Scheme 1.
Defluorinative cyclization of enamides with fluoroalkyl halides through two vicinal C(sp3)-F bonds functionalization.

Organofluorines play a crucial role in medicine, agrochemicals, and materials science. Adding fluorine to molecules creates structures with specific beneficial properties or tunes properties through interactions with their environment. Many popular pharmaceuticals and agrochemicals contain fluorine because it enhances hydrogen bonding at protein's active sites. Replacing hydrogen in polyethylene with fluorine creates high-performance polytetrafluoroethylene (Teflon®), which has excellent thermal stability and mechanical properties. This has led to ongoing efforts to develop fluorine-containing polymers for advanced applications. While fluorine incorporation offers many benefits, synthesizing fluorine-containing compounds is challenging. The carbon-fluorine (C—F) bond is a valuable functional group for chemical modification, but its strong bond strength, hydrophobicity, and high thermal stability also make it chemically inert. This stability leads to environmental persistence, raising concerns about contamination and potential long-term health effects [1].
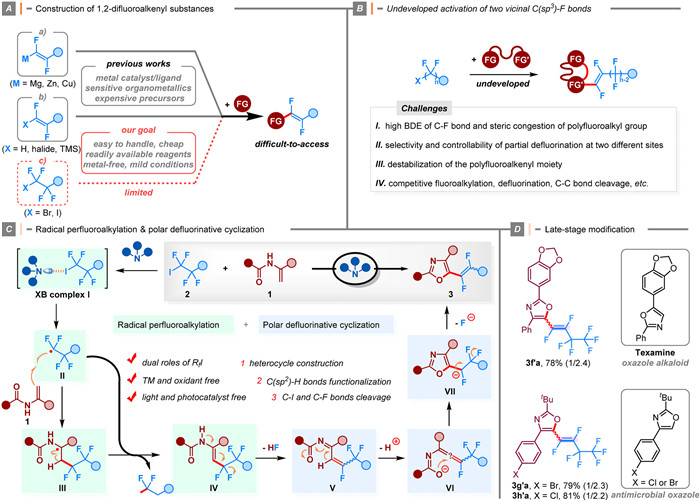
Access to unique 1, 2-difluoroalkenyl structures is limited due to the lack of straightforward and widely applicable synthetic methods from common feedstocks. Most research has focused on coupling reactions using pre-functionalized fluoroalkenyl precursors and/or highly reactive fluorinated organometallic reagents (e.g., organolithium, magnesium, zinc, aluminum, and copper reagents) (Scheme 1A). However, current methods for synthesizing organofluorides using PFAHs as substrates are mostly limited to functionalizing either one or three carbon-halogen bonds at the terminal carbon atom. Despite significant progress, methods for activating multiple C—F bonds on different carbon atoms of perfluoroalkyl groups remain scarce. Furthermore, the hypothetical reaction mode encounters other challenges, including: (a) The difficulty in activating the robust C(sp3)—F bonds in perfluoroalkyl groups, which are intrinsically less reactive and sterically hindered; (b) The existence of non-site-selective transformations and unwanted multi-activation of C—F and other C—C bonds; (c) Competing interference from the generated fluoroalkenyl moiety due to harsh reaction conditions and highly reactive reagents typically required in such reactions (Scheme 1B) [2-4].
Very recently, Chu and co-workers disclosed a defluorinative cyclization reaction of enamides for the construction of fluoroalkenyl oxazoles [5]. The selective and controllable two-fold cleavage of vicinal C(sp3)—F bonds in PFAH not only enables the introduction of a specific 1, 2-difluoroalkenyl moiety with ease but also results in the functionalization of two C(sp2)—H bonds of enamides without the need for metal catalyst, photocatalyst, oxidant, or light. The method could be applied to the late-stage modification of complex molecules, the synthesis of biologically relevant oxazole analogues, and scalable synthesis, all of which further highlight the real-world utility of this protocol. Mechanistic studies revealed that the reaction possibly proceeds through a radical perfluoroalkylation, consecutive C—F bond heterolytic cleavage, and cyclization process. In addition, the in situ formed perfluoroalkyl radical might also serve as an essential hydrogen abstractor.
A possible relay involving radical perfluoroalkylation and downstream polar defluorination process using terminal enamide 1 and PFAH 2 as substrates was proposed (Scheme 1C). When an amine is present, it initially interacts with PFAH to form an encounter complex Ⅰ via halogen bonding (XB). The existence of XB complex was investigated by the 19F NMR titration experiment and UV–vis spectroscopic measurement. Subsequently, an electrophilic perfluoroalkyl radical Ⅱ is formed from the collapse of XB adduct through intramolecular electron transfer (ET), and it can be readily intercepted by enamide 1 to deliver an α-amino radical Ⅲ. Then, the oxidation of the reactive radical intermediate Ⅲ by an excess of perfluoroalkyl radicals or other oxidative species, such as the radical cation from DBU, produced the perfluoroalkylated enamide Ⅳ, which undergoes rapid polar defluorination under basic conditions, leading to the fluoroallene species Ⅵ. Further intramolecular O-nucleophilic cyclization and ensuing β-F elimination at an adjacent carbon site forge the desired 1, 2-difluoroalkyl product 3. The approach also features broad substrate scope, good functional group tolerance, excellent scalability, and useful synthetic applications. Given the increasing importance of heterocycle skeleton containing fluorinated moiety, this method provides a modular and reliable platform for accessing specific fluorinated heterocycles of medicinal and biological interest. The mild process also permitted late-stage modification of more complex molecules, as demonstrated by transformations of tonalid, celestolide, l-menthol, metiapine, diacetonefructose, and (Z)-Nerol derivatives. The power of the present defluorinative cyclization is illustrated by introducing fluoroalkenyl moiety on biologically active-relevant compounds (Scheme 1D).
In summary, Chu and coworkers have successfully developed a defluorinative cyclization reaction of enamides for the construction of fluoroalkenyl oxazoles by using perfluoroalkyl halide (PFAH) as a nontraditional, readily available, and ideal 1, 2-difluoroalkenyl coupling partner. Given the increasing importance of heterocycle skeleton containing fluorinated moiety, this method provides a modular and reliable platform for accessing specific fluorinated heterocycles of medicinal and biological interest.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jun Jiang: Writing – review & editing. Hui Dai: Writing – original draft. Tao Tu: Writing – original draft.
S. Joudan, R.J. Lundgren, Science 377 (2022), 816–817. doi: 10.1126/science.add1813
B.Q. Cheng, D. Ge, X. Wang, X.Q. Chu, Chin. J. Org. Chem. 41 (2021), 1925–1938. doi: 10.6023/cjoc202009035
C. Douvris, O.V. Ozerov, Science 321 (2008) 1188–1190. doi: 10.1126/science.1159979
R. Doi, M. Yasuda, N. Kajita, K. Koh, S. Ogoshi, J. Am. Chem. Soc. (145) (2023), 11449–11456. doi: 10.1021/jacs.3c03471
Y.L. Chen, W. Han, Y.Y. Ren, et al., Adv. Sci. (2025), https://doi.org/10.1002/advs.202404738. doi: 10.1002/advs.202404738

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
 下载:
下载: