Figure 1.
Catalytic methods for preparing cyano-BCHs as bioisosteres for cyano-arenes.

Although benzene rings, which consist entirely of sp2 carbon atoms, have traditionally been important in small-molecule drug discovery, recent findings have shown that a high fraction of sp3 carbon atoms (Fsp3, which is the ratio of the number of sp3 carbons to the total number of carbons) is associated with the success of drug discovery [1-3]. The optimal Fsp3 value is considered to be ≥0.42, and ap-proximately 84% of marketed drugs meet this criterion [4-7]. Moreover, data analysis has revealed that compounds with fewer than three aromatic rings are more likely to be developed as drug molecules. As a result, increasing saturation (referred to as “escape from flatland”) has gained attention in the medicinal chemistry community to meet the goal of improving clinical success in drug development by replacing flat benzenoid moieties with sp3-carbon-rich three-dimensional bioisosteres [2,3,8-15]. Bicyclo[2.1.1]hexanes (BCHs) are among the most important bioisosteres of benzene rings because they not only have a high Fsp3 but also mimic the relationships between ortho-substituents on the benzene rings (Fig. 1a) [16-23]. Recently, catalytic synthesis of BCHs has been intensively studied. For example, in 2020 and 2023, Mykhailiuk et al. reported pioneering work on photochemical intramolecular [2π + 2π] cycloadditions of 1,5-dienes to prepare multi-substutited BCHs [24,25]. Subsequently, methods for [2σ + 2π] cycloadditions of bicyclo[1.1.1]butanes (BCBs) to construct BCHs have emerged and have gradually come to dominate this field [26-40]. Specifically, the groups of Glorius, Bach, Brown and Lu developed a series of elegant, efficient methods for synthesizing BCH scaffolds by means of triplet-energy-transfer-enabled intermolecular [2σ + 2π] cycloadditions of BCBs and 2π substrates, such as coumarins [26], aza-renes [27], cyclobutenones [28], phenols [29], styrenes [30] and cyclic imines [31]. Additionally, the groups of Procter, Li and Wang established methods for SmI2-catalyzed [32] and pyridine diboron–catalyzed [33,34] radical [2σ + 2π] cycloadditions. More recently, Glorius et al. developed a new strategy for single-electron oxidative activation of BCBs via photoredox catalysis to afford BCB radical cations, which can be captured with unactivated olefins to form BCHs [35]. In addition, the groups of Studer, Deng, Feng, Chen and Zhou achieved Lewis acid–catalyzed ionic [2σ + 2π] cycloadditions of BCBs with ketenes [36], indoles [37,38], 3-benzylideneindoline-2-thione derivatives [39], silyl enol ethers [40] and ynamides [41].
BCHs can contain valuable chiral information that has the potential to be significant in drug design and development [16,42-45]. However, the enantioselective synthesis of BCHs is still in its early stages (Fig. 1b). In 2023, Bach reported the earliest success of asymmetric [2σ + 2π] cycloadditions, but stoichiometric chiral reagent was required to maintain high enantioselectivity [46]. In 2024, the Jiang group reported the catalytic asymmetric radical [2σ + 2π] cycloadditions of BCBs with vinylazaarenes to obtain heteroaryl-substituted BCHs by utilizing a chiral photo-sensitive Brønsted acid catalyst [47]. Lately, Yoon achieved a photochemical chromophore activation enabled enantioselective [2σ + 2π] cycloadditions of BCBs with α,β-unsaturated ketones [48]. All these reports are based on photochemical radical mechanism and developing alternative pathways to prepare chiral BCHs with other substitution patterns are still high desirable.
Nitriles play a significant role in medicinal chemistry, with over 60 small molecule drugs currently on the market that contain the cyano functional group (Fig. 1c) [49]. The introduction of the cyano group can enhance a drug candidate’s pharmacodynamic (PD) and pharmacokinetic (PK) profiles, as well as improve water solubility [50]. Additionally, it may facilitate beneficial noncovalent interactions [50-52] or covalent binding [53] with the drug target. Giving the importance of cyano-arenes moiety in drug molecules, it would be attractive to develop cyano-BCHs as the potential bioisosteres for cyano-arenes. However, currently there are no methods available to access this structural interesting motif. Inspired by the fruitful donor-acceptor cyclopropane chemistry [54-64], we recently developed the precursor vinyl-carbonyl-bicyclo[1.1.0]butanes (VC-BCBs) that are capable of undergoing palladium(0)-catalyzed (3 + 2) cycloadditions with aldehydes (Fig. 1d) [65]. As part of our exploration of the potential utility of palladium-catalyzed cycloadditions of VC-BCBs, especially in terms of enantioselective variants, we herein report a palladium-catalyzed [2σ + 2π] cycloadditions of VC-BCBs with arylidenemalononitriles, providing a platform to synthesize enantioenriched cyano-BCHs (Fig. 1e).
We initiated our study by using VC-BCB 1a and arylidenemalononitrile 2a as model substrates to screen various chiral ligands in reactions in THF at 30 ℃ under argon for 12 h (Table 1). Initially, we found that electron-rich phosphorus ligands showed poor enantioselectivity for the desired cycloaddition (for details, see Supporting information). Therefore, we shifted our focus to electron-deficient phosphoramidite ligands. Ligands L1 and L2 showed good activity, yielding desired product 3a in >90% yields, but the enantioselectivity was poor (entries 1 and 2). By switching to ligand L3, we achieved moderate enantioselectivity (43% ee), but the yield of 3a dropped dramatically (entry 3). In previous work, we [66] and others [67] had found that a bidentate bisphosphite ligand can accelerate the allylic substitution step. Therefore, we explored some typical BINOL-derived bisphosphite ligands (L4–L6; Table 1, entries 4–6). We were pleased to find that using L5 allowed us to obtain 3a in 73% yield with 61% ee (entry 5). After carefully exploring various linkages for the bisphosphite, we found that L7, a new ligand featuring a SPINOL linkage, greatly enhanced the enantioselectivity (entry 7). Furthermore, when we replaced the BINOL backbone of L7 with H8BINOL, resulting in ligand L8, 3a was obtained in 62% yield with 80% ee (entry 8). Subsequently, by varying the reaction temperature and solvent, entries 9–14), we established the optimal conditions. Specifically, by using L8 as the ligand, methyl tert-butyl ether as the solvent, and a reaction temperature of −40 ℃, we obtained 3a in 75% yield with 92% ee. Moreover, we discovered that we could reduce the loadings of PdCp(η3-C3H5) and L8 to 2.5 mol% and 3 mol%, respectively, without affecting the yield of 3a (entry 15). However, extending the reaction time to 48 h was needed to achieve completion of the reaction. In the absence of L8 or PdCp(η3-C3H5), this reaction could not occur (entry 16).
 DownLoad:
CSV
DownLoad:
CSV
 |
||||||
| Entry | Ligand | Solv. | Temp (℃) | Time (h) | Yield (%)b | ee (%)c |
| 1 | L1 | THF | 30 | 12 | >95 | 19 |
| 2 | L2 | THF | 30 | 12 | 90 | 21 |
| 3 | L3 | THF | 30 | 12 | 30 | 43 |
| 4 | L4 | THF | 30 | 12 | 90 | 9 |
| 5 | L5 | THF | 30 | 12 | 73 | 61 |
| 6 | L6 | THF | 30 | 12 | 58 | 29 |
| 7 | L7 | THF | 30 | 12 | 46 | 79 |
| 8 | L8 | THF | 30 | 12 | 62 | 80 |
| 9 | L8 | THF | −10 | 24 | 77 | 84 |
| 10 | L8 | DME | −10 | 24 | 65 | 85 |
| 11 | L8 | DCM | −10 | 24 | 74 | 77 |
| 12 | L8 | Toluene | −10 | 24 | 72 | 79 |
| 13 | L8 | MTBE | −10 | 24 | 76 | 88 |
| 14 | L8 | MTBE | −40 | 36 | 75 | 92 |
| 15d | L8 | MTBE | −40 | 48 | 73 (72e) | 92 |
| 16 | w/o L8 or Pd | MTBE | −40 | 48 | 0 | – |
| a Reaction conditions, unless otherwise noted: 1a (0.075 mmol), 2a (0.05 mmol), PdCp(η3-C3H5) (5 µmol, 10 mol%), ligand (11 µmol [22 mol%] for L2 and L3, 6 µmol [12 mol%] for L1, L4–L8), solvent (0.5 mL, [ 2a] = 0.1 mol/L) argon atmosphere. b The yields were determined from the1H NMR spectra of the crude products, with CH2Br2 as the internal standard. c The ee values were determined by chiral HPLC. d PdCp(η3-C3H5) (1.25 µmol, 2.5 mol%), L8 (1.5 µmol, 3 mol%), methyl tert-butyl ether (MTBE, 0.5 mL). e An isolated yield is given in parentheses. |
||||||
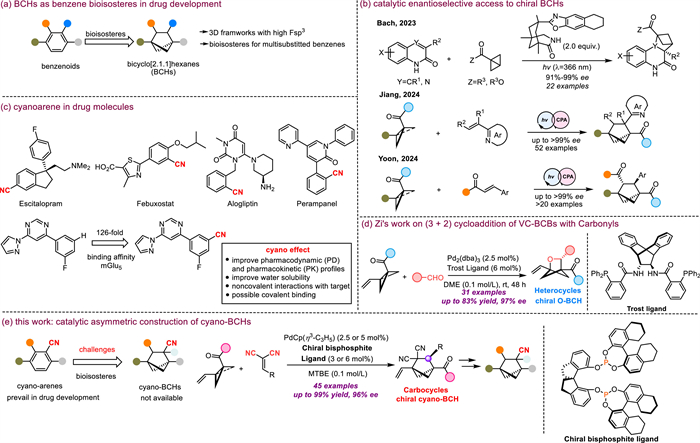
Having established the optimal conditions, we next turned our attention to the substrate scope, beginning with the arylidenemalononitrile (Scheme 1). The catalytic system proved to efficiently mediate reactions of malononitrile derivatives with either an electron-withdrawing or electron-donating substituent on the benzene ring, smoothly delivering chiral BCHs 3a–3j in 63%–93% yields with 87%–96% ee. Notably, substrates with a sterically hindered ortho-methyl or 3,5-dimethyl-substituted benzene ring or a naphthyl motif also gave the target products (3k–3n) with satisfactory yields and enantioselectivities. In addition, substrates with a thiophene, benzothiophene, benzofuran, quinoline, or pyridine motif were tolerated, furnishing 3o–3u in 71%–95% yields with 77%–95% ee. Alkylidenemalononitrile could also be used as a reaction partner in this cycloaddition; however, it produced poor enantioselectivity, as shown by the formation of 3v with 15% ee. This unsatisfying outcome could be caused by the smaller steric hindrance of 2v. The structure and stereochemistry of the products were confirmed by single-crystal X-ray diffraction analysis of 3h.
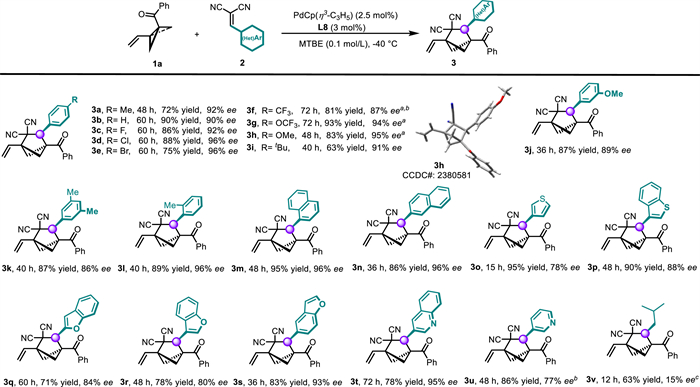
With 2n as the reaction partner, we next explored the substrate scope with respect to the VC-BCB (Scheme 2). Specifically, the carbonyl moiety of the VC-BCB was systematically varied. We found that benzoyl groups with various substituents, such as an electron-donating alkyl or methoxy group or electron-withdrawing halogen atoms or a trifluoromethyl group, were well tolerated, affording 3ab–3ak in 60%–95% yields with 89%–95% ee. Moreover, naphthyl ketones (3al, 3am) and heteroaryl ketones (3an–3as) were suitable substrates, demonstrating the generality and adaptability of this method. An n-butyl ketone–substituted substrate was compatible with the catalytic system as well, furnishing corresponding product 3at in 90% yield with 88% ee. In addition to ketones, acyl pyrazole (3au) proved to be competent substrates for this catalytic asymmetric cycloaddition. For carboxylate-derived VC-BCBs, the ligand L8 yielded relatively low ee values. Specifically, the phenyl and tert-butyl carboxylate products 3av and 3aw were obtained with enantioselectivities of 69% ee and 60% ee, respectively, under the standard conditions. However, following further optimization, ligand L9 was identified as a highly effective ligand for these challenging substrates, improving the enantioselectivities of 3av and 3aw to 81% ee and 77% ee, respectively. Additionally, the VC-BCB containing a (l)-menthol-moeity produced 3ay in 78% yield with >20:1 dr by using L9 as the ligand. Regardless of whether L8 or L9 was used, no cycloaddition products were observed for VC-BCBs bearing an internal alkene (3ax).
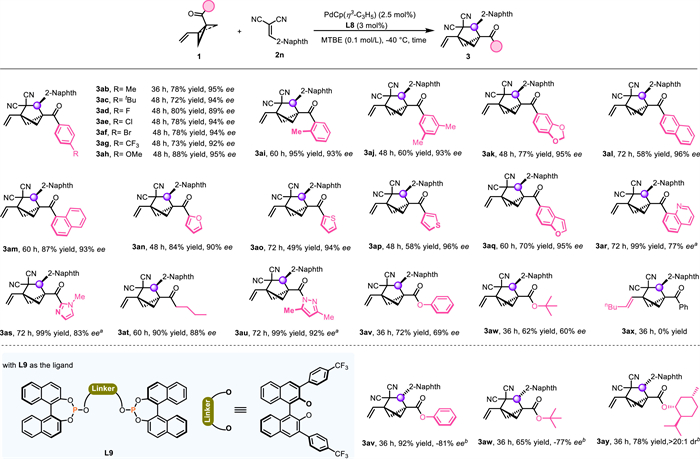
To illustrate the synthetic utility of our method, we performed a gram-scale synthesis of chiral BCH 3b and found that the yield and ee value were well-maintained upon scale up, making this method practical for large-scale production (Scheme 3a). Subsequently, transformations of 3b were carried out. Aiming to overcome the limitation posed by the geminal cyano groups of the substrates, we set out to transform the cyano groups into other functionality (Scheme 3b). One cyano group could be selectively hydrolyzed by using copper(Ⅱ) acetate as a catalyst for the generation of amide 4, which was obtained in 80% yield with the formation of a new quaternary stereocenter with a diastereomer ratio (dr) of 8:1 [68]. Decyanative coupling of 3b with chlorosilanes enabled by nickel/titanium dual catalysis [69] allowed us to construct a tertiary C(sp3)–Si bond to afford 5 in 19% yield with 90% ee and >20:1 dr, together with decyanative protonation product cis-6 (21% yield, >20:1 dr, 90% ee). The structure and stereochemistry of cis-6 were confirmed by X-ray crystallography (CCDC 2380580). Interestingly, treatment of 5 with tetra-n-butylammonium fluoride at −20 ℃ selectively afforded trans-6 with >20:1 dr. That is, stereodivergent synthesis of cyano-BCHs bearing vicinal stereocenters was achievable. Next, the vinyl group of 3b was converted to various other functional groups (Scheme 3c). Specifically, oxidative cleavage of the double bond by RuCl3/NaIO4 and subsequent esterification furnished ester 7 in 90% yield. An iridium-catalyzed hydroboration/oxidation procedure transformed 3b to alcohol 8 in 58% yield. Additionally, the pyrazole moiety of 3au could be hydrolyzed with aqueous LiOH, then underwent esterification reaction to afford a 66% yield of redox-active ester 9 (NHPI = N-hydroxyphthalimide) which could be converted to heteroaryl-substituted BCH 10 by means of a decarboxylative Minisci reaction (Scheme 3d) [70].
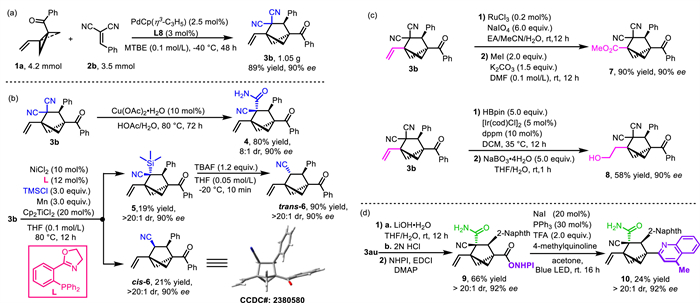
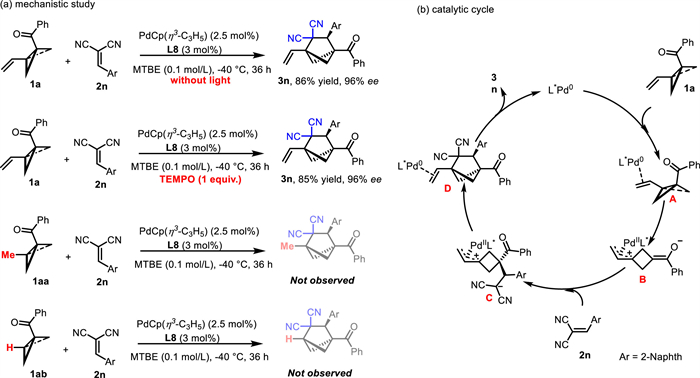
To get insight into the reaction mechanism, we carried out some control experiments (Scheme 4a). First, the reaction of 1a and 2n was performed in the dark, which had little to no effect on the yield or enantiopurity of product 3n, a result that rules out a pathway involving catalysis by an excited-state palladium species. Moreover, addition of the radical scavenger TEMPO (2,2,6,6-tetramethylpiperidine 1-oxyl, 1.0 equiv.) did not notably affect the yield, suggesting that a radical pathway may not be involved. Furthermore, when methyl-substituted BCB ketone 1aa or des-methyl BCB ketone 1ab was subjected to reaction with 2n under the standard conditions, formation of the corresponding cycloaddition product was not observed. This result indicates that the vinyl group of the VC-BCB is crucial for the success of the cycloaddition with the arylidenemalononitrile. On the basis of these results, along with results of previous studies [65,71,72] we propose that the reaction proceeds via the mechanism shown in Scheme 4b. Initially, a chiral palladium(0) complex coordinates with the vinyl group of 1a to form complex A. The strong π-donating ability of the palladium helps to cleave the strained C–C bond of the BCB, resulting in the formation of palladium-zwitterionic enolate B. Subsequently, B undergoes a stereoselective Michael addition to 2n to produce species C, which then undergoes an intramolecular allyl substitution reaction to generate complex D. Finally, disassociation of LPd from D generates product 3n and releases the palladium(0) catalyst.
In summary, we have developed a palladium-catalyzed enantioselective [2σ + 2π] cycloadditions of VC-BCBs with arylidenemalononitriles. The method has a wide substrate scope and provides rapid access to cyano-BCHs with good enantioselectivity. Various transformations of the cyano-BCHs were accomplished to give diverse functionality for the products. Mechanistic experiments supported a pathway involving a zwitterionic intermediate generated by palladium-mediated ring opening of the VC-BCBs followed by stereoselective 1,2-addition and allylic substitution reactions. We believe this work not only demonstrates a platform for the preparation of chiral cyano-BCHs, but also provides a good complement to the rare asymmetric BCBs cycloaddition reactions [46-48,73-80].
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Tianzhu Qin: Methodology, Investigation. Weiwei Zi: Writing – review & editing, Writing – original draft, Project administration, Conceptualization.
We gratefully acknowledge funding support from the National Key R&D Program of China (Nos. 2022YFA1503703, 2023YFA1506700) the National Natural Science Foundation of China (Nos. 22071118, 22271162). We thank the Haihe Laboratory of Sustainable Chemical Transformations and the Fundamental Research Funds for the Central Universities for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
P. de Sena M. Pinheiro, D.A. Rodrigues, R. do Couto Maia, et al., Curr. Top. Med. Chem. 19 (2019) 1712–1733. doi: 10.2174/1568026619666190712205025
P.K. Mykhailiuk, Org. Biomol. Chem. 17 (2019) 2839–2849. doi: 10.1039/c8ob02812e
M.A.M. Subbaiah, N.A. Meanwell, J. Med. Chem. 64 (2021) 14046–14128. doi: 10.1021/acs.jmedchem.1c01215
R.D. Taylor, M. MacCoss, A.D.G. Lawson, J. Med. Chem. 57 (2014) 5845–5859. doi: 10.1021/jm4017625
P. Das, M.D. Delost, M.H. Qureshi, et al., J. Med. Chem. 62 (2019) 4265–4311. doi: 10.1021/acs.jmedchem.8b01610
A. Nilova, L.C. Campeau, E.C. Sherer, et al., J. Med. Chem. 63 (2020) 13389–13396. doi: 10.1021/acs.jmedchem.0c00915
N.A. McGrath, M. Brichacek, J.T.A. Njardarson, et al., J. Chem. Ed. 87 (2010) 1348–1349. doi: 10.1021/ed1003806
F. Lovering, J. Bikker, C. Humblet, J. Med. Chem. 52 (2009) 6752–6756. doi: 10.1021/jm901241e
T.J. Ritchie, S.J.F. Macdonald, Drug Discov. Today 14 (2009) 1011–1020. doi: 10.1016/j.drudis.2009.07.014
F. Lovering, MedChemComm 4 (2013) 515–519. doi: 10.1039/c2md20347b
D. Trauner, Angew. Chem. Int. Ed. 57 (2018) 4177–4191. doi: 10.1002/anie.201708325
R. Gianatassio, J.M. Lopchuk, J. Wang, et al., Science 351 (2016) 241–246. doi: 10.1126/science.aad6252
R. T X. Zhang, R.T. Smith, C. Le, et al., Nature 580 (2020) 220–226. doi: 10.1038/s41586-020-2060-z
M.P. Wiesenfeldt, J.A. Rossi-Ashton, I.B. Perry, et al., Nature 618 (2023) 513–518. doi: 10.1038/s41586-023-06021-8
Y. Yang, J. Tsien, R. Dykstra, et al., Nat. Chem. 16 (2024) 285–293. doi: 10.1038/s41557-023-01342-7
J. Tsien, C. Hu, R.R. Merchant, et al., Nat. Rev. Chem. 8 (2024) 605–627. doi: 10.1038/s41570-024-00623-0
A. Cairncross, E.P. Blanchard, J. Am. Chem. Soc. 88 (1996) 496–504.
A.D. Meijere, H. Wenck, F. Seyed-Mahdavi, et al., Tetrahedron 42 (1986) 1291–1297. doi: 10.1016/S0040-4020(01)87348-8
P. Wipf, M.A.A. Walczak, Angew. Chem. Int. Ed. 45 (2006) 4172–4175. doi: 10.1002/anie.200600723
T.V.T. Nguyen, A. Bossonnet, M.D. Wodrich, et al., J. Am. Chem. Soc. 145 (2023) 25411–25421. doi: 10.1021/jacs.3c09789
P. Bellotti, F. Glorius, et al., J. Am. Chem. Soc. 145 (2023) 20716–20732. doi: 10.1021/jacs.3c08206
S.J. Sujansky, X. Ma, Asian J. Org. Chem. 13 (2024) e202400045. doi: 10.1002/ajoc.202400045
L. Zheng, Y.M. Yang, Z.P. Liu, et al., Org. Lett. 27 (2025) 229–234. doi: 10.1021/acs.orglett.4c04224
A. Denisenko, P. Garbuz, S.V. Shishkina, et al., Angew. Chem. Int. Ed. 59 (2020) 20515–20521. doi: 10.1002/anie.202004183
A. Denisenko, P. Garbuz, Y. Makovetska, et al., Chem. Sci. 14 (2023) 14092. doi: 10.1039/d3sc05121h
R. Kleinmans, T. Pinkert, S. Dutta, S, et al., Nature 605 (2022) 477–483. doi: 10.1038/s41586-022-04636-x
R. Kleinmans, S. Dutta, K. Ozols, et al., J. Am. Chem. Soc. 145 (2023) 12324–12332. doi: 10.1021/jacs.3c02961
S. Dutta, Y.L. Lu, J.E. Erchinger, et al., J. Am. Chem. Soc. 146 (2024) 5232–5241. doi: 10.1021/jacs.3c11563
S. Dutta, D. Lee, K. Ozols, et al., J. Am. Chem. Soc. 146 (2024) 2789–2797. doi: 10.1021/jacs.3c12894
R. Guo, Y.C. Chang, L. Herter, et al., J. Am. Chem. Soc. 144 (2022) 7988–7994. doi: 10.1021/jacs.2c02976
M. Yan, Z. Ye, P. Lu, Chin. Chem. Lett. 36 (2025) 110540. doi: 10.1016/j.cclet.2024.110540
S. Agasti, F. Beltran, E. Pye, et al., Nat. Chem. 15 (2023) 535–541. doi: 10.1038/s41557-023-01135-y
M. Xu, Z. Wang, Z. Sun, et al., Angew. Chem. Int. Ed. 61 (2022) e202214507. doi: 10.1002/anie.202214507
Y. Liu, S. Lin, Y. Li, et al., ACS Catal. 13 (2023) 5096–5103. doi: 10.1021/acscatal.3c00305
J.L. Tyler, F. Schäfer, H. Shao, et al., J. Am. Chem. Soc. 146 (2024) 16237–16247. doi: 10.1021/jacs.4c04403
N. Radhoff, C.G. Daniliuc, A. Studer, Angew. Chem. Int. Ed. 62 (2023) e202304771. doi: 10.1002/anie.202304771
L. Tang, Y. Xiao, F. Wu, et al., Angew. Chem. Int. Ed. 62 (2023) e202310066. doi: 10.1002/anie.202310066
D. Ni, S. Hu, X. Tan, et al., Angew. Chem. Int. Ed. 62 (2023) e202308606. doi: 10.1002/anie.202308606
J.J. Wang, L. Tang, Y. Xiao, et al., Angew. Chem. Int. Ed. 63 (2024) e202405222. doi: 10.1002/anie.202405222
S. Hu, Y. Pan, D. Ni, et al., Nat. Commun. 15 (2024) 6128. doi: 10.1038/s41467-024-50434-6
Q.Q. Hu, L.Y. Wang, X.H. Chen, et al., Angew. Chem. Int. Ed. 63 (2024) e202405781. doi: 10.1002/anie.202405781
W.H. Brooks, W.C. Guida, K.G. Daniel, et al., Curr. Top. Med. Chem. 11 (2011) 760–770. doi: 10.2174/156802611795165098
Food and Drug Administration. FDA’s Policy Statement For The Development of New Stereoisomeric Drugs, Chirality 4 (1992) 338–340. doi: 10.1002/chir.530040513
Agency E.M. Investigation of chiral active substances. 1994.
J.X. Zhao, Y.X. Chang, C. He, et al., Proc. Natl. Acad. Sci. U. S. A. 118 (2021) e2108881118. doi: 10.1073/pnas.2108881118
M. De Robichon, T. Kratz, F. Beyer, et al., J. Am. Chem. Soc. 145 (2023) 24466–24470. doi: 10.1021/jacs.3c08404
Q. Fu, S. Cao, J. Wang, et al., J. Am. Chem. Soc. 146 (2024) 8372–8380. doi: 10.1021/jacs.3c14077
E.F. Plachinski, R.Z. Qian, R. Villanueva, et al., J. Am. Chem. Soc. 146 (2024) 31400–31404. doi: 10.1021/jacs.4c13596
X. Wang, Y. Wang, X. Li, et al., RSC Med. Chem. 12 (2021) 1650–1671. doi: 10.1039/d1md00131k
F.F. Fleming, L. Yao, P.C. Ravikumar, et al., J. Med. Chem. 53 (2021) 7902–7917.
Y.X. Wang, Y.F. Du, N. Huang, Future Med. Chem. 10 (2018) 2713–2727. doi: 10.4155/fmc-2018-0252
J.Y. Le Questel, M. Berthelot, C. Laurence, et al., J. Phys. Org. Chem. 13 (2000) 347–358. doi: 10.1002/1099-1395(200006)13:6<347::AID-POC251>3.0.CO;2-E
V. Bonatto, R.F. Lameiro, F.R. Rocho, et al., RSC Med. Chem. 14 (2023) 201–217. doi: 10.1039/d2md00204c
H.U. Reissig, Top. Curr. Chem. 144 (1988) 73–135.
H.U. Reissig, R. Zimmer, Chem. Rev. 103 (2003) 1151–1196. doi: 10.1021/cr010016n
C. Carson, M.A. Kerr, Chem. Soc. Rev. 38 (2009) 3051–3060. doi: 10.1039/b901245c
T.F. Schneider, J. Kaschel, D.B. Werz, et al., Angew. Chem. Int. Ed. 53 (2014) 5504–5523. doi: 10.1002/anie.201309886
M.A. Cavitt, L.H. Phun, S. France, Chem. Soc. Rev. 43 (2014) 804–818. doi: 10.1039/C3CS60238A
F. De Nanteuil, F. De Simone, R. Frei, et al., Chem. Commun. 50 (2014) 10912–10928. doi: 10.1039/C4CC03194F
H.K. Grover, M.R. Emmett, M.A. Kerr, Org. Biomol. Chem. 13 (2015) 655–671. doi: 10.1039/C4OB02117G
O.A. Ivanova, I.V. Trushkov, Chem. Rec. 19 (2019) 2189–2208. doi: 10.1002/tcr.201800166
J. Wang, S.A. Blaszczyk, X. Li, et al., Chem. Rev. 121 (2021) 110. doi: 10.1021/acs.chemrev.0c00160
Y. Xia, X. Liu, X. Feng, Angew. Chem. Int. Ed. 60 (2021) 9192–9204. doi: 10.1002/anie.202006736
V. Pirenne, B. Muriel, J. Waser, Chem. Rev. 121 (2021) 227–263. doi: 10.1021/acs.chemrev.0c00109
T. Qin, M. He, W. Zi, Nat. Synth. 4 (2025) 124–133.
J. Han, R. Liu, Z. Lin, et al., Angew. Chem. Int. Ed. 62 (2023) e202215714. doi: 10.1002/anie.202215714
X. Pan, L. Yu, S. Wang, et al., Org. Lett. 24 (2022) 2099–2103. doi: 10.1021/acs.orglett.2c00290
E.B. Obregón, L.G. Rost, T.R. Kocemba, et al., Angew. Chem. Int. Ed. 63 (2024) e202410524.
Z.H. Chen, Y.Q. Zheng, H.G. Huang, et al., J. Am. Chem. Soc. 146 (2024) 14445–14452. doi: 10.1021/jacs.4c04495
M.C. Fu, R. Shang, B. Zhao, et al., Science 363 (2019) 1429–1434. doi: 10.1126/science.aav3200
C. Yang, Z.X. Yang, C.H. Ding, et al., Chem. Rec. 21 (2021) 1442–1454. doi: 10.1002/tcr.202000177
B. Xu, Q. Wang, C. Fang, et al., Chem. Soc. Rev. 53 (2024) 883–971. doi: 10.1039/d3cs00489a
J. Zhou, Y. Xiao, L. He, et al., J. Am. Chem. Soc. 146 (2024) 19621–19628. doi: 10.1021/jacs.4c01851
X. Wang, R. Gao, X. Li, et al., J. Am. Chem. Soc. 146 (2024) 21069–21077. doi: 10.1021/jacs.4c06436
F. Wu, W.B. Wu, Y. Xiao, et al., Angew. Chem. Int. Ed. 63 (2024) e202406548. doi: 10.1002/anie.202406548
M. Zhang, M. Chapman, B.R. Sarode, et al., Nature 633 (2024) 90–95. doi: 10.1038/s41586-024-07865-4
W.B. Wu, B. Xu, X.C. Yang, et al., Nat. Commun. 15 (2024) 8005. doi: 10.1038/s41467-024-52419-x
X.G. Zhang, Z.Y. Zhou, J.X. Li, et al., J. Am. Chem. Soc. 146 (2024) 27274–27281. doi: 10.1021/jacs.4c10123
J. Jeong, S. Cao, H.J. Kang, et al., J. Am. Chem. Soc. 146 (2024) 27830–27842. doi: 10.1021/jacs.4c10153
T. Li, Y. Wang, Y. Xu, et al., ACS Catal. 14 (2024) 18799–18809. doi: 10.1021/acscatal.4c06660

Scheme 1 Substrate scope with respect to the arylidenemalononitrile. Reaction conditions, unless otherwise noted: 1a (0.26 mmol), 2 (0.2 mmol), PdCp(η3-C3H5) (5 µmol, 2.5 mol%), L8 (6 µmol, 3 mol%) methyl tert-butyl ether (MTBE, 2 mL), −40 ℃, argon atmosphere. Isolated yields are given. The ee values were determined by chiral HPLC. a PdCp(η3-C3H5) (10 µmol, 5 mol%) and L8 (12 µmol, 6 mol%) were used. b The reaction temperature was −20 ℃. c The reaction temperature was 30 ℃.

Scheme 2 Substrate scope with respect to the VC-BCB. Reaction conditions, unless otherwise noted: 1 (0.26 mmol), 2n (0.2 mmol), PdCp(η3-C3H5) (5 µmol, 2.5 mol%), L8 (6 µmol, 3 mol%), methyl tert-butyl ether (MTBE, 2 mL), −40 ℃, argon atmosphere. Isolated yields are given. The ee values were determined by chiral HPLC. a The reaction was carried out at −30 ℃. b L9 (6 µmol, 3 mol%) and 1,4-dioxane (2 mL) were used instead of L8 and MTBE, and the reaction was carried out at room temperature.
Table 1. Optimization of reaction conditions.a
|
||||||
| Entry | Ligand | Solv. | Temp (℃) | Time (h) | Yield (%)b | ee (%)c |
| 1 | L1 | THF | 30 | 12 | >95 | 19 |
| 2 | L2 | THF | 30 | 12 | 90 | 21 |
| 3 | L3 | THF | 30 | 12 | 30 | 43 |
| 4 | L4 | THF | 30 | 12 | 90 | 9 |
| 5 | L5 | THF | 30 | 12 | 73 | 61 |
| 6 | L6 | THF | 30 | 12 | 58 | 29 |
| 7 | L7 | THF | 30 | 12 | 46 | 79 |
| 8 | L8 | THF | 30 | 12 | 62 | 80 |
| 9 | L8 | THF | −10 | 24 | 77 | 84 |
| 10 | L8 | DME | −10 | 24 | 65 | 85 |
| 11 | L8 | DCM | −10 | 24 | 74 | 77 |
| 12 | L8 | Toluene | −10 | 24 | 72 | 79 |
| 13 | L8 | MTBE | −10 | 24 | 76 | 88 |
| 14 | L8 | MTBE | −40 | 36 | 75 | 92 |
| 15d | L8 | MTBE | −40 | 48 | 73 (72e) | 92 |
| 16 | w/o L8 or Pd | MTBE | −40 | 48 | 0 | – |
| a Reaction conditions, unless otherwise noted: 1a (0.075 mmol), 2a (0.05 mmol), PdCp(η3-C3H5) (5 µmol, 10 mol%), ligand (11 µmol [22 mol%] for L2 and L3, 6 µmol [12 mol%] for L1, L4–L8), solvent (0.5 mL, [ 2a] = 0.1 mol/L) argon atmosphere. b The yields were determined from the1H NMR spectra of the crude products, with CH2Br2 as the internal standard. c The ee values were determined by chiral HPLC. d PdCp(η3-C3H5) (1.25 µmol, 2.5 mol%), L8 (1.5 µmol, 3 mol%), methyl tert-butyl ether (MTBE, 0.5 mL). e An isolated yield is given in parentheses. |
||||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: