School of Chemistry, South China Normal University, Guangzhou 510006, China
b.
Jiangsu Engineering Laboratory for Environmental Functional Materials, School of Chemistry and Chemical Engineering, Huaiyin Normal University, Huaian 223300, China
Received Date:
24 January 2025 Accepted Date:
18 March 2025 Revised Date:
07 March 2025 Available Online:
01 July 2026
Abstract:
Near-infrared (NIR) light offers significant advantages in photocatalytic reactions due to its excellent penetration ability and low phototoxicity. However, the development and utilization of NIR light for efficient photocatalysis continue to encounter several challenges. In this work, we designed and synthesized two stable iron-oxo clusters functionalized with 1, 1-ferrocene dicarboxylic acid (Fcdc), Fe11-Fcdc and BiFe10-Fcdc, both of which can effectively utilize full-spectrum light to realize efficient oxidative coupling reaction of benzylamine (BA) with a product of selectivity over 90% and a conversion up to 97%. Particularly, under the NIR light, Fe11-Fcdc shows significantly better photocatalytic performance (a conversion of 82.3%) than BiFe10-Fcdc (41.0%), which may be responsible for the stronger metal-ligand charge transfer effect in Fe11-Fcdc. This work reports for the first time the study of ferrocene-modified crystalline clusters achieving effective utilization of NIR light.
In recent years, ferrocene has been used as a functional ligand to modify various photoelectric materials due to its inherent good photoelectric response, excellent redox properties, and superior multi-electron transfer capabilities [1,2]. The modified compounds typically exhibit a variety of structural types, extended light absorption, and rapid charge transfer rates [3,4], which can be applied in photocatalytic [5-7] electrocatalytic [8,9], and photo-electrocatalytic reactions [10], showing excellent catalytic activity. Research shows that the primary reason for the improved performance of these materials is the strong metal-ligand charge transfer effect between the metal atoms and ligands after coordination with ferrocene [11-13]. However, to our knowledge, the reported ferrocene-modified photocatalytic materials exhibit limited or negligible light absorption in the near-infrared (NIR) region, which greatly limits the utilization of light in photocatalytic reactions.
NIR light constitutes about 50% of the sunlight, which is comparable to the ultraviolet-visible (UV–vis) range [14]. Due to the unique advantages of NIR light such as excellent penetration ability and low phototoxicity [15-18], it has a broad range of applications in biomedicine [17], spectroscopy [19], photodetectors [20], and photocatalysis [21-27]. Especially in the field of photocatalysis, effectively harnessing the NIR region can maximize the utilization of solar energy and improve light efficiency [22,23,25]. Additionally, the lower energy of NIR light minimizes disturbances to the structure of photocatalysts and the reaction medium in the reaction system [17]. However, precisely because of the low energy of NIR light, it cannot provide the energy required to excite most photocatalysts, which greatly limits its application in photocatalytic reactions [28]. Based on the reported ferrocene-modified catalysts, we found that the type of metal coordinated with ferrocene in the structure of the catalyst can significantly influence the optical properties of the modified materials. We speculate that by selecting the appropriate coordinating metal atoms, the light absorption of ferrocene-modified materials can not only be extended to the NIR region but also combined with the rapid multi-electron transfer characteristics of ferrocene itself, thus effectively improving the photocatalytic reaction activity. However, this understanding necessitates the modeling of catalysts for further validation.
Based on the aforementioned considerations, we designed and synthesized two isomorphic iron-oxo clusters modified with 1, 1-ferrocene dicarboxylic acid (Fcdc) through a simple solvothermal method, named Fe11-Fcdc (Fe11(µ4-O)4(µ-O)(Fcdc)8(µ2-OCH3)3·DMF), and BiFe10-Fcdc (BiFe10(µ4-O)6(µ2-O)(µ-OH)(Fcdc)8(µ3-OCH3)2·H2O). BiFe10-Fcdc was synthesized through hetero-metal doping, which involves replacing one iron atom in the structure of Fe11-Fcdc with a bismuth (Bi) atom. Experiments show that these two clusters exhibit strong light absorption in the NIR region, along with good capabilities for photo-generated charge transfer and separation. It is worth noting that Fe11-Fcdc shows markedly higher absorption than BiFe10-Fcdc in the NIR region. Based on these, the oxidative coupling reaction of benzylamine (BA) was employed as a model reaction to investigate the photocatalytic performance of the two clusters. The results show that both photocatalysts exhibit high catalytic activity with a high conversion of substrate (> 94%) under full-spectrum light. Notably, under NIR light, the catalytic performance of Fe11-Fcdc (82.3%) is better than that of BiFe10-Fcdc (41.0%), which may be attributed to the stronger metal-ligand charge transfer effect between the metal atoms and ligands in Fe11-Fcdc. Importantly, this is the first report on the use of ferrocene-modified photocatalytic materials to catalyze the conversion of organic molecules in the NIR region.
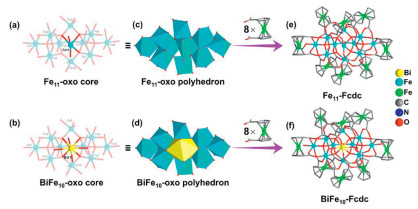
The black Fe11-Fcdc and BiFe10-Fcdc crystals were synthesized by the one-step solvothermal synthesis method (Figs. S1 and S2 in Supporting information). Single-crystal X-ray diffraction (SCXRD) analysis reveals that Fe11-Fcdc and BiFe10-Fcdc both crystallize in the triclinic space group P1¯ (Table S1 in Supporting information). The molecule of Fe11-Fcdc consists of an undeca-nuclear Fe-oxo core, eight Fcdc ligands, three µ2–OCH3, one terminal-coordinated N,N-dimethylformamide (DMF) molecule, and a free DMF molecule present (Fig. 1 and Fig. S3 in Supporting information). The Fe atoms in the undeca-nuclear Fe-oxo core are connected by five µ4-O, one µ3-O and three µ2-OCH3 (Fig. 1a). As shown in the Fig. S3a, except for the penta-coordinated Fe9, Fe10, and Fe11, the rest of the Fe atoms are hexa-coordinated. The structure of BiFe10-Fcdc is essentially similar to that of Fe11-Fcdc (Fig. 1b), but the difference is that Bi11 replaces the original position of Fe11, which is coordinated with eight O atoms to form a dodecahedral structure (Figs. 1c and d, Figs. S3a and S4a in Supporting information). At the same time, the Bi atom is connected to Fe atoms through four µ4-O, one µ2-O and two µ3-OCH3. Additionally, Fe4 and Fe5 are connected by a µ2-O, and the terminal O atom coordinated to Fe1 and Fe8 come from hydroxide and water molecules, respectively (Fig. S4a). It is worth noting that the terminal-coordinated solvent molecule in BiFe10-Fcdc and Fe11-Fcdc can easily dissociate, potentially becoming catalytically active sites for absorbing activated small molecules during the catalytic process (Figs. 1e and f).
Figure 1
Figure 1.
Molecular structures of Fe11-Fcdc and BiFe10-Fcdc. (a, b) The structures of two Fe-oxo cores. (c, d) The polyhedron structures of two Fe-oxo cores. (e, f) The entire structures of Fe11-Fcdc and BiFe10-Fcdc. All hydrogen atoms are omitted for clarity.
As shown in Figs. S5 and S6 (Supporting information), the powder X-ray diffraction (PXRD) patterns are highly consistent with the simulated patterns derived from SCXRD data, which indicates that Fe11-Fcdc and BiFe10-Fcdc possess high crystallinity and purity. Subsequently, the solvent stability of the two clusters was assessed. After both materials were soaked in CH3CN for 24 h, the PXRD patterns still matched well with the experimentally obtained PXRD patterns, indicating that these two clusters have good stability in CH3CN, which provides the possibility for heterogeneous catalysis (Figs. S7 and S8 in Supporting information). The Fourier-transform infrared spectroscopy (FT-IR) spectrum shows distinct peaks at 1489, 915, and 513 cm-1, corresponding to the δ(C=C), ν(O-H), and ν(Fe-C) vibrations of Fcdc [29], respectively, which confirms the successful modification of Fcdc into structures of the two clusters (Figs. S9 and S10 in Supporting information). Additionally, thermogravimetric analysis (TGA) was performed to evaluate the thermal stabilities of the two clusters. As shown in Figs. S11 and S12 (Supporting information), both Fe11-Fcdc and BiFe10-Fcdc can maintain their basic structural frameworks before ca. 250 ℃ under an N2 atmosphere, which provides a good foundation for further performance applications.
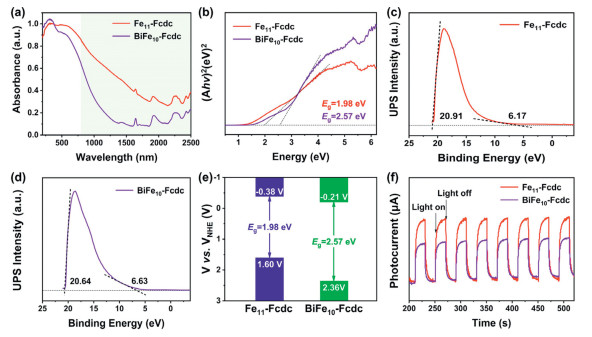
As the photosensitivity of ferrocene, the UV–vis diffuse reflectance spectroscopy was conducted to evaluate the light absorption capabilities of Fe11-Fcdc and BiFe10-Fcdc. Fig. 2a shows that both Fe11-Fcdc and BiFe10-Fcdc possess strong light absorption capabilities and a wide absorption range, which can be extended into the NIR region. Significantly, Fe11-Fcdc exhibits superior light absorption ability in the NIR region. Based on the UV–vis diffuse reflectance spectra, the optical band gaps (Eg) of Fe11-Fcdc and BiFe10-Fcdc were further calculated using the Kubelka-Munk function, which are about 1.98 and 2.57 eV, respectively (Fig. 2b). Subsequently, to evaluate the band structure of the compounds, the Mott-Schottky electrochemical measurements were conducted to determine their lowest unoccupied molecular orbital (LUMO). As shown in Figs. S13 and S14 (Supporting information), the Mott-Schottky plots of Fe11-Fcdc and BiFe10-Fcdc exhibit a positive slope, indicating that they exhibit characteristics similar to n-type semiconductors. Thus, their flat band potential (Efb) is −0.58 V and −0.41 V vs. Ag/AgCl, respectively, and the LUMO positions of Fe11-Fcdc and BiFe10-Fcdc are estimated to be −0.38 V and −0.21 V vs. NHE, respectively, due to the bottom of LUMO nearly equals to the Efb in the n-type semiconductors [30]. Based on the equation EHOMO = ELUMO − Eg [31,32], the corresponding highest occupied molecular orbital (HOMO) positions of Fe11-Fcdc and BiFe10-Fcdc are ascertained to be 1.60 V and 2.36 V vs. NHE, respectively, which is consistent with the results of ultraviolet photoelectron spectroscopy (UPS) (Figs. 2c and d). Obviously, these two clusters have more positive HOMO levels, which makes them suitable for applications in certain oxidation reactions, such as the oxidation of amines (Fig. 2e). The photocurrent response was subsequently used to characterize the separation efficiency of photo-induced carriers. As shown in Fig. 2f, the photocurrent response intensity of Fe11-Fcdc is significantly stronger than that of BiFe10-Fcdc, indicating that Fe11-Fcdc has a higher electron migration efficiency and would provide more available surface carriers in the photocatalytic process [33-35].
Figure 2
Figure 2.
(a) UV-visible diffuse reflectance spectra of Fe11-Fcdc and BiFe10-Fcdc. (b) Tauc plots of Fe11-Fcdc and BiFe10-Fcdc. (c) UPS spectra of Fe11-Fcdc. (d) UPS spectra of BiFe10-Fcdc. (e) The energy band structures diagram for Fe11-Fcdc and BiFe10-Fcdc. (f) Transient photocurrent curves of Fe11-Fcdc and BiFe10-Fcdc measured in 0.5 mol/L Na2SO4 aqueous solution.
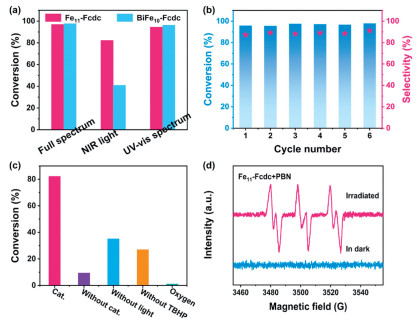
Based on the aforementioned excellent photophysical properties, the performance of photocatalytic oxidative coupling reaction of BA with the two clusters was investigated, respectively. The qualitative analysis of products was determined by gas chromatography-mass spectrometry (GC-MS), and the conversion of substrate and selectivity of products were determined by gas chromatography (GC) (Figs. S15-S17 in Supporting information). As shown in Fig. 3a and Fig. S18 (Supporting information), under full-spectrum light irradiation for 18 h, both Fe11-Fcdc and BiFe10-Fcdc achieved high conversion of the substrate (97.0% and 97.7%) and selectivity of the product (90.3% and 91.5%). The exceptional light absorption capability of the two clusters in the NIR region prompts an investigation into their performance in catalyzing the oxidation of BA to N-benzylidenebenzylamine (BDA) under NIR light. The experimental results show that under NIR light, Fe11-Fcdc can also demonstrate excellent performance, which can reach a conversion of 82.3% with a selectivity of 80.2%. However, under the same conditions, BiFe10-Fcdc shows poor catalytic performance with a conversion of 41.0% and a selectivity of 73.4%. To explore the relationship between the conversion of substrate and time under NIR light conditions, the reaction solution was extracted and analyzed every 3 h (Fig. S19 in Supporting information). The results show that Fe11-Fcdc, as a photocatalyst, has better reaction activity than BiFe10-Fcdc under NIR light. This difference can be explained by the varying strengths of light absorption in the NIR region and the interfacial charge transfer rate of the two photocatalysts. Moreover, electrochemical impedance spectroscopy (EIS) was performed to investigate the impedance characteristics of these two photocatalysts. In Fig. S20 (Supporting information), Fe11-Fcdc has a smaller electronic transmission resistance, indicating that it has a faster interfacial charge transport rate than BiFe10-Fcdc, which is consistent with the results of the photocurrent response tests. To further investigate the utilization of full-spectrum light by the two clusters, the photocatalytic performance under UV–vis spectrum light was further evaluated. As displayed in Fig. 3a and Fig. S18, both clusters can achieve high conversion of the substrate (94.5% and 96.3%) and good selectivity of the product (89.2% and 90.7%) under UV–vis spectrum light to realize a great utilization of light.
Figure 3
Figure 3.
(a) Conversion of BA under light sources with different wavelengths. (b) Conversion of BA and selectivity of product for oxidative coupling reaction with Fe11-Fcdc in 6 cycles experiments under full-spectrum light. (c) Control experiments of the oxidative coupling reaction of BA under NIR light. (d) EPR spectra of Fe11-Fcdc under irradiated and in the dark in the presence of PBN.
Recycling experiments were conducted to demonstrate the tolerance of the photocatalysts in the reaction. After 6 cycles of experiments, Fe11-Fcdc consistently maintains stable and efficient reaction activity (Fig. 3b), proving that Fe11-Fcdc maintains good stability during the catalytic process. Besides, PXRD and FT-IR characterizations of tested photocatalysts were performed after the photocatalytic reaction to further verify the stability of photocatalysts. Figs. S21-S24 (Supporting information) show that the PXRD patterns and IR spectra of the catalyst after the reaction match well with that before the reaction, which confirms that Fe11-Fcdc and BiFe10-Fcdc maintain good structural integrity during the photocatalytic reaction. X-ray photoelectron spectroscopy (XPS) analysis confirms that the valence states of the metal elements remained consistent before and after the reaction (Figs. S25 and S26 in Supporting information). Subsequently, to determine the indispensable reaction conditions, a series of control experiments were conducted. As shown in Table S4 (Supporting information) and Fig. 3c, photocatalyst, light, and tert‑butyl hydroperoxide (TBHP) are the basic conditions for the oxidation coupling reaction of BA, otherwise, only a small or negligible amount of conversion can be observed.
To study the mechanism of the reaction, electron paramagnetic resonance (EPR) tests were conducted at room temperature with N-tert-butyl-α-phenylnitrone (PBN) as the trapping agent to identify the reactive radicals involved in the reaction. The yielded clear EPR peaks for the PBN-spin adducts indicate that TBHP may generate tBuO•/tBuOO• radical during the reaction, which could be the active species in the reaction (Fig. 3d) [36-38]. We also conducted quenching experiments to distinguish the contributions of specific active species. As shown in Fig. S27 (Supporting information), the addition of p-benzoquinone (BQ) and 1, 4-diazabicyclo[2.2.2]octane (DABCO) results in only a marginal reduction in reaction conversion, ruling out the presence of O2•- and 1O2 radicals during the photocatalytic process. Subsequently, butylated hydroxytoluene (BHT) is used as a scavenger for tBuO•/tBuOO• radical and added to the reaction. The significant decrease in the conversion of benzylamine (BA) indicates that tBuO•/tBuOO• radical plays a crucial role in the amine coupling reaction. Moreover, holes are likely involved in the oxidative amine coupling process because the addition of KI as a hole scavenger during the photocatalytic reaction results in a reduced conversion to BDA (60.8%). Furthermore, when CCl4 was introduced as an electron scavenger, the conversion of BA to BDA decreased to 51.5%, indicating that the transfer of photogenerated electrons plays a significant role in the reaction (Table S4).
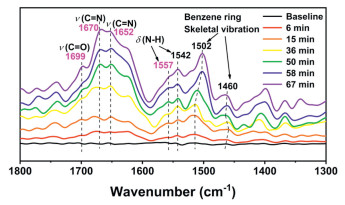
To gain a deeper understanding of the reaction mechanism involved in the photocatalytic conversion of BA to BDA, the in-situ diffuse reflectance infrared Fourier transform spectroscopy (DRIFTS) measurement was conducted (Fig. 4) [39,40]. Under a dark condition, no new IR band/peak is observed at 0 min after achieving adsorption equilibrium for 20 min, indicating the photocatalytic nature of the conversion of BA to BDA. Upon light stimulation, the new IR peak appears at 1670 cm-1 attributed to the C=N stretching vibration of the benzyl imine intermediate. Over time, the product BDA generates, as evidenced by the appearance of the C=N stretching vibration (1652 cm-1) for BDA. This phenomenon suggests that BDA can form via the pathway that the benzyl imine intermediate is nucleophilically attacked by BA molecules. Simultaneously, the peak at 1557 cm-1 for N—H bending vibration gradually intensifies, indicating the formation of NH3 alongside the production of BDA. Additionally, we observe the characteristic IR peak of the C=O bond at 1699 cm-1, indicating the presence of another pathway involving a benzaldehyde intermediate for the photocatalytic conversion of BA to BDA in this photocatalytic system.
Figure 4
Figure 4.In-situ DRIFTSR on cluster complexes under dark and irradiation conditions using BA as the reactants.
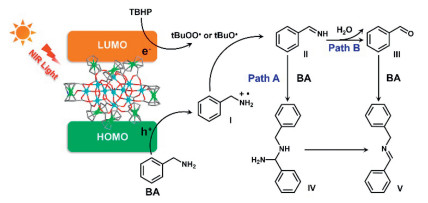
Based on all the foregoing discoveries, a possible mechanism for the oxidative coupling of BA is proposed. As shown in Fig. 5, upon light irradiation, Fe11-Fcdc is photoexcited to generate electrons and holes. The adsorbed BA molecules are oxidized by photo-generated holes to form BA radical cations (Ⅰ), while TBHP reacts with photo-generated electrons to produce tBuO•/tBuOO•. In the process of generating the final coupling products from BA radical cations (Ⅰ), there are two distinct pathways [41-43]. In path A, intermediate Ⅰ reacts with tBuO•/tBuOO•, further transforming into intermediate Ⅱ. Then, Ⅱ can be readily attacked by another free BA molecule. After the release of ammonia, the final product BDA (Ⅴ) is produced. In path B, Ⅲ is hydrolyzed to obtain benzaldehyde (Ⅴ), which is then condensed with another BA to produce the coupling product
Figure 5
Figure 5.
Proposed reaction mechanisms for photocatalytic oxidative amine coupling reaction by Fe11-Fcdc.
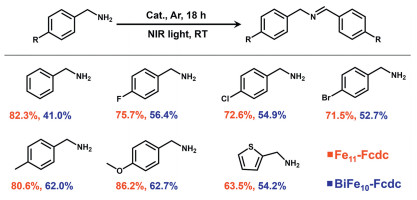
To demonstrate the generality of the reaction, the substrate scope was investigated (Scheme 1). Various benzylamine derivatives were tested as substrates under the same conditions. Benzylamines with different substituents (such as Me-, MeO-, F-, Cl-, and Br-) can be effectively converted into the desired product and the conversion of substrate follows the trend of electron-donating groups being superior to electron-withdrawing groups. When the substrates contain the electron-donating group, p-Me, and p-OMe, the conversion of amines can reach 80.6% and 86.2%, respectively. However, when the substrates contain the electron-withdrawing groups p-F, p-Cl, and p-Br, the conversion of amines is determined to be 75.7%, 72.6%, and 71.5%, respectively (Figs. S28-S33 in Supporting information). This may be due to the electron-withdrawing group, after resonance with the benzene ring, induces more positive charge on the carbon atom adjacent to the nitrogen atom in the amine, making it difficult for the hydrogen atom on the carbon to depart, thereby resulting in a decrease in conversion rate [44-47].
Scheme 1
Scheme 1.
Heterogeneous photocatalytic oxidative coupling of diverse amines. Reagents: substrate (0.1 mmol), photocatalyst (20.0 mg), solvent (1 mL CH3CN), TBHP (0.27 mmol), 300 W Xenon lamp (200 mW/cm2) with a cutoff filter (> 800 nm).
In summary, we synthesized two stable iron-oxo clusters modified with ferrocene ligands (Fe11-Fcdc and BiFe10-Fcdc), which contain different metal atoms (Fe or Bi) at the center of the cluster and exhibit excellent full-spectrum utilization efficiency. When they were used as photocatalysts for photo-induced oxidative coupling reaction of BA, high conversion of 97% and great selectivity over 90% can be achieved under full-spectrum light. Especially, compared to BiFe10-Fcdc, Fe11-Fcdc demonstrates superior photocatalytic activity under NIR light, achieving a conversion of 82.3% and a product selectivity of 80.2%. The excellent photocatalytic performance under NIR may be attributed to the stronger metal-ligand charge transfer effect between the metal atoms and ligands in Fe11-Fcdc. This study broadens the application of crystalline materials in photocatalytic reactions in the NIR region.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Li-Ling He: Writing – original draft, Investigation, Data curation. Sheng-Nan Sun: Writing – original draft, Project administration, Investigation. Jing-Wen Shi: Software, Investigation, Data curation. Jiang Liu: Writing – review & editing, Supervision, Funding acquisition. Ning Li: Writing – review & editing, Supervision, Funding acquisition.
Acknowledgments
This work was supported by the National Key R & D Program of China (No. 2023YFA1507201), the National Natural Science Foundation of China (No. 22201046), Guangzhou Basic and Applied Basic Research Fund Project (No. 2025A04J0092), Young Top Talents of Pearl River Talent Program of Guangdong Province (No. 2021QN02L617), Guangdong Basic and Applied Basic Research Foundation (No. 2023B1515120060).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111111.
G. He, Y. Lai, Y. Guo, et al., ACS Appl. Mater. Interfaces14 (2022) 53724–53735. doi: 10.1021/acsami.2c14554
[46]
S. Liu, Q. Su, W. Qi, et al., Catal. Sci. Technol. 12 (2022) 2837–2845. doi: 10.1039/d2cy00167e
[47]
F. Su, S.C. Mathew, L. Möhlmann, et al., Angew. Chem. Int. Ed. 50 (2011) 657–660. doi: 10.1002/anie.201004365
Figure 1
Molecular structures of Fe11-Fcdc and BiFe10-Fcdc. (a, b) The structures of two Fe-oxo cores. (c, d) The polyhedron structures of two Fe-oxo cores. (e, f) The entire structures of Fe11-Fcdc and BiFe10-Fcdc. All hydrogen atoms are omitted for clarity.
Figure 2
(a) UV-visible diffuse reflectance spectra of Fe11-Fcdc and BiFe10-Fcdc. (b) Tauc plots of Fe11-Fcdc and BiFe10-Fcdc. (c) UPS spectra of Fe11-Fcdc. (d) UPS spectra of BiFe10-Fcdc. (e) The energy band structures diagram for Fe11-Fcdc and BiFe10-Fcdc. (f) Transient photocurrent curves of Fe11-Fcdc and BiFe10-Fcdc measured in 0.5 mol/L Na2SO4 aqueous solution.
Figure 3
(a) Conversion of BA under light sources with different wavelengths. (b) Conversion of BA and selectivity of product for oxidative coupling reaction with Fe11-Fcdc in 6 cycles experiments under full-spectrum light. (c) Control experiments of the oxidative coupling reaction of BA under NIR light. (d) EPR spectra of Fe11-Fcdc under irradiated and in the dark in the presence of PBN.

 DownLoad:
DownLoad:

 下载:
下载:







 下载:
下载:
