing the growth of adapted and non-adapted C. necator under different Cd2+ concentrations (25, 50, and 75 μmol/L). (c) Comparison of Cd2+ leaching between CdS and CdS@Au@PDDA composites during photocatalytic reactions. (d, e) CLSM images of C. necator/CdS (d) and C. necator/CdS@Au@PDDA (e), respectively. The living C. necator cells were stained with a LIVE/DEAD BacLight kit, which consists of green fluorescent SYTO 9 (live cells) and red fluorescent propidium iodide (dead cells). (f) The changes in ROS concentrations before and after photocatalytic reactions.
We employed a Live/Dead bacterial viability assay to examine their survival rates after 12 h of simulated solar irradiation to assess bacterial activity during photocatalytic reactions. CLSM images revealed that green fluorescence (representing live bacteria) was significantly reduced in the C. necator/CdS system, while red fluorescence (representing dead bacteria) dominated, indicating widespread bacterial death (Fig. 4d). In contrast, the C. necator/CdS@Au@PDDA system exhibited predominantly green fluorescence, signifying that CdS@Au@PDDA provides a more favorable catalytic environment due to its lower toxicity and higher biocompatibility (Fig. 4e). Notably, reactive oxygen species (ROS) levels are critical in determining bacterial survival rates and the stability of reaction systems. Previous studies have shown that Au NCs effectively suppress ROS generation during photocatalysis through multiple mechanisms [42,43]. First, Au NCs act as electron reservoirs, capturing photogenerated electrons from the semiconductor and reducing electron-hole recombination, thereby preventing the formation of superoxide and hydroxyl radicals. Second, they can catalyze the reduction of oxygen through multi-electron pathways, favoring the formation of less reactive species like H2O2 instead of harmful radicals. Their strong affinity for ROS also allows them to scavenge and neutralize oxidative species directly. Moreover, AuNCs influence the semiconductor's band structure, shifting the Fermi level and lowering the energy available for ROS generation. These combined effects not only mitigate oxidative damage but also enhance photocatalytic efficiency and material stability. Therefore, we measured ROS levels in different biohybrid systems after 12 h of light irradiation (Fig. 4f). The results showed significantly elevated ROS levels in C. necator/CdS, while ROS levels in C. necator/CdS@Au@PDDA were notably reduced, indicating the latter effectively suppressed ROS generation. The substantial reduction in ROS not only decreased cytotoxicity within the biohybrid system but also improved bacterial survival rates, ultimately enhancing system stability and increasing PHB production. However, further increasing the Au loading did not enhance the system's ROS suppression capability (Fig. S10 in Supporting information). In summary, C. necator/CdS@Au@PDDA demonstrated significant advantages in charge transfer capability, environmental stability, and biocompatibility. The Au modification and PDDA coating not only effectively minimized Cd2+ release but also reduced ROS levels during photocatalysis, thereby enhancing bacterial survival rates and system stability.
Compared to previous PBSs utilizing semiconductor-bacteria hybrids [23,25], our system demonstrates key technological advancements in the following aspects: (1) Enhanced electron transfer efficiency. The incorporation of Au NCs extends the visible light absorption range of CdS, reducing charge recombination and improving photogenerated electron transfer. EIS confirms a Rct compared to CdS-only systems, enabling more efficient electron delivery to bacterial metabolic pathways. (2) Improved stability and photocorrosion resistance. CdS photocorrosion, a major limitation in PBS applications, is mitigated through the synergistic effects of Au NCs and PDDA modification. PDDA functions as a protective layer, reducing Cd2+ leaching from CdS nanorods. Experimental results indicate that Cd2+ release in the CdS@Au@PDDA system is reduced to 1.3 µg/L, compared to 29 µg/L for unmodified CdS, ensuring prolonged bacterial viability. (3) Superior PHB yield and quantum efficiency. The PHB production of 53.6 ± 5.2 mg/L achieved in our system represents the highest reported yield among C. necator-based artificial PBSs. The quantum efficiency of 2.76 ± 0.22% surpasses many existing biohybrid platforms, demonstrating the effectiveness of our material modifications in optimizing microbial CO2 reduction efficiency. (4) Structural and functional integration for improved biocompatibility. Unlike previous semiconductor-bacteria hybrids, where limited bacterial adhesion reduces charge transfer efficiency, PDDA-modified CdS@Au exhibits strong electrostatic interactions with C. necator, ensuring stable biohybrid formation. This structural optimization enables prolonged operational stability under continuous illumination conditions.
In summary, we have developed a high-performance C. necator/CdS@Au@PDDA biohybrid system for photocatalytic CO2 conversion into bioplastics. By integrating Au NCs and PDDA coating onto CdS, C. necator/CdS@Au@PDDA achieves enhanced light absorption, efficient charge transfer, and reduced photocorrosion, enabling in situ H2 production that drives microbial CO2 fixation pathways. The biohybrid system demonstrates a record-breaking PHB yield of 53.6 ± 5.2 mg/L and a quantum efficiency of 2.76% ± 0.22%, representing the highest reported production for C. necator-based artificial photosynthetic systems. Moreover, the incorporation of Au and PDDA effectively suppresses ROS generation and minimizes Cd2+ leaching, ensuring environmental stability and microbial viability. This study provides a scalable and environmentally sustainable platform for CO2 reduction and bioplastic production, offering a promising pathway toward mitigating climate change and plastic pollution.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
W. Zhu and Y. Jiang would like to acknowledge the support from National Natural Science Foundation of China (No. 22176086), the Fundamental Research Funds for the Central Universities - Cemac "GeoX" Interdisciplinary Program (No. 021114380217), the Fundamental Research Funds for the Central Universities (Nos. 021114380222, 021114380214), Frontiers Science Center for Critical Earth Material Cycling of Nanjing University (No. 2024QNXZ07), Research Funds for Jiangsu Distinguished Professor, Carbon Peaking and Carbon Neutrality Technological Innovation Foundation of Jiangsu Province (No. BE2022861), the Research Funds from Frontiers Science Center for Critical Earth Material Cycling of Nanjing University and State Key laboratory of Pollution Control and Resource Reuse. R. Lin would like to acknowledge the support from National Natural Science Foundation of China (No. 52276177). Y. Jiang would like to acknowledge the support from the China Postdoctoral Science Foundation (No. 2024M761388), Postdoctoral Fellowship Program of CPSF (No. GZC20231105) and the Jiangsu Funding Program for Excellent Postdoctoral Talent (No. 2023ZB226).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111115.
[1]
S. Cestellos-Blanco, H. Zhang, J.M. Kim, et al., Nat. Catal. 3 (2020) 245–255. doi: 10.1038/s41929-020-0428-y
[2]
N. Kornienko, J.Z. Zhang, K.K. Sakimoto, et al., Nat. Nanotechnol. 13 (2018) 890–899. doi: 10.1038/s41565-018-0251-7
Y. Jv, B. Li, R. Cao, Chem. Commun. 46 (2010) 8017–8019. doi: 10.1039/c0cc02698k
Figure 1
Preparation and photocatalytic mechanism of the C. necator/CdS@Au@PDDA biohybrid system. Au NCs are synthesized by the reduction of AuCl₄⁻ with NaBH₄ and loaded onto CdS nanorods to form CdS@Au, followed by surface modification with PDDA. Subsequently, CdS@Au@PDDA adheres to the surface of C. necator via electrostatic adsorption. Under light irradiation, it generates photogenerated electrons, catalyzing the reduction of H⁺ to H2 and facilitating CO2 fixation. Through the synergistic action of carbonic anhydrase and hydrogenase, H2 and CO2 are converted into 3HB-CoA, which is further utilized for the biosynthesis of PHB and biomass.
Figure 2
Characterization and performance analysis of CdS@Au@PDDA nanocomposite. (a) TEM image of CdS nanorods. (b) High-angle annular dark field (HAADF) STEM image of CdS@Au, showing Au clusters distributed uniformly across the surface of CdS nanorods. (c) High-resolution transmission electron microscopy (HRTEM) image of CdS@Au, illustrating the lattice spacings of CdS and Au crystals, measured as 0.316 nm for CdS and 0.118 nm for Au. (d) XRD patterns of CdS and CdS@Au. (e) XPS spectra of Cd 3d, demonstrating the influence of Au modification on the chemical state of Cd atoms. (f) XPS spectra of Au 4f, indicating the chemical state of Au on the CdS surface. (g) Zeta potential comparison showing the surface charge characteristics of various samples. (h) Photocurrent response under light on-off cycles. (i) Time-dependent H2 production curves from cycling photocatalytic tests under light irradiation (AM 1.5 G).
Figure 3
Morphology, elemental distribution, and photocatalytic performance for PHB synthesis of C. necator/CdS@Au@PDDA. (a) SEM image of C. necator. (b) SEM image of the C. necator/CdS@Au@PDDA. (c) HAADF-STEM image of dividing C. necator/CdS@Au@PDDA. (d-f) Energy-dispersive X-ray spectroscopy (EDS) elemental mapping images showing the distribution of S, Cd, and Au, indicating that CdS@Au@PDDA are uniformly distributed on the bacterial surface. (g) PHB concentration changes under different CdS@Au@PDDA dosages for the photocatalytic synthesis of PHB using C. necator/CdS@Au@PDDA. (h) The effect of light-dark cycles on PHB production. (i) Comparison of PHB production performance in this work with previously reported studies.
Figure 4
Electrochemical properties, biocompatibility, and ROS quenching capacity of C. necator/CdS@Au@PDDA. (a) EIS spectra illustrating the interfacial charge transfer resistance of CdS, CdS@Au, and CdS@Au@PDDA composites. (b) Cd2+ tolerance assays comparing the growth of adapted and non-adapted C. necator under different Cd2+ concentrations (25, 50, and 75 μmol/L). (c) Comparison of Cd2+ leaching between CdS and CdS@Au@PDDA composites during photocatalytic reactions. (d, e) CLSM images of C. necator/CdS (d) and C. necator/CdS@Au@PDDA (e), respectively. The living C. necator cells were stained with a LIVE/DEAD BacLight kit, which consists of green fluorescent SYTO 9 (live cells) and red fluorescent propidium iodide (dead cells). (f) The changes in ROS concentrations before and after photocatalytic reactions.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
