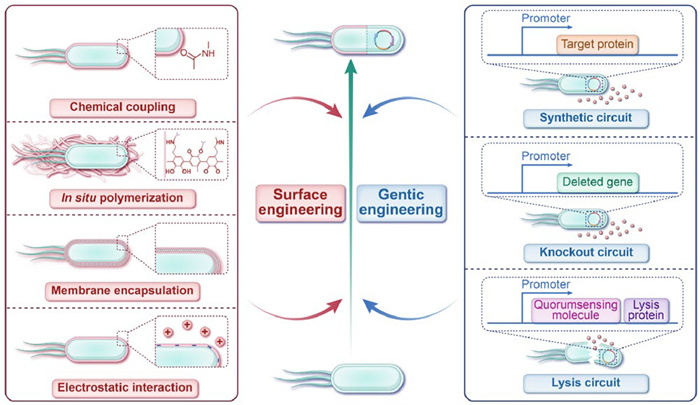
Figure 1.
Strategies for engineering bacteria, including surface engineering and genetic engineering.
Engineered bacteria potentiate cancer immunotherapy
Meng Sun , Jiazhen Yang , Leijiao Li , Yunhui Li , Wenliang Li , Jianxun Ding

The interactions between the immune system and tumor cells begin in the initial stages of tumorigenesis and continue throughout cancer progression [1]. Once tumor cells are discovered, the innate immune system is activated and serves as the first line of defense against these cells. This process involves the recognition of pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs) by pattern recognition receptors (PRRs) on the surface of innate immune cells [2-4]. The innate immune response helps activate adaptive immunity, with dendritic cells (DCs) as central hubs for transferring information. DCs present pathogen-related signals to T cells and B cells, enabling the establishment of long-term immune memory [5,6]. Cancer immunotherapy enhances the body's immune response, offering significant clinical efficacy and advantages. Currently, strategies, such as programmed death-1 (PD-1) inhibitors, cytotoxic T lymphocyte antigen-4 (CTLA-4) inhibitors, and chimeric antigen receptor T cells (CAR-T), are widely used in cancer immunotherapy [7]. These strategies have demonstrated potent anti-cancer activity in a broad range of malignant tumors, including melanoma, non-small-cell lung cancer, and renal cell carcinoma [8-10].
However, in some immunosuppressive cancers, immunotherapy is ineffective. The low response rate of clinical immunotherapy is a significant limitation to its therapeutic impact [11,12]. To address this issue, researchers have employed nanotechnology to target drug delivery and enhance the infiltration of tumor-killing T cells [13,14]. Although nanotechnology has somewhat improved immunotherapy efficacy, the clinical translation of nanomedicine delivery systems has been unsatisfactory [15,16]. This is primarily due to variability in the enrichment capacity within tumor tissues across different patients [17]. In the search for more effective treatments, the complex interactions between the bacterial microbiota and the immune systems of cancer patients have opened new avenues for therapeutic intervention [18]. Bacteria can be engineered for targeted enrichment in tumor regions and possess unique physiological structures that stimulate immunity. Furthermore, they are easily modified for therapeutic purposes [19,20]. Using engineered bacteria as tumor immunotherapeutic agents enables multiple immunizations, induces highly functional antigen-specific cellular immune responses, and improves the therapeutic benefit of immunotherapy.
Bacteria are highly efficient in targeting the tumor area. This may be due to their ability to significantly increase the level of tumor necrosis factor-α (TNF-α) in the bloodstream, which leads to rupture of the tumor's blood vessels. As a result, bacteria are flushed into the tumor with the influx of blood and become trapped in the disordered vascular system [21]. Additionally, the specialized anaerobic and partially anaerobic biology of certain bacteria enables them to preferentially colonize hypoxic or necrotic areas within solid tumors following systemic administration [22]. Bacteria targeting the tumor region enhance the effectiveness of immunotherapy through their immunogenetic material.
Immunogenic components and metabolites of bacteria are recognized by immune cells in the tumor microenvironments (TMEs) and participate in the immune response. One of the earliest studies explored the relationship between bacterial flora and the efficacy of immunotherapy. It found that the elevated relative abundance of Bifidobacterium in the intestinal microbiota increases the level of DCs in tumors and promotes the efficacy of immune checkpoint blockade (ICB) therapy [23].
In addition, metabolites produced by specific bacterial flora play a role in regulating the immune response. For instance, the cyclic di-adenosine monophosphate (c-di-AMP) produced by intestinal bacteria, such as Akkermansia muciniphila (AKK), has been shown to activate the stimulator of interferon genes (STING) pathway. The cyclic guanosine monophosphate-adenosine monophosphate synthase (cGAS)-STING signaling pathway is an intracellular set of early warning systems. When DNA from pathogenic or cancer cell chromosomes enters the cytoplasm, cGAS directly recognizes pathogen-derived DNA and activates STING proteins, which activate downstream immune signaling pathways that contribute to the tumor immune cycle in several ways [24]. This microbial-derived metabolite leads to the activation and proliferation of classical innate immune cells that express pro-inflammatory agents, potentiating immunotherapy [25]. However, balancing the therapeutic efficacy and safety of natural bacteria is challenging. Manipulating microbial compositions in the organism disrupts balance of the bacterial ecosystem, leading to systemic toxicity, which limits the development of bacteria as immunotherapeutic agents. To avoid the limitations of natural bacteria in vivo and preserve their ability to modulate innate immunity, a promising strategy is to carry out rational engineering transformations.
Engineered bacteria combine the characteristics of biology, genetics, chemistry, and materials science. Various bacterial species, including Escherichia coli (E. coli), Bifidobacterium, Lactobacillus (LAC), Salmonella, Listeria, and Clostridium, have been engineered to potentiate cancer immunotherapy [26]. Currently, these strategies fall into two main categories: (1) Surface engineering, which involves modifying bacterial surfaces through chemical coupling, in situ polymerization, membrane encapsulation, and electrostatic interactions, and (2) genetic engineering, which involves selectively expressing genes in bacteria via synthetic circuits, lysis circuits, inducible promoters, and gene knockout (Fig. 1). These strategies enable bacteria to reduce biotoxicity, synergize with other immunotherapy treatments to reverse the tumor immunosuppressive TMEs, and enhance the efficiency of immunotherapy responses [27]. This review summarizes the diverse effects of engineered bacteria on innate and adaptive immune responses, discusses the potential immunomodulatory mechanisms of different engineered bacteria as microbial therapeutic agents (Fig. 2), and predicts the future development and challenges of engineered bacteria for clinical of cancer immunotherapy applications.
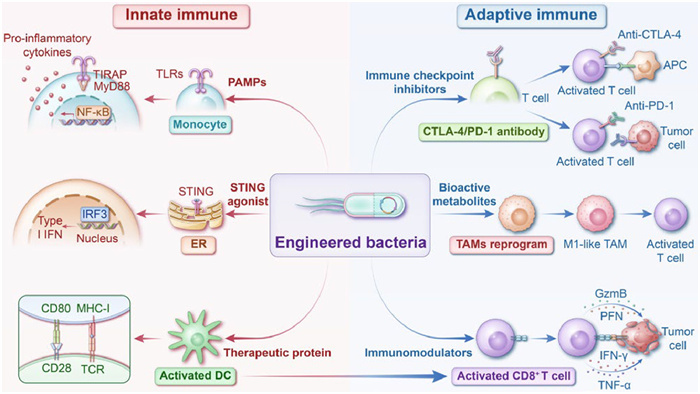
The innate immune system responds to infectious agents and induces anti-cancer immunity [28]. Specific bacterial species enhance immunotherapy efficiency. For example, E. coli Nissle 1917 (EcN) and Bacillus Calmette-Guérin (BCG) improve immune surveillance and reduce immunosuppression in the TMEs [29,30]. As infectious agents with a unique innate genetic background (lipoproteins, lipopolysaccharides, flagellin, and bacterial RNA), bacteria colonizing the TMEs bind to toll-like receptors (TLRs) expressed by antigen-presenting cells (APCs), thereby activating innate immunity [31-33]. When TLRs recognize bacteria, they initiate sequential signaling through two proteins, myeloid differentiation primary response 88 (MyD88) and tumor necrosis factor receptor-associated factor 6 (TRAF6). These proteins regulate downstream mediators, inflammatory cytokines production, and immune tolerance modulation, stimulating the recruitment and activation of monocytes, tumor-associated macrophages (TAMs), and neutrophils to promote anti-cancer immunity [34,35].
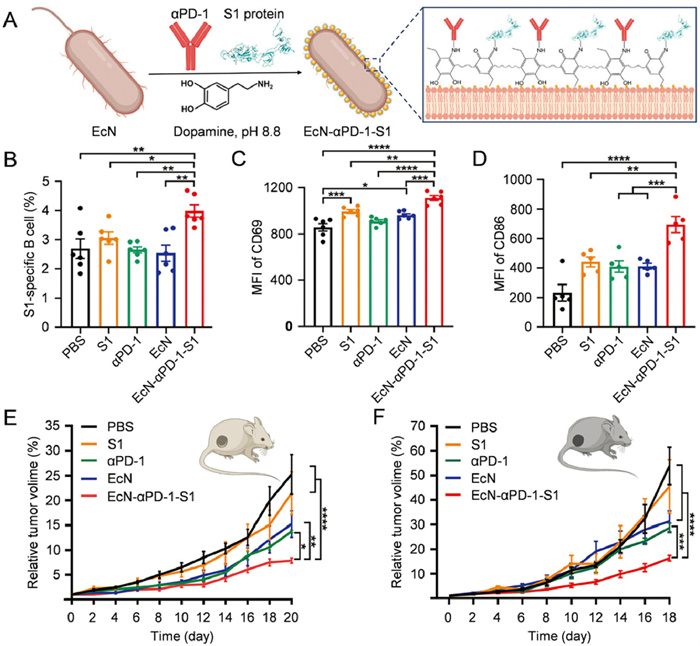
Surface-engineered bacteria interact efficiently with artificially selected surface structures and targets in vivo. Different surface-engineering strategies enable bacteria to perform various functions, including enhancing cellular binding efficiency, delivering drugs to activate the innate immune system, or providing a platform for immunotherapy with other therapeutic approaches [36-40]. For example, Chen et al. designed a nonpathogenic EcN for tumor-targeted delivery of STING agonist (MSA-2), by integrating MSA-2 onto a polydopamine shell [41]. During the oxidation and self-polymerization of dopamine, MSA-2 and EcN were added, and after 4 h of stirring PDMN was obtained. The study showed that the polydopamine-coated bacteria exhibited similar physiological functions to uncoated bacteria but provided a more significant opportunity to induce intra-tumoral immunity. The engineered bacteria activated the STING-interferon-I (IFN-I) pathway, induced IFN-β production in the TMEs, and stimulated the activation and infiltration of tumor-specific effector T cells, NK cells, and M1-type TAMs in immune-associated tissues. This led to long-lasting anti-cancer efficacy in vivo. Similarly, in another study, anti-programmed cell death protein-1 antibody (αPD-1) and severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) spike 1 (S1) protein were covalently linked to nanoparticular polydopamine (Fig. 3). After anchoring onto the EcN surface, EcN-αPD-1-S1 acted as an immune adjuvant, triggering innate immunity by providing inherent PAMPs. In vivo experiments showed that EcN-αPD-1-S1 promoted antigen processing and presentation by DCs, enhancing the production of S1 protein-specific immunoglobulin G (IgG) antibodies in serum while blocking the immune checkpoint (n = 6, **P < 0.01). This combination triggered strong humoral and cellular immune responses [42]. It is also possible to generate EcN-cypate by modifying a photothermal fluorophore (Cypate) on the surface of EcN via an amidation reaction. Under near-infrared (NIR) laser irradiation, EcN-cypate achieves photothermal therapy and triggers immunogenic cell death (ICD), thereby triggering a systemic immune response [43]. The combination of multiple therapeutic modalities provides bacterial fragments and tumor-associated antigens, such as PAMPs and DAMPs, which activate innate immune cells, enhance cell infiltration in a positive feedback manner, and reduce bacterial-induced side effects. This significantly inhibits tumor growth and recurrence through immune stimulatory responses.
In addition to surface modifications, genetically engineered bacteria offer unique advantages in activating innate immune responses [44]. For instance, to specifically activate the STING pathway in APCs within the tumor, an engineered bacterium, SYNB1891, was developed by incorporating the dacA gene (encoding the cyclic di-AMP [CDA]-producing enzyme) into EcN and using the hypoxia-inducible PfnrS promoter to control dacA expression [45]. The biotherapy derived from SYNB1891 showed higher levels of CDA and IFN-I production in vivo. CDA stimulates the TAMs, leading to a dose-dependent secretion of IFN-β1 and enhanced expression of T cell-associated cytokines (e.g., interleukin-2 (IL-2), granzyme B, and IFN-γ). Moreover, the engineered bacteria were preferentially phagocytosed by APCs, effectively avoiding "off-target" STING pathway activation and preventing unintended apoptosis in non-target cells, such as effector T cells. After intra-tumoral injection, SYNB1891 activated APCs, triggered a persistent anti-cancer immune response, and significantly improved the survival in both B16F10 and A20 tumor-bearing mice. For enhanced immune response and improved spatio-temporal controllability, an ultrasound promoter could be introduced in the engineered plasmid construction. Li et al. designed an expression vector encoding the antigen AH1, the adjuvant CDA, and the targeting peptide Co1 for EcN engineering [46]. Upon spatiotemporal ultrasound irradiation, the engineered bacteria were activated to secrete bacterial outer membrane vesicles (OMVs) containing a tumor-rejecting epitope for AH1, an enzyme that produces the STING agonist CDA, and the microfolded cell-targeting peptide Co1. Activation of the innate immune signaling pathways of TLR4 and STING was achieved, triggering a prolonged immune response. In the mouse subcutaneous CT26 colon tumor model, the engineered bacteria as a vaccine resulted in complete tumor regression in 60% of mice after three inoculations.
Modifying bacteria to express tumor antigens is another straightforward and effective genetic design approach [47]. Yue et al. fused plasmids expressing OMV surface proteins with Fc fragments of murine immunoglobulin G. The engineered bacteria were obtained via plasmid transformation. As an oral therapeutic agent, when delivered to the gut, these bacteria were captured by the colonic mucus layer, eliciting a mucosal immune response. This allows the in situ production of OMVs bearing tumor antigens in the gut, penetrating the intestinal epithelial barrier, promoting DC maturation and lymph node drainage for tumor antigen presentation. In the MC38 subcutaneous tumor model, this microbial therapeutic agent led to a tumor inhibition rate of 81.2%, higher than the 63.9% achieved with the subcutaneous poly(I: C)+Adpgk vaccine positive control and induced adequate immune memory. Zhang et al. designed a synchronous lysis circuit to enable spatiotemporally controlled antigen release from engineered bacteria. A fragment derived from the envelope protein (MuLV GP70), was displayed on bacterial micro-components (BMCs) [48]. The released nanovaccine was internalized by DCs, activating CD8+ T cells in the mesenteric lymph nodes and ultimately triggering systemic immunity. In addition to expressing tumor antigens, bacteria can be engineered to express active components, such as therapeutic proteins, to activate innate immunity. Chung et al. isolated Pediococcus pentosaceus SL4 from Korean fermented vegetable food kimchi and engineered it with dual gene cassettes driven by a strongly inducible promoter [49]. These cassettes encoded the therapeutic protein P8, fused with a secreted signal peptide and a complementary system. P8 is a unique therapeutic protein against colorectal cancer (CRC) with a molecular weight of 8 kDa, isolated from LAC rhamnosus CBT LR5. Oral administration of the engineered bacterium in a mouse colitis-associated CRC model inhibited the p53-p21 signaling pathway, inhibiting colon cancer cell proliferation.
Engineered bacteria have shown great potential in activating innate immunity for cancer therapy through surface and genetic modifications (Table 1) [37-43,45-49]. More engineered bacteria are often designed with both strategies combined to improve their therapeutic efficiency. Surface engineering is used to optimize delivery and interact with host immunity, and genetic engineering is used to enable programmable, environment-aware therapeutic output. This dual strategy addresses off-target effects, immune escape, and safety concerns. In addition, activation of adaptive immunity is equally essential to improve immunotherapy efficacy further. Therefore, developing engineered bacteria that enhance adaptive immunity is crucial.
 DownLoad:
CSV
DownLoad:
CSV
| Bacterial | Engineering method | Administration | Immunity enhancement | Ref. |
| Salmonella VNP20009 | Surface engineering of loaded glucose polymer and photo-sensitive nanoparticles | i.v. | Provoke innate immunity due to PAMPs | [37] |
| Bifidobacterium bifidum | Surface engineering of encapsulated organic dye molecules | i.t. | Provoke innate immunity due to PAMPs | [38] |
| Rhodobacter capsulatus | Surface engineering of encapsulated poly(ethylene glycol) derivatives | i.t. | Provoke innate immunity due to PAMPs and photo-thermal | [39] |
| Clostridium novyi-NT | Surface engineering of loaded nano-bio emulsion | i.t. | Provoke innate immunity due to PAMPs | [40] |
| EcN | Surface engineering of loaded STING agonist | i.v. | Provoke innate immunity due to PAMPs and STING pathway activation | [41] |
| Surface engineering of loaded αPD-1 antibody and SARS-CoV-2 spike 1 | i.t. | Provoke innate immunity due to PAMPs | [42] | |
| Surface engineering of loaded photothermal fluorophore | i.v. | Provoke innate immunity due to PAMPs and photo-thermal | [43] | |
| Genetic engineering for expressing STING-agonist cyclic diAMP | i.t. | Provoke innate immunity due to PAMPs and STING pathway activation | [45] | |
| Genetic engineering for expressing OMVs | p.o. | Provoke innate immunity due to PAMPs and STING pathway activation | [46] | |
| Genetic engineering for expressing nanovaccines | p.o. | Provoke innate immunity due to PAMPs and mucosal immunity activation | [48] | |
| E. coli | Genetic engineering for expressing OMVs | p.o. | Provoke innate immunity due to PAMPs and stimulate DC maturation | [47] |
| P. pentosaceus SL4 | Genetic engineering for expressing therapeutic protein | p.o. | Provoke innate immunity due to PAMPs and mucosal immunity activation | [49] |
The adaptive immune system is activated during the physiological colonization of bacteria, mediated by DCs, which elicit an effective T cell immune response and enhance immune cell infiltration. Bacteria-derived peptides are more readily recognized by CD8+ T cells than host self-derived peptides. When microbial epitopes are similar to tumor antigens and form a "molecular mimic", they induce symbiotic-specific memory T cells to cross-react with tumor antigens, promoting the recognition and destruction of tumor cells [50,51]. In addition to the direct immune response induced by colonization, intra-tumor microbes secrete bioactive metabolites that reprogram the immunosuppressive TMEs and indirectly modulate host adaptive immunity [52]. Therefore, screening and applying natural methods and exploiting the easily modifiable engineering properties of bacteria are essential to developing safer and more efficient tumor therapeutic agents that enhance adaptive immunity.
Surface engineering strategies provide bacteria with multiple functional properties. Modified bacteria exhibit advanced functions, such as enhanced targeting and colonization, improved immune cell infiltration, and more potent ICD to induce stronger anti-cancer immunity [53-55]. Liu et al. used ultraviolet light to induce pre-colonized Salmonella Typhimurium VNP20009 cells to form apoptotic bodies, which were automatically wrapped with an extra tumor cell-derived nanoshell. The tumor cell-mimetic bacteria showed improved biosafety and higher homologous targeting ability with this membrane support. Specifically, the number of bacteria localized to the tumor was 10 times higher than that of uncoated Salmonella Typhimurium VNP20009 on the fifth day after administration—the engineered bacteria enhanced T cell immune responses and induced apoptosis of tumor cells [56]. In addition to membrane encapsulation, electrostatic interactions can also be used to modify the surface of bacteria. For example, bacteria can be attached to TAMs using cationic polymer chitosan to interact with the negative charges on the bacterial surface. This strategy is a promising candidate for adoptive cell therapy [57]. Experimental results demonstrated that the nanocoating delayed the growth phase of EcN, improved biosecurity, and enabled macrophage attachment. The engineered bacteria proliferated in tumor tissues, leading to sustained activation of the M1 phenotype, repolarizing endogenous TAMs, stimulating infiltrating cytotoxic T cells, and remodeling the immunosuppressive TMEs. In the subcutaneous 4T1 breast tumor model, the engineered bacteria achieved a 64.5% tumor inhibition rate compared with the PBS group. The abundant functional groups on the bacterial surface also allow flexible combinations with multiple treatment modalities through chemical bonding strategies. For example, LAC co-loaded lactate oxidase (LOD) and tirazamine (TPZ) onto its surface via an amide condensation reaction [58]. The nanomedicines synergistically amplified the tumor-inhibitory effects of designed microbial drugs by inducing significant tumor cell apoptosis through in situ ROS generation and LAC metabolism-based TPZ activation, promoting anti-cancer M1 polarization of TAMs, and modulating CD8+ T cell infiltration.
Unlike surface-modified bacteria, genetic engineering enables more direct modulation of adaptive immune responses by programming bacterial nucleic acids or genetic circuits to express immunomodulators [59]. The genetic engineering bacteria break through the functional limitations of surface-engineered bacteria and enable the bacteria to synthesize and secrete immunomodulators autonomously in specific environments (e.g., hypoxia, high lactate) and achieve intelligent, dynamic, and targeted expression of immunomodulators. For example, engineered probiotics guide CAR-T cells to enhance CAR-T cell recruitment and therapeutic response [60]. EcN strains were genetically modified to include synchronized lysis and ProCAR-Target synthesis circuits in this approach. ProCAR-Target was designed as a dimer of sfGFP fused to HBD (PlGF123–144). Upon reaching a critical population density, the engineered probiotic selectively colonized tumors and triggered a lysis event that released ProCAR-Target in situ under a constitutive tac promoter. CAR-T cells (ProCARs) then recognize and respond to the target, enabling in situ lysis of labeled tumor tissues and driving systemic anticancer immunity. This strategy showed strong anti-cancer effects in CT26, A20, and MC38 tumor-bearing mouse models. Inspired by the natural sulfur cycle and sulfur-metabolizing bacteria, engineered bacteria regulate sulfur-containing amino acid metabolism, such as cysteine, in cells. Qiao et al. constructed engineered bacteria using E. coli MG1655 as a base strain by introducing a loop containing a hypoxic promoter and the engineered cyst(e)inase enzyme (CGL) gene [61]. The strain effectively blocked cysteine metabolism by catabolizing intracellular cysteine and extracellular cystine, inducing ferroptosis. Ferroptosis-induced ICD releases DAMPs and exposes tumor antigens to APCs, triggering robust adaptive immunity. The engineered bacteria demonstrated significant tumor suppression in the pancreatic ductal adenocarcinoma mouse model.
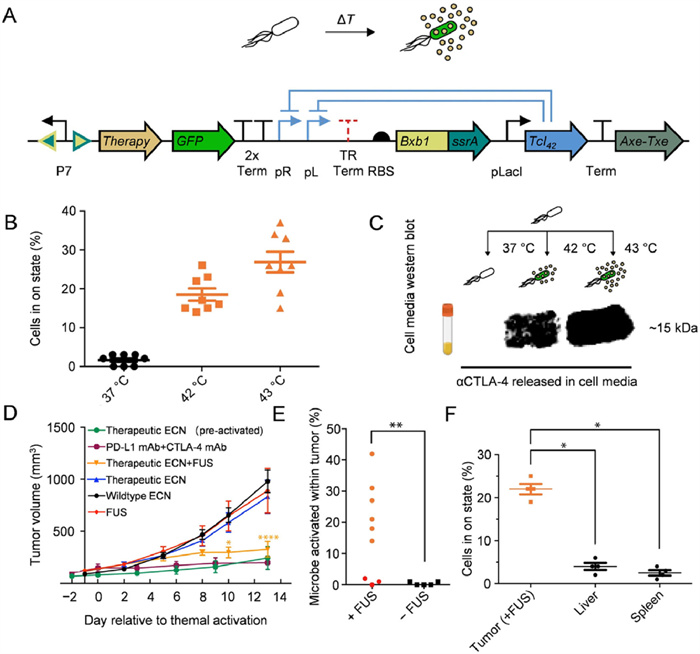
Another design approach for genetic modification is to modulate bacterial metabolism through synthetic biology, locally altering the concentration of immunomodulatory metabolites in the TMEs to improve the efficacy of immunotherapy [62]. It has been reported that L-arginine and programmed cell death-ligand 1 (PD-L1) blockers cooperate to restrict tumor growth in mice. Increasing the concentration of L-arginine in tumors increases the number of tumor-infiltrating T cells and the survival of activated CD4+ and CD8+ T cells and improves their anti-tumor activity in mice [63]. Based on this finding, an EcN knockout strain, deleting the arginine repressor gene (ArgR), was designed [64]. The L-arginine knockout strain was able to colonize the tumor and continuously convert ammonia accumulated in the tumor into high local concentrations of arginine, increasing tumor-infiltrating T cells and enhancing therapeutic effect of the PD-L1 blocking antibodies. In the MC38 model, tumors were completely eradicated in 74% of mice treated with L-arginine bacteria combined with anti-PD-L1 antibody (αPD-L1). In contrast, tumor eradication rates in mice treated with αPD-L1 alone or αPD-L1 antibody combined with non-engineered EcN were only 44% and 39%, respectively. In addition to these strategies, bacteria can be engineered to directly secrete active ingredients, such as immune checkpoint inhibitors, to target immune escape and inhibit tumor growth [65]. For example, the genetic circuitry of EcN was modified to enable the secretion of anti-cytotoxic T-lymphocyte-associated protein-4 antibody (αCTLA-4) and αPD-L1, controlled by the Tcl42 repressor (Fig. 4). The engineered bacteria sustained expression of αCTLA-4 and αPD-L1 in response to focused ultrasound (FUS) activation, which inhibited tumor growth in the A20 model.
Engineered bacteria activate adaptive immunity through multiple pathways (Table 2) [56-62,64,65]. A more effective anti-cancer response can be achieved by modulating bacterial interactions with adaptive immune cells. However, bacteria must be carefully engineered for the sequential activation of innate and adaptive immunity to ensure satisfactory therapeutic efficacy. Enabling engineered bacteria first to activate innate immune cells, such as TAMs and DCs, to build the foundation for subsequent activation of adaptive immunity. Then, further direct the activation and proliferation of adaptive immune cells, such as T cells and B cells to ensure that they provide the appropriate therapeutic effect at each stage of the immune response.
DownLoad:
CSV
| Bacterial | Engineering method | Treatment | Immunity enhancement | Ref. |
| Salmonella VNP20009 | Surface engineering of encapsulated tumor cell derived nanoshell coatings | i.v. | Enhance the adaptive immunity by the antigen-specific immune response activation | [56] |
| EcN | Surface engineering of loaded TAMs | i.v. | Enhance the adaptive immunity by increasing immune cell infiltration, reshaping TAM into M1 | [57] |
| Genetic engineering for producing nanovaccine | p.o. | Enhanc the adaptive immunity by activating CD8+ T in mesenteric lymph nodes | [59] | |
| Genetic engineering for expressing synthetic targets | i.t. | Enhance the adaptive immunity by labeling tumor tissue for CAR-mediated lysis | [60] | |
| Genetic engineering for expressing OMVs carrying therapeutic agents | i.v. | Enhance the adaptive immunity by remodeling tumor stroma | [62] | |
| Genetic engineering for producing high local concentrations of arginine | i.t. | Enhance the adaptive immunity by increasing the number of tumor-infiltrating T cells | [64] | |
| Genetic engineering for expressing CTLA-4 and PD-L1 | i.v. | Enhance the adaptive immunity by releasing immune checkpoint inhibitors | [65] | |
| LAC | Surface engineering of loaded natural enzyme and chemotherapeutic prodrugs | i.v. | Enhance the adaptive immunity by promoting macrophage M1 polarization and CD8+ T cell infiltration | [58] |
| E. coli MG1655 | Genetic engineering for expressing CGL | i.v. | Enhance the adaptive immunity by blocking cysteine metabolism and inducing ferroptosis | [61] |
The history of tumor immunotherapy dates back to early bacterial therapy investigations. Various bacterial species have been shown to activate the host immune system, and bacterial therapy has been explored for treating multiple malignancies due to their immunogenic properties and favorable colonization in tumor core regions. However, despite the significant theoretical advantages of bacterial therapies, most clinical trials have failed to achieve the desired outcomes. Advances in molecular biology and sequencing technologies have reignited interest in this field. Researchers tailor these microorganisms to meet specific immunomodulatory needs by surface-modifying and genetically engineering natural bacteria. Engineered bacteria locally produce and release immune checkpoint inhibitors or other immunotherapeutic agents, thereby enhancing the delivery of immunotherapy to target sites, reducing the need for repeated dosing, and improving immune efficacy. Combining different engineering strategies allows the development of novel bacterial therapies with spatiotemporal control and increased tumor specificity. Additionally, individualized treatment plans can be designed by modulating the expression levels of therapeutic agents based on the patient's tumor type and disease stage to optimize therapeutic outcomes. Engineered bacteria hold considerable potential in cancer immunotherapy, whether as monotherapies or combined with other therapeutic modalities.
Despite this potential, the complexity of using bacteria as therapeutic agents presents both distinct biological advantages and uncertain risks, and clinical applications have yet to meet expectations. Challenges related to ethics, regulatory, and potential safety risks must also be addressed in the clinical translation of engineered bacteria. For example, accidental release of genetically modified bacteria might disrupt environmental microbial equilibrium, traditional drug regulatory frameworks are not directly applicable to engineered bacteria, and off-target regulation of engineered bacteria potentially triggers systemic inflammatory responses. Strategies, such as incorporating temperature-sensitive or chemically inducible suicide genes, which activate bacterial self-destruction via external signals, or designing antibiotic systems to eliminate residual engineered bacteria post-treatment mitigate these risks. Additionally, engineered bacteria face challenges, including poor precision regulation, proliferation variability, and expression inconsistencies. Integrating multi-omics data analysis to predict colonization patterns in different individuals and developing personalized treatment protocols may enhance therapeutic efficacy. A deeper understanding of the interactions between bacteria, tumors, and the immune system is critical. The use of engineered bacteria as a multi-functional platform to mobilize immune responses, in combination with other therapies, represents a promising avenue for enhancing cancer treatment outcomes. By integrating basic research with clinical applications and implementing more rational, targeted designs, bacterial treatment is poised to become a crucial tool in cancer immunotherapy shortly.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Meng Sun: Writing – original draft, Writing – review & editing. Jiazhen Yang: Writing – review & editing. Leijiao Li: Writing – review & editing. Yunhui Li: Conceptualization, Writing – review & editing, Supervision, Funding acquisition, Project administration. Wenliang Li: Conceptualization, Writing – review & editing, Supervision, Funding acquisition, Project administration. Jianxun Ding: Conceptualization, Writing – review & editing, Supervision, Funding acquisition, Project administration.
This work was supported by the Science and Technology Research Project of Jilin Education Bureau (No. JJKH20230804KJ).
D. Pardoll, Annu. Rev. Immunol. 21 (2003) 807–839. doi: 10.1146/annurev.immunol.21.120601.141135
X. Cao, A.F. Cordova, L. Li, Chem. Rev. 122 (2021) 3414–3458.
A.J. Wolf, D.M. Underhill, Nat. Rev. Immunol. 18 (2018) 243–254. doi: 10.1038/nri.2017.136
C.A. Janeway, R. Medzhitov, Annu. Rev. Immunol. 20 (2002) 197–216. doi: 10.1146/annurev.immunol.20.083001.084359
M.G. Netea, A. Schlitzer, K. Placek, et al., Cell Host Microbe 25 (2019) 13–26. doi: 10.1016/j.chom.2018.12.006
L.L. Cao, J.C. Kagan, Immunity 56 (2023) 2206–2217. doi: 10.1016/j.immuni.2023.07.018
Y.Y. Xie, X.T. Li, J.Y. Wu, et al., Chin. Chem. Lett. 34 (2023) 108202. doi: 10.1016/j.cclet.2023.108202
P. Sharma, S. Hu-Lieskovan, J.A. Wargo, A. Ribas, Cell 168 (2017) 707–723. doi: 10.1016/j.cell.2017.01.017
D.R. Wang, X.L. Wu, Y.L. Sun, Sign. Transduct. Target. Ther. 7 (2022) 331.
A. Asrir, C. Tardiveau, J. Coudert, et al., Cancer Cell 40 (2022) 318–334. doi: 10.1016/j.ccell.2022.01.002
Y.Z. Chen, J.J. Zhu, J.S. Ding, W.H. Zhou, Chin. Chem. Lett. 35 (2024) 108706. doi: 10.1016/j.cclet.2023.108706
S.Y. Han, K.Q. Huang, Z.P. Gu, J. Wu, Nanoscale 12 (2020) 413–436. doi: 10.1039/c9nr08086d
S. Xu, Y. Xu, N.C. Solek, et al., Adv. Mater. 36 (2024) 2400307. doi: 10.1002/adma.202400307
M.M. Rahman, J. Wang, G. Wang, et al., Nat. Nanotechnol. 19 (2024) 818–824. doi: 10.1038/s41565-024-01620-6
Y.M. Wu, Z. Zhang, Y.Q. Wei, et al., Chin. Chem. Lett. 34 (2023) 108098. doi: 10.1016/j.cclet.2022.108098
L. Fu, X.B. Ma, Y.T. Liu, et al., Chin. Chem. Lett. 33 (2022) 1718–1728. doi: 10.1016/j.cclet.2021.10.074
M. Chehelgerdi, M. Chehelgerdi, O.Q.B. Allela, et al., Mol. Cancer 22 (2023) 169.
S.Y. Kwon, H.T.T. Ngo, J. Son, et al., Nat. Rev. Clin. Oncol. 21 (2024) 569–589. doi: 10.1038/s41571-024-00908-9
X. Dai, Z. Liu, X. Zhao, et al., Adv. Mater. 36 (2024) 202407927.
J. Cong, P. Liu, Z. Han, et al., Immunity 57 (2024) 876–889.
J.H. Fritz, S. Leschner, K. Westphal, et al., PLoS One 4 (2009) e6692. doi: 10.1371/journal.pone.0006692
T. Danino, A. Prindle, G.A. Kwong, et al., Sci. Transl. Med. 7 (2015) 289-284.
A. Sivan, L. Corrales, N. Hubert, et al., Science 350 (2015) 1084–1089. doi: 10.1126/science.aac4255
S. Liang, J.J. Yao, D. Liu, et al., Chin. Chem. Lett. 36 (2025) 109856.
K.D. McCoy, M.B. Geuking, Cell 184 (2021) 5301–5303.
M.R. Charbonneau, V.M. Isabella, N. Li, C.B. Kurtz, Nat. Commun. 11 (2020) 1738.
H. Pan, M. Zheng, A. Ma, et al., Adv. Mater. 33 (2021) 2100241.
S. Strobl, D. Zucchetta, T. Vašícek, et al., Angew. Chem. Int. Ed. 63 (2024) ˇ e202408421.
W. Lin, Y. Liu, J. Wang, et al., Angew. Chem. Int. Ed. 62 (2023) e202310158.
E. Moreo, A. Jarit-Cabanillas, I. Robles-Vera, et al., Nat. Commun. 14 (2023) 6090.
F. Hayashi, K.D. Smith, A. Ozinsky, et al., Nature 410 (2001) 1099–1103.
A.O. Aliprantis, R.B. Yang, M.R. Mark, et al., Science 285 (1999) 736–739.
X.M. Liu, M.Y. Sun, F. Pu, et al., J. Am. Chem. Soc. 145 (2023) 26296–26307. doi: 10.1021/jacs.3c09472
S. Uematsu, M.H. Jang, N. Chevrier, et al., Nat. Immunol. 7 (2006) 868–874. doi: 10.1038/ni1362
C.H. Lee, C.L. Wu, A.L. Shiau, Clin. Cancer Res. 14 (2008) 1905–1912.
Y. Xiao, D. Wang, B. Luo, et al., Nano Today 47 (2022) 101632.
R. Sun, M. Liu, J. Lu, et al., Nat. Commun. 13 (2022) 5127.
S. Reghu, E. Miyako, Nano. Lett. 22 (2022) 1880–1888. doi: 10.1021/acs.nanolett.1c04037
S. Reghu, S. Iwata, S. Komatsu, et al., Nano Today 52 (2023) 101966.
W. Park, S. Cho, D. Kang, et al., Adv. Healthc. Mater. 9 (2020) 1901812.
W. Chen, Y. Zhu, J. Chen, et al., Adv. Funct. Mater. 33 (2023) 2307001.
Y. Liu, M. Zhang, X. Wang, et al., Adv. Mater. 35 (2023) e2210949.
K.F. Xu, S.Y. Wu, Z.H. Wang, et al., Nat. Commun. 15 (2024) 5147.
H. Wang, F. Xu, C.L. Yao, et al., Proc. Natl. Acad. Sci. U. S. A. 121 (2024) e2412070121.
D.S. Leventhal, A. Sokolovska, N. Li, et al., Nat. Commun. 11 (2020) 2739.
J.X. Li, R.Q. Yang, Y.H. Yuan, et al., Adv. Funct. Mater. 35 (2024) 2414994.
Y. Yue, J. Xu, Y. Li, et al., Nat. Biomed. Eng. 6 (2022) 898–909. doi: 10.1038/s41551-022-00886-2
Y. Zhang, R. Kang, X. Zhang, et al., Biomaterials 299 (2023) 122147.
Y. Chung, Y. Ryu, B.C. An, et al., Microbiome 9 (2021) 122.
A. Fluckiger, R. Daillère, M. Sassi, et al., Science 369 (2020) 936–942. doi: 10.1126/science.aax0701
B. Routy, E. Le Chatelier, L. Derosa, et al., Science 359 (2018) 91–97. doi: 10.1126/science.aan3706
Q. Chen, C. Liu, C. Liu, et al., Nano Today 41 (2021) 101311.
E. Ylosmaki, M. Fusciello, B. Martins, et al., J. Immunother. Cancer 9 (2021) e002707. doi: 10.1136/jitc-2021-002707
H. Yue, Y. Li, T. Yang, et al., Nat. Nanotechnol. 20 (2024) 167–176. doi: 10.1097/shk.0000000000002278
M. Fidelle, L. Zitvogel, Nat. Biotechnol. (2024), doi: 10.1038/s41587-024-02429-3.
R. Liu, Z. Cao, L. Wang, et al., Nano Today 45 (2022) 101537.
J.X. An, Z.Y. Han, Y.T. Qin, et al., Adv. Mater. 36 (2023) 2305384.
W. Wu, Y. Pu, H. Yao, et al., Nano Today 42 (2022) 101377.
T. Harimoto, J. Hahn, Y.Y. Chen, et al., Nat. Biotechnol. 40 (2022) 1259–1269. doi: 10.1038/s41587-022-01244-y
R.L. Vincent, C.R. Gurbatri, F.D. Li, et al., Science 382 (2023) 211–218. doi: 10.1126/science.add7034
C.Q. Qiao, L.X. Wang, C.T. Huang, et al., Adv. Mater. 37 (2024) 2412982.
S.C. Thomas, T. Madaan, N.S. Kamble, et al., Adv. Healthc. Mater. 11 (2021) 2101487.
R. Geiger, J.C. Rieckmann, T. Wolf, et al., Cell 167 (2016) 829.
F.P. Canale, C. Basso, G. Antonini, et al., Nature 598 (2021) 662–666. doi: 10.1038/s41586-021-04003-2
M.H. Abedi, M.S. Yao, D.R. Mittelstein, et al., Nat. Commun. 13 (2022) 1585.

Figure 1 Strategies for engineering bacteria, including surface engineering and genetic engineering.

Figure 3 Engineering bacteria with a hybrid immunoreactive substance. (A) Decoration of EcN with a hybrid immunoreactive surface-bound αPD-1 and SARS-CoV-2 S1 proteins by one-step in situ polymerization of dopamine. (B) Percentage of SARS-CoV-2 S1-specific B cells analyzed by flow cytometry. (C) MFI on B cells activated with the CD69. (D) MFI on DCs co-stimulated with CD86. (E) Tumor suppression effect of CT26. (F) Tumor suppression effect of B16. All statistical data are represented as means ± standard error of mean (SEM; n = 6; P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001). Reproduced with permission [42]. Copyright 2023, Wiley Publishing Group.

Figure 4 Engineering bacterial heat activation for sustained release of immune checkpoint inhibitors. (A) Temperature responsive state switch modified to release αCTLA-4 or αPD-L1 nanobody. (B) Percent activation 24 h after 1-h thermal induction at 37, 42, or 43 ℃ for the circuit described in (A). (C) Western blot against hexahistidine-tagged αCTLA-4 nanobody. (D) Tumor suppression effect of A20. (E) Percent activation of therapeutic EcN isolated from FUS-treated and non-FUS-treated tumors two weeks after FUS treatment. (F) Background activation in bystander tissues following FUS activation of tumors. Percent activation of therapeutic EcN isolated from FUS-treated tumors and bystander organs (liver, spleen). All statistical data are represented as means ± SEM (n = 8 for B and C; n = 8 (therapeutic EcN, FUS), n = 10 (wildtype EcN), n = 4 (therapeutic EcN (pre-activated)); n = 6 (PD-L1 mAb+CTLA-4 mAb) and n = 7 (therapeutic EcN+FUS) for D; n = 9 (+FUS) and n = 5 (FUS) for E; n = 4 for F; P < 0.05, **P < 0. 01, ****P < 0.0001). Reproduced with permission [65]. Copyright 2022, Springer Nature.
Table 1. Engineering bacteria with enhanced innate immunity. i.v., intravenous injection; i.t., intratumoral injection; p.o., oral administration.
| Bacterial | Engineering method | Administration | Immunity enhancement | Ref. |
| Salmonella VNP20009 | Surface engineering of loaded glucose polymer and photo-sensitive nanoparticles | i.v. | Provoke innate immunity due to PAMPs | [37] |
| Bifidobacterium bifidum | Surface engineering of encapsulated organic dye molecules | i.t. | Provoke innate immunity due to PAMPs | [38] |
| Rhodobacter capsulatus | Surface engineering of encapsulated poly(ethylene glycol) derivatives | i.t. | Provoke innate immunity due to PAMPs and photo-thermal | [39] |
| Clostridium novyi-NT | Surface engineering of loaded nano-bio emulsion | i.t. | Provoke innate immunity due to PAMPs | [40] |
| EcN | Surface engineering of loaded STING agonist | i.v. | Provoke innate immunity due to PAMPs and STING pathway activation | [41] |
| Surface engineering of loaded αPD-1 antibody and SARS-CoV-2 spike 1 | i.t. | Provoke innate immunity due to PAMPs | [42] | |
| Surface engineering of loaded photothermal fluorophore | i.v. | Provoke innate immunity due to PAMPs and photo-thermal | [43] | |
| Genetic engineering for expressing STING-agonist cyclic diAMP | i.t. | Provoke innate immunity due to PAMPs and STING pathway activation | [45] | |
| Genetic engineering for expressing OMVs | p.o. | Provoke innate immunity due to PAMPs and STING pathway activation | [46] | |
| Genetic engineering for expressing nanovaccines | p.o. | Provoke innate immunity due to PAMPs and mucosal immunity activation | [48] | |
| E. coli | Genetic engineering for expressing OMVs | p.o. | Provoke innate immunity due to PAMPs and stimulate DC maturation | [47] |
| P. pentosaceus SL4 | Genetic engineering for expressing therapeutic protein | p.o. | Provoke innate immunity due to PAMPs and mucosal immunity activation | [49] |
 下载: 导出CSV
下载: 导出CSV
Table 2. Engineering bacteria with enhanced adaptive immunity.
| Bacterial | Engineering method | Treatment | Immunity enhancement | Ref. |
| Salmonella VNP20009 | Surface engineering of encapsulated tumor cell derived nanoshell coatings | i.v. | Enhance the adaptive immunity by the antigen-specific immune response activation | [56] |
| EcN | Surface engineering of loaded TAMs | i.v. | Enhance the adaptive immunity by increasing immune cell infiltration, reshaping TAM into M1 | [57] |
| Genetic engineering for producing nanovaccine | p.o. | Enhanc the adaptive immunity by activating CD8+ T in mesenteric lymph nodes | [59] | |
| Genetic engineering for expressing synthetic targets | i.t. | Enhance the adaptive immunity by labeling tumor tissue for CAR-mediated lysis | [60] | |
| Genetic engineering for expressing OMVs carrying therapeutic agents | i.v. | Enhance the adaptive immunity by remodeling tumor stroma | [62] | |
| Genetic engineering for producing high local concentrations of arginine | i.t. | Enhance the adaptive immunity by increasing the number of tumor-infiltrating T cells | [64] | |
| Genetic engineering for expressing CTLA-4 and PD-L1 | i.v. | Enhance the adaptive immunity by releasing immune checkpoint inhibitors | [65] | |
| LAC | Surface engineering of loaded natural enzyme and chemotherapeutic prodrugs | i.v. | Enhance the adaptive immunity by promoting macrophage M1 polarization and CD8+ T cell infiltration | [58] |
| E. coli MG1655 | Genetic engineering for expressing CGL | i.v. | Enhance the adaptive immunity by blocking cysteine metabolism and inducing ferroptosis | [61] |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: