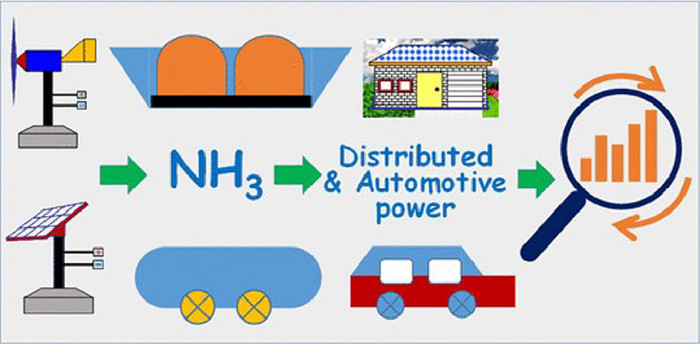
Figure 1.
The synthesis and application of ammonia in various areas. Reproduced with permission [5]. Copyright 2020, American Chemical Society.

Electroreduction of N2 to ammonia (NH3) has been extensively investigated in the pioneering works due to the great significance for industry and agriculture [1,2]. This is because NH3 can be served as an important source for both fertiliser and fuel [3,4]. According to the statistics, 117 million metric tons of ammonia was produced globally in 1996, which has increased to 175 million metric tons in 2016. In 2016, the ammonia utilization has reached to 88% for fertilizer in America. The forms of ammonia fertilizer mainly include anhydrous ammonia, urea, ammonium nitrates, ammonium phosphates, and various nitrogen compounds (Fig. 1) [5-7]. Ammonium is also a promising hydrogen carrier utilized in vehicles. It can be treated as an ideal fuel because the reaction products contain only water. As we know, the storage of hydrogen is always a challenging issue. However, it can be solved via transporting of liquid ammonia and hydrogen can be extracted from ammonia by thermal decomposition [8].
The traditional method for ammonia synthesis should be performed at high pressure (150–200 atm) and high temperature (300–500 ℃), which is called the Haber-Bosch method [9,10]. This industrial ammonia synthesis has satisfied the demand of human beings. In the mid-1990s, the Haber-Bosch method has supplied 40% of the world's dietary protein [11,12]. However, the rigorous condition may not only lead to a high energy input but also release large amount of greenhouse gases into the atmosphere [13]. Moreover, this industrial process significantly depends on fossil fuel, whose formation takes millions of years. On the other hand, the fossil fuels will be depleted in about 100 years at the current consumption rate [14]. Therefore, it is urgently needed to find renewable energy sources to realize environmentally friendly processes. Above all, the reduction of N2 in the electrochemical condition is treated as a promising strategy which can be performed at ambient conditions.
The electroreduction of N2 is performed on the surface of an electrode in electrochemical cell, which can provide a small-scale ammonia synthesis device [15-17]. Besides, it can directly utilize renewable sources, including solar or wind. The water in the electrolyte is used as the hydrogen source, which is produced via the oxidation of water in the electrochemical cell [18,19]. The N2 molecules are first adsorbed on the electrode and then protonated by an electron-proton pair, supplied from the electrolyte and external circuit [20,21]. Compared to the traditional Haber-Bosch method, the N2 is activated via electrical energy under low temperature and pressures [22,23]. In addition, the electrochemical condition can modulate the product distribution via altering the potential and temperature [24,25].
Although extensive studies on N2 electrochemical reduction (NRR), the NRR is still hindered by many factors: (1) The adsorption strength of N2 is always very weak, leading to a sluggish activation of N$\equiv$N triple bond. This may eventually lead to the high overpotential of NRR [26]; (2) The theoretical limiting potential (UL) of hydrogen evolution reaction (HER), which is the main competing path of NRR, is smaller than that of NRR. In addition, the protons and electrons tend to go towards the evolution of H2 compared to NH3 from kinetic aspect. Thus, H2 is always dominant in the final products during N2 reduction, leading to low Faradaic efficiency (FE) of NRR [27]. Thus, design of catalysts with low overpotential and high FE is necessary to enable the electrocatalytic NRR at ambient conditions.
In pioneering works, the NRR has been investigated on many kinds of catalysts, including metal [28,29], metal compound [30,31], single atom catalyst [32,33], and metal-free catalysts [34]. In addition, many strategies are also proposed to improve NRR performance, such as introducing vacancies [35-39], heteroatom doping [40-45], increasing magnetic moment [46], and regulation of valence [47,48]. These strategies mainly focus on the improving of intrinsic activity. Besides, there also exists many schemes with the purpose of increasing the apparent activity, depending on the number of exposed active sites [49]. Compared to the apparent activity, the intrinsic activity can be promoted via tailoring the electronic configuration of the catalyst to alter the properties of each active site. Nevertheless, there still lacks a universal criterion for catalyst design. This may impede the quick screen of promising catalysts among large pools of candidates. In theory, the adsorption energies of the key intermediates have been adopted as an effective descriptor for quick screen of NRR catalysts. The adsorption energy of *N is generally used, which can be plotted with the NRR activity and the scaling relationship has been obtained in pioneering works [50-55]. In order to reflect the intrinsic properties of active center, many pioneering works have focused on other descriptors such as d-band center [56], magnetic moment [57], and dipole [58,59]. These screening criterions of promising catalysts can be denoted as energy-derived descriptors. These descriptors are always defined with electronic properties of the elements in active centers, and manifest intimate correlations with the adsorption energies. Compared to the descriptors of pure energies, the energy-derived descriptors can accurately predict the reactivity of catalysts without large pools of calculations, and can be generalized into extensive systems.
This review article focuses on the summarization of effect strategies and universal criterions in pioneering works on NRR catalysts. Firstly, the fundamental mechanisms of NRR are depicted. Then, various strategies of improving the intrinsic activity of NRR are presented. Considering that the commonly used catalysts have been systematically summarized in pioneering review articles, we finally place emphasis on the high-throughput screen of promising catalysts based on descriptors.
The mechanism of NRR electroreduction generally includes two situations: associative and dissociative mechanisms (Fig. 2). The detailed reaction pathway is somewhat complicated, which largely depends on the adsorption behavior of N2. As N2 is adsorbed via end-on pattern, the NRR prefers to proceed through distal and alternating pathways. In contrast, NRR will occur through enzymatic and consecutive pathways as N2 adsorbed via side-on pattern [60-67].
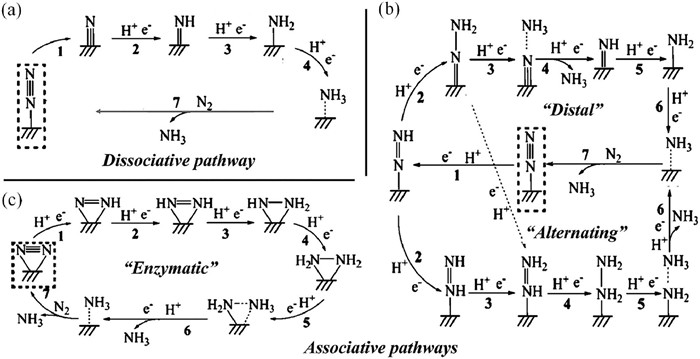
The traditional Haber-Bosch method generally follows the dissociative pathway: The N$\equiv$N triple bond will cleave at high temperature condition, leaving a single N atom on the catalyst surface. This N atom can be protonated successively to produce NH3. However, this dissociative mechanism is significantly difficult for electrochemical condition due to the lack of energy input. For the alternating mechanism, the two N atoms in the adsorbed N2 is protonated alternatively until the first ammonia formed accompanied with the N$\equiv$N bond dissociated. The other ammonia is produced via successive protonation of the remaining N atom. For the distal mechanism, The N atom far from the catalyst is preferentially protonated until the first ammonia obtained. Then, the N atom adsorbed directly on the catalyst is protonated successively to produce another ammonia. The alternating and distal mechanisms are preferred to occur in the ambient condition. The common rate controlling steps (RCS) of the mechanisms mentioned above include two aspects: The adsorption and activation of N2 to form *N2H; The protonation of *NH2 and the desorption of *NH3 [60-67].
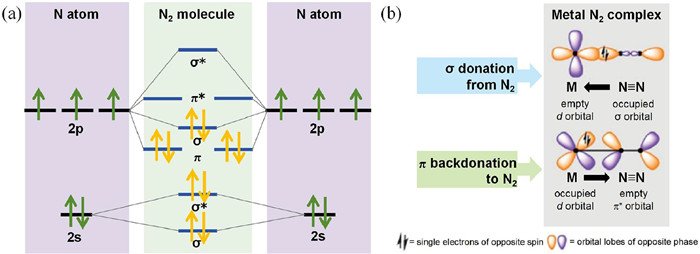
The activation of N2 is an important topic for NRR, which has been extensively investigated in the pioneering works especially on transition metals (TM). On transition metals, N2 is interacted on the surface via N-metal bonds, where the N$\equiv$N triple bond can be weakened via the "acceptance-donation path" (Fig. 3). The electrons transferred between N2 and TM includes two orientations: The vacant d orbitals of TM accept the lone-pair electrons of N2; The occupied d orbitals of TM provide electrons to the π* antibonding orbitals [68-72]. The transfer of electrons from catalysts to the adsorbed N2 plays an important role for N2 activation. This is because that the occupation of N2's antibonding orbitals can effectively weaken the N$\equiv$N triple bond, leading to the formation of dangling bonds on the N atoms of adsorbed N2. This character can eventually decrease the reaction energy of N2 protonation processes.
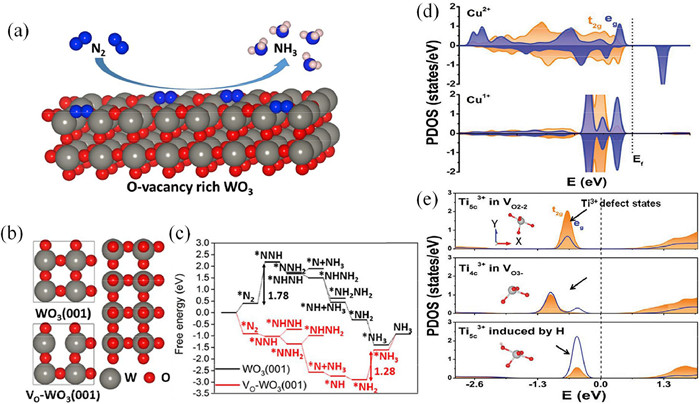
As mentioned above, the RCS of NRR is the activation of N$\equiv$N triple bonds. Introducing vacancy defects, which can be served as electron-rich sites, is assumed as an effective strategy. The electron-rich character is beneficial for charge transfer between catalyst and N2, leading to an enhanced reactivity of the active sites (Figs. 4a-c) [35]. Hirakava et al. have introduced oxygen vacancies (OV) into TiO2 and successfully produced NH3 at room temperature [36]. The OV can effectively tailor the electronic structures and surface properties, leading to the effective adsorption of N2 at Ti3+ site. Qu et al. have introduced OVs into layered MoO2 and boosted the N2 adsorption via electron donation on Mo3+ site. The activation energy barrier is reduced due to the stabilization of *N2H and destabilization of *N2H2. The favorable reaction mechanism exists in the distal/alternating hybrid path [37]. Qiao et al. have investigated N2 activation on W2N3 surface and found that there exists little charge transfer between perfect W2N3 and adsorbed N2. Thus, the perfect W2N3 cannot activate N2 effectively. However, as nitrogen vacancies (NV) introduced, the charge transfer will increase significantly and N2 is activated effectively with a protonation energy barrier of only 0.55 eV [38]. Compared to the O vacancies, the N vacancies are more essential in determining the NRR reactivity. This may be because that the adsorbed NRR intermediates can form chemical bonds with the catalysts on the N vacancies. The newly formed chemical bonds are identical to the pristine state of catalysts. In addition to oxygen and nitrogen vacancies, the sulfur vacancy (SV) is also an effect strategy as reported by Zeng et al. They have introduced SV into MoS2 catalyst, which can facilitate electron transfer from Mo 3d orbitals to N2. The transferred electrons mainly originate from S 2p and N 2p orbitals [39]. Vacancy defects have obtained great achievements in pioneering works for NRR. However, the adsorption strength of this kind of active site is strong, leading to the poisoning of catalysts. In comparison, doping heteroatoms at the vacancy sites can alleviate the poisoning effect.
Doping heteroatoms is a commonly used strategy to promote the NRR reactivity. Among numerous dopants, both metal and nonmetal atoms are extensively investigated. Fe is a commonly used dopant due to the intrinsic property of high magnetism. This character may facilitate the generation of lone-pair electrons in the adsorbed *N2. Sun et al. have attempted to dope TiO2 with Fe atom and the results have demonstrated that Fe can increase the number of oxygen vacancies and decrease the valence of Ti4+ to Ti3+. The bi-Ti3+ and oxygen vacancy is responsible for the high catalytic performance [40]. Azofra et al. have also designed a Fe doped MoS2 catalyst, where can establish a perfect donation-back donation process during N2 adsorption. The lone pair electrons in N2 can form σ bonds with Fe and the d-orbital of Fe can back donate electrons to N2 with π* bonds [41]. In addition to Fe, Mo is also an effective dopant with high spin density. Wand et al. have doped Mo into MnO2, where N2 can be activate over Mo site with side-on configuration. This is mainly attributed to the dz2 occupied states of Mo, which can interact with 1π* orbital of N2 [42]. For nonmetal dopants, atoms, including B, O, and S, are commonly used in pioneering works. Unlike the metal dopants, the active centers of nonmetal atom doped systems generally delocalized from the dopants. Sun et al. have designed the sulfur-doped graphene as NRR catalyst and demonstrated that the carbon atoms close to substituted sulfur atoms can act as the underlying active sites for NRR [43]. Dai et al. have also proved that O doping in the GDY can lead to the redistribution of electron density. The sp-states of C atoms nearest to the O atom can provide enhanced capture to N2 molecules [44]. Kong et al. have proposed that the sp-substitution of B can moderate the local spin and charge densities on the graphene surface, which is convenient for N2 adsorption and activation [45].
Considering the inert character of bulk materials, many researches have focused on decreasing the size of catalysts. This strategy can increase the atomic utilization of atoms and provide more active sites for NRR. In addition, the decrease of size can generate more free electrons at corner and edge sites, leading to an enhanced capture ability of N2 molecules. Among numerous catalysts, the clusters and single atom catalyst (SAC) are commonly used. Qiao et al. have loaded Co clusters onto the N doped carbon and the Co cluster is proved to act as electron-donating promoter. This character can successfully capture the N2 molecules and change the RCS of NRR to the first N2 protonation [73]. Li et al. have loaded Fe3 cluster on Al2O3 and achieved an enhanced spin polarization for the catalyst. The *N2 is easily hydrogenated to form *NNH, which is easily to dissociate during the following reaction process [74]. Luo et al. have also attempted to load Fe4 cluster on graphene. This catalyst is superior to the Fe3 and Fe2 clusters on graphene, displaying an outstanding NRR performance with side-on N2 adsorption [75]. About the single atom catalysts, Chen et al. have embedded various kinds of single metal atoms into BN monolayer and screened Mo@BN as the favorable catalyst. This is because that Mo@BN possesses the favorable performance for N2 activation and NH2 stabilization [76]. Huang et al. have designed a graphene-based Fe single atom catalyst and uncovered the importance of Fe's magnetic moment in N2 activation. The increased Fe magnetic moment can strengthen the charge transfer between the N2 and the substrate, resulting in a lower overpotential for NRR [77]. Tao et al. have compared Ru SAC on both ZrO2 and NC2 substrates. They all shows improved catalytic reactivity compared to bulk Ru. N2 is more favorable to adsorb on Ru@ZrO2 compared to Ru@NC2. This is because that the O vacancies in ZrO2 can attribute to the stabilization of *NNH, destabilization of *H and adsorption of N2 [78].
As mentioned above, the electronic properties of the active centers can determine the adsorption of NRR intermediates especially N2 molecule. The valence and spin density are important criteria for electronic configuration. Sun et al. have investigated the modulation effect of mixed-valence Cu ions in TiO2 for NRR. The mixed-valence Cu can effectively modulate the contribution of O vacancies, leading to the formation of Ti3+ defect states. This phenomenon is in accordance with the discussions in Part 3.1. Therefore, there exists intimate relevance between vacancy and valence of active centers, which is essential for the charge transfer between adsorbed N2 and catalysts. The Ti3+-d states can split into eg and t2g orbitals upon the influence of the coordination environment (Figs. 4d and e) [47]. Ding et al. have regulated the valence of Nb into negative state upon embedded into C3N4 substrate. The negative valence can effectively host the empty d orbitals of N2 and provide single d-orbitals to N2. The d-electrons back donation to N2 can activate the N$\equiv$N triple bond, leading to the enhanced adsorption of N2 [48]. An et al. have embedded a variety of metal atoms into defective BN and a scaling relationship is plotted between adsorption energies of *N2 and *NH2. However, the candidates with high spin density are deviated away from the scaling line, leading to enhanced NRR reactivity. Thus, the spin states of active sites play an important role in modulating the reactivity of NRR [46]. The regulation of valence can also be realized via electronic field as reported by Jiang et al. [79]. The results showed that the partially occupied pz orbital of a B atom can form B-to-N π-back bonding with the antibonding state of N2, thus weakening the N$\equiv$N triple bond. The proper electric field on the BN surface can promote this process and eventually realize N2 adsorption and activation.
The discovery of advanced catalysts in large pools of candidates should be time-consuming and expensive due to large amounts of trial and errors. Many computational methods have been explored to provide convenient methods to screen catalysts with low cost, which can propose many design principles for advanced catalysts and effectively guide the experimental works. Density functional theory is a state-of-art method for theoretical calculation, which is accurate enough to agree well with the experiments [79-81].
Norskov et al. have proposed a "computational hydrogen electrode" (CHE) model, which has become an important theoretical tool for the investigation of electro-catalysts [82]. This method can calculate the adsorption energies of NRR intermediates, the reaction free energies, the rate controlling steps and the ULs. It can also explain the tendency of catalytic reactivity from one catalyst to the next and provide new insights into the mechanism of NRR.
The adsorption energy (Ead) is an important property, which can determine the reactivity of NRR. The adsorption energy of a molecule (Ead[M]) can be described as follows (Eq. 1):
|
|
(1) |
where E[M-sub], E[M], and E[sub] denote the energies of substrate with molecules adsorbed on it, singel atoms in gas phase, and the pure substrate.
From the reaction-pathway diagram, reaction UL and RCS should be identified to evaluate the reactivity, which are determined based on the largest free-energy change (Eq. 2):
|
|
(2) |
where, the ΔGi is the free-energy change associated with step i at potential of 0 V vs. RHE; n is the number of electrons transferred in step i; e is the charge of an electron. The RCS was defined as the step with maximum free energy change, which determine the reactivity of NRR.
CHE calculations have provided key insights into various kinds of catalysts, mainly about the influence of geometric and electronic properties for NRR reactivity.
As revealed in the pioneering studies, the adsorption energies of intermediates can be plot into scaling relationships due to the d-band center theory. For oxygen evolution and reduction, there exists scaling relationships between adsorption energies of O-species, including *O, *OH, and *OOH [83,84]. Similarly, the C-species, including *CO, *COOH, *CHO, can also be plot into scaling lines for CO2 electroreduction [85,86]. Therefore, for NRR, the adsorption energies of N-species, including *N, *NH2, *N2H, are not independent [87,88]. Thus, the adsorption energies of important intermediates can be treated as an effective descriptor, which can facilitate the quick screen of efficient catalysts. However, the adsorption-energy based descriptor cannot reveal the intrinsic properties of the catalyst. The adsorption behavior of molecules on the catalyst significantly determined by the electronic properties of active centers. Thus, many studies have focused on many adsorption-energy derived descriptors, including d-band center, orbital hybridization, spin density, dipole moment, valence electrons, and electronegativity [56-59].
The originate of the scaling lines stems from the surface of transition metals (TMs). Norskov et al. have performed DFT calculations on TMs systematically and found a perfect linear scaling between two key adsorbates: *N2H and NH2. In the CHE model, the UL is a criterion for reactivity, which is defined as the applied potential required to make every reaction step exergonic. Thus, they have also plotted the adsorption energies of *N with ULs on TMs and a perfect volcano model is established. The volcano model has assumed that the RCSs of NRR occur at either the protonation of N2 to form *N2H or the protonation of *NH2 to form NH3. This is a classical descriptor that proposed by Norskov et al. and it has great referential value for the following works. In addition, the conclusions revealed on TMs have also been utilized into other systems like SACs, metal clusters and metal compounds [50].
About SACs, Dai et al. have plotted the adsorption energies of *N with the ULs on graphdiyne based SACs. The metal atoms embedded in the substrates are treated as the active centers. The perfect linear correlation has demonstrated that the descriptor of *N adsorption energy can be extended successfully into SAC systems [51]. Similar works have also been performed by Qiao et al. on C3N4 based SACs [52]. They have not only found the linear relationship between the adsorption energies of *N with the ULs, but also obtained similar volcano model proposed by Norskov et al. In addition to SACs, this classical theory is also hold on double atom catalysts (DACs) reported by Huang et al. Considering the bidentate adsorption behavior of N-species on DACs, they have treated the descriptor as adsorption energies of *N2H, unlike *N, on DACs. They have also established a volcano model on their designed catalysts with identical RCSs: protonation of *N2 to *N2H and protonation of *NH2 to *NH3 [53]. About metal clusters, Vegge et al. have constructed various TM nanoclusters with 12 atoms. They have established linear scaling relations for dissociative reaction intermediates, including *NH, *NH2, and NH3. In addition, they have also determined linear scaling relations for the associative reaction intermediates, including *N2H, *N2H2, and *N2H3 [54]. About metal compounds, Huang et al. have designed a two-dimensional metal-borides catalysts (denoted as MBenes). They have taken 16 MBenes as representatives and screened 7 candidates as promising NRR catalysts based on DFT calculation. The adsorption energies of *N2H can be fitted into scaling lines with both ULs and d-band centers. The CrMnB2 locates nearest to the top of the volcano model established in MBene systems [55]. These works have illustrated that the d-band center theory is also hold in many other systems, including SACs, DACs, metal clusters and metal compounds. This can be treated as the origin of the perfect linear relationships between the adsorption energies of NRR intermediates.
However, the adsorption energy is not a general property and it cannot demonstrate the intrinsic properties of the active centers of catalysts. In addition, it cannot be generalized into extensive systems. These are the limitations of adsorption-energy descriptors and should be corrected based on some electronic properties, which can effectively reflect the intrinsic properties of the catalysts.
In order to demonstrate the intrinsic characters of catalysts, many studies attempt to transfer the adsorption energies into other parameters correlated to electronic properties. The properties, such as magnetic moments, d-band centers, and dipole moments, are proved to possess intimate correlations with adsorption strength of NRR intermediates. This can make it possible to propose new descriptors derived from adsorption energies (Fig. 5).
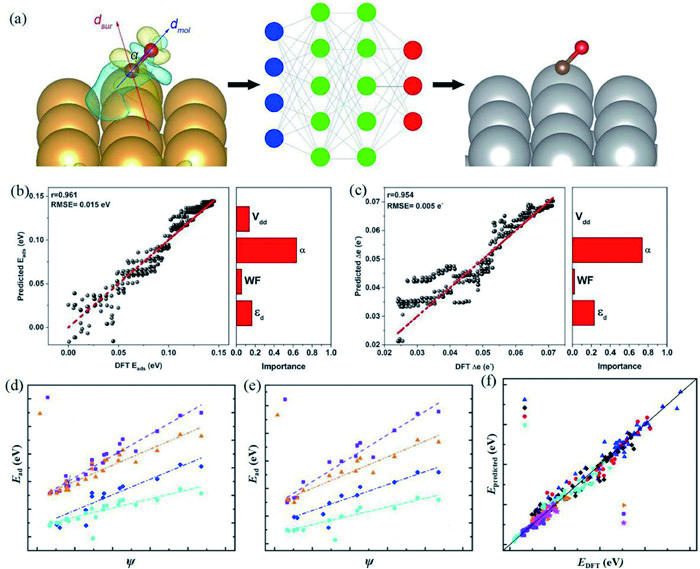
For descriptor of d-band centers, Deng et al. have designed 20 kinds of TM atom-pair catalysts anchored on N-doped graphene. They have studied the correlations between ULs, free energies, and d-band centers and furthermore evaluated the activity tendency of the designed catalysts. The results demonstrated that the enzymatic pathway is the most suitable and the d-band center can be used as the descriptor to describe NRR performance [56]. About the descriptor of magnetic moments, Huang et al. have designed Fe SACs based on N-doped graphene. Through the change of coordination environment of Fe, they have uncovered that the magnetic moment of the embedded Fe atom plays an important role in N2 activation. In addition, a linear relationship between the adsorption energies of *N and the magnetic moments of Fe has been established, indicating that the magnetic moment can be treated as an effective descriptor [57]. For descriptor of dipole moments, Li et al. first proposed that dipole of N$\equiv$N triple bond is an accurate indicator for screening NRR catalysts. They have performed DFT calculations on phthalocyanine based SACs and Mo is screened as the optimal candidate due to the lowest dipole moment. This character can promote the donation of electrons from the Mo atom to the 2π* antibonding orbitals of N2 [58]. Wang et al. have also performed similar work on the descriptor of dipole moment via machine leaning techniques. Among the descriptors selected, the included angle between surface dipole and molecule dipole have shown the better predictive performance compared to d-band centers. This is because that the dipole-based descriptor can incorporate information of both electronic structure and geometric detail. Wang et al. have extended the works of Li et al. into more deep level and pointed out the deficiency of d-band center descriptor (Figs. 5a-c) [59].
As we know, the real reaction of NRR generally happened in the electrochemical environments. Therefore, the solvent models can dramatically influence the adsorption strength of NRR intermediates. The perspective modeling techniques mainly include implicit and mixed implicit/explicit solvation models [89,90]. Lv et al. have applied solvation correction on Fe2/CN system for NRR. The solvation effects were treated by using the implicit solvation model implemented in VASPsol. The author has also demonstrated that the solvation model would change the RCSs and UL of the most promising candidate [91].
Although various kinds of descriptors have been successfully established, the data collection of these properties need numerical calculations and cannot be generalized into multiple systems. This eventually hampers the quick screen of efficient catalysts from a large pool of candidates. Thus, the establishment of universal descriptors is a challenge for the researchers.
Xu et al. have proposed a universal descriptor based on the valence electrons in d orbital and electronegativity, which exhibits good correlation with the HER, OER, and ORR reactivity (Figs. 5d-f) [92]. In addition, Zeng et al. have also presented a universal design principle to evaluate the activity of graphene-based single-atom catalysts towards the oxygen reduction, oxygen evolution and hydrogen evolution reactions. Moreover, the catalytic activity of the active centers is highly correlated with the local coordination environment of the metal center with the neighbor atoms. The proposed principle can also be extended to metal-macrocycle complexes [93]. Inspired by these works, Liu et al. proposed a similar descriptor φ as follows (Eqs. 3 and 4):
|
|
(3) |
|
|
(4) |
where, θd represents the valance electrons in d-orbital of TM atom, ETM, EN and EC denote the electronegativity of TM, N and C elements, respectively. nN and nC represent the number of N and C atoms of the 2 × 2 × 1 C9N4 substrate.
Through this descriptor, they have plotted a universal volcano relationship between φ and various properties, including charge transfer, adsorption energy, and N—N bond length. More surprisingly, the candidates located at the left side of volcano are all early TMs and the right-side candidates are all late TMs. This work has provided a predictive power to guide the rational design of new and high-performance NRR catalysts [94].
Gao et al. have also proposed a predictive model for quantitative determination of the adsorption energies of small molecules on metallic materials and oxides and successfully applied into NRR area. The descriptor can be described as follows (Eq. 5):
|
|
(5) |
where Sv is the number of valence electrons; Χ denote the electronegativity; β is an index determined by the role of d- and s-orbitals in valence descriptions and electronegativity [95].
In addition, there always exists various elements in the active centers for real situations. Therefore, the correction of the descriptor is necessary to accurately describe the reactivity of the catalysts. The author has performed the method of geometric mean to correct the electronic properties, including the valence electrons and electronegativities.
Through this descriptor, Gao et al. have systematically studied NRR on SACs and BACs with diverse coordinate environments. They have qualified the influence of coordinate environments on the adsorption properties and uncovered the difference and similarities between SACs, homonuclear BACs, and heteronuclear BACs. In addition, this descriptor can also reveal the role of N dopant [95].
The pioneering works have proposed various design strategies for efficient NRR catalysts. The lack of design principles makes the investigations expensive and time consuming. Thus, various descriptors have been proposed to accelerate the screen of promising NRR catalyst. However, the conventional descriptors based on adsorption and electronic properties cannot reflect the intrinsic properties of the catalyst. Thus, some researches have attempted to propose some universal principles for the design of catalysts. In our further study, we should propose more effective descriptors for NRR referenced to the pioneering works. Based on the new theory proposed, we will clarify the mechanism in depth and further discover more proper candidates for NRR. There is still a long way to go.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jinzhou Zheng: Writing – original draft, Resources, Methodology, Investigation. Chaozheng He: Writing – review & editing, Resources, Funding acquisition, Data curation. Chenxu Zhao: Visualization, Supervision, Methodology, Formal analysis, Data curation.
This work was supported by the National Natural Science Foundation of China (No. 21603109), the Henan Joint Fund of the National Natural Science Foundation of China (No. U1404216), the Special Fund of Tianshui Normal University, China (No. CXJ2020-08) and the Scientific Research Program Funded by Shaanxi Provincial Education Department (No. 20JK0676). This work was also supported by Natural Science Basic Research Program of Shanxi (Nos. 2022JQ-108, 2022JQ-096).
L. Wang, M. Xia, H. Wang, et al., Joule 2 (2018) 1055-1074.
N. Cao, G. Zheng, Nano Res. 11 (2018) 2992-3008. doi: 10.1007/s12274-018-1987-y
D. Liu, M. Chen, X. Du, et al., Adv. Funct. Mater. 31 (2021) 2008983.
S. Giddey, S. P. S. Badwal, A. Kulkarni, Int. J. Hydrogen Energ. 38 (2013) 14576-14594.
S. Giddey, S. P. S. Badwal, C. Munnings, M. Dolan, ACS Sustainable Chem. Eng. 5 (2017) 10231-10239. doi: 10.1021/acssuschemeng.7b02219
U. B. Demirci, P. Miele, J. Cleaner Prod. 52 (2013) 1-10.
Z. Ma, Y. Luo, P. Wu, et al., Adv. Funct. Mater. 33 (2023) 2302475.
G. Ercolino, M. A. Ashraf, V. Specchia, S. Specchia, Appl. Energy 143 (2015) 138-153.
J. W. Erisman, M. A. Sutton, J. Galloway, Z. Klimont, W. Winiwarter, Nat. Geosci. 1 (2008) 636-639. doi: 10.1038/ngeo325
K. Wang, D. Smith, Y. Zheng, Carbon Resour. Convers. 1 (2018) 2-31. doi: 10.3847/2515-5172/aaa4c2
H. P. Jia, E. A. Quadrelli, Chem. Soc. Rev. 43 (2014) 547-564.
C. J. M. van der Ham, M. T. M. Koper, D. G. H. Hetterscheid, Chem. Soc. Rev. 43 (2014) 5183-5191.
K. Nagaoka, T. Eboshi, Y. Takeishi, et al., Sci. Adv. 3 (2017) e1602747.
S. L. Foster, S. I. P. Bakovic, R. D. Duda, et al., Nat. Catal. 1 (2018) 490-500. doi: 10.1038/s41929-018-0092-7
L. Malavasi, C. A. J. Fisher, M. S. Islam, Chem. Soc. Rev. 39 (2010) 4370. doi: 10.1039/b915141a
D. J. L. Brett, A. Atkinson, N. P. Brandon, S. J. Skinner, Soc. Rev. 37 (2008) 1568. doi: 10.1039/b612060c
G. Zhai, D. Xu, S. Zhang, et al., Adv. Funct. Mater. 30 (2020) 2005779.
M. Yuan, Q. Li, J. Zhang, et al., Adv. Funct. Mater. 30 (2020) 2004208.
W. Liao, L. Qi, Y. Wang, et al., Adv. Funct. Mater. 31 (2021) 2009151.
J. D. Ramirez, V. Kyriakou, I. Garagounis, et al., ACS Sustainable Chem. Eng. 5 (2017) 8844-8851.
G. Qing, R. Ghazfar, S. T. Jackowski, et al., Chem. Rev. 120 (2020) 5437-5516. doi: 10.1021/acs.chemrev.9b00659
D. Liu, J. Wang, S. Bian, et al., Adv. Funct. Mater. 30 (2020) 2002731.
H. Liu, X. Cao, L. Ding, H. Wang, Adv. Funct. Mater. 32 (2022) 2111161.
X. Shen, S. Liu, X. Xia, et al., Adv. Funct. Mater. 32 (2022) 2109422.
H. Zou, L. J. Arachchige, W. Rong, et al., Adv. Funct. Mater. 32 (2022) 2200333.
J. H. Montoya, C. Tsai, A. Vojvodic, J. K. Nørskov, ChemSusChem 8 (2015) 2180-2186. doi: 10.1002/cssc.201500322
R. Aayush, S. Brian, A. R. Jay, et al., ACS Catal. 7 (2017) 706-709.
W. Fu, Y. Cao, Q. Feng, et al., Nanoscale 11 (2019) 1379. doi: 10.1039/c8nr08724e
L. Fu, L. Yan, L. Lin, et al., Alloys Compd. 875 (2021) 159907.
Y. Ying, K. Fan, X. Luo, H. Huang, J. Mater. Chem. A 7 (2019) 11444. doi: 10.1039/c8ta11605a
X. Liu, Z. Wang, J. Zhao, J. Zhao, Y. Liu, Appl. Surf. Sci. 487 (2019) 833-839.
F. Li, Q. Tang, Nanoscale 11 (2019) 18769. doi: 10.1039/c9nr06469a
P. Ou, X. Zhou, F. Meng, et al., Nanoscale 11 (2019) 13600. doi: 10.1039/c9nr02586c
C. Ji, A. A. Adeleke, L. Yang, et al., Sci. Adv. 6 (2020) eaba9206.
Z. Sun, R. Huo, C. Choi, et al., Nano Energy 62 (2019) 869-875.
H. Hirakawa, M. Hashimoto, Y. Shiraishi, T. Hirai, J. Am. Chem. Soc. 139 (2017) 10929-10936. doi: 10.1021/jacs.7b06634
G. Zhang, Q. Jia, K. Zhang, et al., Nano Energy 59 (2019) 10-16.
H. Jin, L. Li, X. Liu, et al., Adv. Mater. 31 (2019) 1902709.
L. Zeng, S. Chen, J. van der Zalm, X. Li, A. Chen, Chem. Commun. 55 (2019) 7386. doi: 10.1039/c9cc02607j
T. Wu, X. Zhu, Z. Xing, et al., Angew. Chem. Int. Ed. 58 (2019) 18449-19453. doi: 10.1002/anie.201911153
L. M. Azofra, C. H. Sun, L. Cavallo, et al., Chem. Eur. J. 23 (2017) 8275-8279. doi: 10.1002/chem.201701113
L. Wang, M. Wu, X. Lang, S. Gao, W. Wang, ChemCatChem 12 (2020) 3937-3945. doi: 10.1002/cctc.202000185
L. Xia, J. Yang, H. Wang, et al., Chem. Commun. 55 (2019) 3371. doi: 10.1039/c9cc00602h
Z. Feng, Y. Tan, W. Chen, et al., Mol. Catal. 483 (2019) 110705.
Q. Liu, S. Wang, G. Chen, Q. Liu, X. Kong, Inorg. Chem. 58 (2019) 11843-11849. doi: 10.1021/acs.inorgchem.9b02280
C. Fang, W. An, Nano Res. 14 (2021) 4211-4219. doi: 10.1007/s12274-021-3373-4
T. Wu, H. Zhao, X. Zhu, et al., Adv. Mater. 32 (2020) 20200299.
C. Ren, Q. Jiang, W. Lin, et al., ACS Appl. Nano Mater. 3 (2020) 5149-5159. doi: 10.1021/acsanm.0c00512
Y. Sun, Z. Deng, X. Song, et al., Nano Micro. Lett. 12 (2020) 133.
E. Skulason, T. Bligaard, J. K. Norskov, et al., Phys. Chem. Chem. Phys. 14 (2012) 1235-1245.
Z. Feng, Y. Tang, W. Chen, et al., Phys. Chem. Chem. Phys. 22 (2020) 9216. doi: 10.1039/d0cp00722f
X. Liu, Y. Jiao, Y. Zheng, M. Jaroniec, S.Z. Qiao, J. Am. Chem. Soc. 141 (2019) 9664-9672. doi: 10.1021/jacs.9b03811
X. Guo, J. Gu, S. Lin, et al., J. Am. Chem. Soc. 142 (2020) 5709-5721. doi: 10.1021/jacs.9b13349
J. G. Howalt, T. Bligaard, J. Rossmeislb, T. Vegge, Phys. Chem. Chem. Phys. 15 (2013) 7785. doi: 10.1039/c3cp44641g
X. Guo, S. Lin, J. Gu, et al., Adv. Funct. Mater. 31 (2020) 202008056.
T. Deng, C. Cen, H. Shen, et al., J. Phys. Chem. Lett. 11 (2020) 6320-5329. doi: 10.1021/acs.jpclett.0c01450
X. Li, Q. Li, J. Cheng, et al., J. Am. Chem. Soc. 138 (2016) 8706-8709. doi: 10.1021/jacs.6b04778
F. Liu, L. Song, Y. Liu, et al., J. Am. Chem. Soc. 8 (2020) 3598-3605. doi: 10.1039/c9ta12345h
X. Wang, S. Ye, W. Hu, et al., J. Am. Chem. Soc. 142 (2020) 7737-3343. doi: 10.1021/jacs.0c01825
X. Cui, C. Tang, Q. Zhang, Adv. Energy Mater. 8 (2018) 1800369.
J. Hou, M. Yang, J. Zhang, Nanoscale 12 (2020) 6900-6920. doi: 10.1039/d0nr00412j
H. Xu, K. Ithisuphalap, Y. Li, et al., Nano Energy 69 (2020) 104469.
L. Chen, C. He, R. Wang, et al., Chin. Chem. Lett. 32 (2021) 53-56.
D. Ma, Z. Zeng, L. Liu, Y. Jia, J. Energy Chem. 54 (2021) 501-509.
W. Song, J. Wang, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 3137-3142.
R. Wang, C. He, W. Chen, C. Zhao, J. Huo, Chin. Chem. Lett. 32 (2021) 3821-3824.
J. Yang, W. Weng, W. Xiao, J. Energy Chem. 43 (2020) 195-207.
C. Ling, X. Niu, Q. Li, A. Du, J. Wang, J. Am. Chem. Soc. 140 (2018) 14161-14168. doi: 10.1021/jacs.8b07472
L. Shi, Y. Yin, S. Wang, H. Sun, ACS Catal. 10 (2020) 6870-6899. doi: 10.1021/acscatal.0c01081
M. A. Légaré, G. Bélanger-Chabot, R. D. Dewhurst, et al., Science 359 (2018) 896-900. doi: 10.1126/science.aaq1684
C. Lv, L. Zhong, Y. Yao, et al., Chem 6 (2020) 2690-2702. doi: 10.1016/j.chempr.2020.07.006
Y. Sun, Y. Wang, H. Li, et al., J. Energy Chem. 62 (2021) 51-70.
S. Liu, M. Wang, H. Ji, et al., Natl Sci Rev 8 (2021) nwaa136. doi: 10.1093/nsr/nwaa136
J.C. Liu, X.L. Ma, Y. Li, et al., Nat. Commun. 9 (2018) 1610.
C. Cui, H. Zhang, Z. Luo, Nano Res. 13 (2020) 2280-2288. doi: 10.1007/s12274-020-2847-0
J. Zhao, Z. Chen, J. Am. Chem. Soc. 139 (2017) 12480-12487. doi: 10.1021/jacs.7b05213
X. Guo, S. Huang, Electrochim. Acta 284 (2018) 392-399.
H. Tao, C. Choi, L.X. Ding, et al., Chem 5 (2019) 204-214.
S. Tang, Q. Dang, T. Liu, et al., J. Am. Chem. Soc. 142 (2020) 19308-19315. doi: 10.1021/jacs.0c09527
C. Zhao, M. Xi, J. Huo, C. He, L. Fu, Chin. Chem. Lett. 34 (2023) 107213.
C. He, Y. Yu, C. Zhao, J. Huo, Chin. Chem. Lett. 34 (2023) 107897.
C. He, H. Yang, L. Fu, Chin. Chem. Lett. 34 (2023) 107581.
V. Stamenkovic, B. S. Mun, K. J. J. Mayrhofer, et al., Angew. Chem. Int. Ed. 45 (2006) 2897-2901. doi: 10.1002/anie.200504386
J. K. Nørskov, J. Rossmeisl, A. Logadottir, et al., J. Phys. Chem. B 108 (2004) 17886-17892. doi: 10.1021/jp047349j
A. Vasileff, C. Xu, Y. Jiao, Y. Zheng, S.Z. Qiao, Chem 4 (2018) 1809-1831.
W. Ju, A. Bagger, G. Hao, et al., Nat. Commun. 8 (2017) 944.
M. Wang, S. Liu, T. Qian, et al., Nat. Commun. 10 (2019) 341. doi: 10.1038/s41467-018-08120-x
F. Lu, S. Zhao, R. Guo, et al., Nano Energy 61 (2019) 420-427.
Y. Basdogan, A. M. Maldonado, J. A. Keith, et al., WIREs Comp. Mol. Sci. 10 (2020) e1446. doi: 10.1002/wcms.1446
J. A. Gauthier, S. Ringe, C. F. Dickens, et al., ACS Catal. 9 (2019) 920-931. doi: 10.1021/acscatal.8b02793
X. Lv, W. Wei, B. Huang, Y. Dai, T. Frauenheim, Nano Lett. 21 (2021) 1871-1878. doi: 10.1021/acs.nanolett.0c05080
H. Xu, D. Cheng, D. Cao, X.C. Zeng, Nat. Catal. 1 (2018) 339-348. doi: 10.1038/s41929-018-0063-z
Z. Xue, X. Zhang, J. Qin, R. Liu, Nano Energy 80 (2021) 105527. doi: 10.1016/j.phrs.2021.105527
W. Gao, Y. Chen, B. Li, et al., Nat. Commun. 11 (2020) 1196. doi: 10.1038/s41467-020-14969-8
X. Liu, L. Qi, E. Song, W. Gao, Catal. Lett. 153 (2022) 300-310.

Figure 1 The synthesis and application of ammonia in various areas. Reproduced with permission [5]. Copyright 2020, American Chemical Society.

Figure 2 The mechanisms of NRR including (a) dissociative pathway, (b) distal and alternating pathways, and (c) enzymatic pathway. Reproduced with permission [60]. Copyright 2018, Wiley.

Figure 3 (a) The hybridization of N2 molecular orbitals. (b) The donation-back donation mechanism between metal and N2. Reproduced with permission [72]. Copyright 2021, Elsevier.

Figure 4 (a) Schematic illustration of NRR on O-vacancy rich WO3 and (b) the corresponding top view. (c) Reaction pathway of the pristine WO3 and O-vacancy rich WO3. (d) The PDOS of Cu1+ and Cu2+ in Cu doped TiO2. (e) The PDOS of different Ti3+ defect states in Cu doped TiO2. (a-c) Reproduced with permission [35]. Copyright 2019, Elsevier. (d, e) Reproduced with permission [47]. Copyright 2020, Wiley.

Figure 5 (a) Diagram of the NN dipole model on Au(111) and Ag(111) surfaces. Comparison of (b) adsorption energies and (c) charge transfer between the dipole predicted and calculated values. The fitted scaling lines between the descriptor ψ and the adsorption energies of N-species on (d) close packed and (e) stepped surfaces of TM. (f) The comparison of adsorption energies between the ψ predicted and calculated values. (a-c) Reproduced with permission [59]. Copyright 2020, American Chemical Society. (d-f) Reproduced with permission [92]. Copyright 2020, Nature.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: