Received Date:
22 January 2025 Accepted Date:
05 March 2025 Revised Date:
26 February 2025 Available Online:
15 June 2026
Abstract:
Searching for new oxide-ion conductors is of great significance in energy-related technologies. Here we identified a novel barium tellurate, Ba10.55Te4.45O23.90, by chemical screening for superstructural oxide-ion conductors. Its crystal structure, solved from polycrystalline specimen by the combination of three-dimension electron diffraction, X-ray diffraction, and neutron diffraction, adopts a quadruple (4 × 4 × 4) cubic superstructure (Fm-3m, a = 17.30612(2) Å) and can be regarded as a derivative of ABO3 perovskite like (Ba1.75□0.25)ABaBWB'O5.75□0.25. The ordered A-site metal vacancies and disordered oxygen vacancies are responsible for the enlarged superstructure. The titled compound is indeed an oxide-ion conductor but shows rather low ionic conductivity, owing to the high inter-polyhedral energy barrier of ionic migration. The discoveries unveil a new structural type for oxide-ion conductor exploration, and will evoke performance improvement by chemical modification such as aliovalent substitution.
Oxide-ion conductors are of significant importance for energy related technologies [1,2], such as solid oxide fuel cells (SOFCs) [3–7], oxygen separation pumps and sensors [8], and solid oxide electrolysis cells (SOECs) [6]. The charge carrier in these materials is mostly oxygen vacancies, as discovered in perovskites [9,10], borates [11], fluorites, and scheelites [12], or oxygen interstitials, such as melilites [13,14], apatites [15], and La2Mo2O9-based systems [16,17], or a mixture of oxygen vacancies and interstitials as in exotic perovskites [18]. The electrical conductivity of these materials is mainly governed by the defected oxygen sublattice to enable proper ionic diffusion at high temperature. Nowadays, a long-standing drawback of oxide-ion conductors is the relatively poor ionic transport in commercial materials at intermediate temperature [3], thus, it is essential to understand the mechanism for oxide ion charge carrier creation and mobility at atomic scale. The commonly applied strategy is to generate oxygen deficiency by aliovalent substitution in open-framework structures. For examples, partial replacement of Zr4+/Ce4+ by Y3+/Gd3+ in ZrO2/CeO2 creates oxygen vacancies in the well-known YSZ (Zr1-xYxO2-δ) and GDC (Ce1-xGdxO2-δ) [1,8]; paired co-doping of Sr2+-Mg2+ at the La3+-Ga3+ sites induces oxygen vacancy in LaGaO3 to form LSGM (La0.9Sr0.1Ga0.8Mg0.2O3-δ) [19]. In contrast to the oxygen-deficient fluorites and perovskites by lower valence doping, higher-valence doping can generate oxygen interstitials as exemplified in the apatite and melilite series, which hold open-framework structures and adaptable coordination environment from tetrahedron to trigonal bipyramid [13–15]. The La-enriched non-stoichiometric La1+xSr1-xGa3O7+δ melilite demonstrates excellent oxygen-interstitial conductivity of 0.02–0.1 S/cm over 600–900 ℃. The incorporation of oxygen interstitial modifies local tetragonal GaO4 to trigonal bipyramid GaO5 [13]. In addition to the doping policy, other approaches, such as cationic geometrical size-mismatch, can induce metal-deficiency and accordingly oxygen defect under charge balance.
Previous studies on the cryolite-type Ba11W4O23 (BWO) unveiled the first quadruple perovskite-related 4 × 4 × 4 superstructure phase, which accommodates ordered A-site metal vacancies, disordered oxygen, and anionic vacancies, and can be written as [(Ba1.75□0.25)ABaBWB'O5.75□0.25]4 derived from ABO3 perovskite [20]. The cubic (Fd-3m) BWO contains statistically distributed oxygen and anionic vacancies, with three disordered oxygen sites in occupancy varying between 0.204 and 0.408, rendering average WO18/3 coordination, indicating promising oxide-ion conductivity. However, BWO is metastable and can only be retained by temperature-quenching to liquid nitrogen from 1100 ℃, which hinders further exploration of its electrical conductivity. Thermal stability analyses on the quenched BWO indicated that, it decomposed above 400 ℃ and recovered back at 1100 ℃ upon heating; while at the cooling process, although the superstructural phase can be retained to ambient temperature, impurity peaks appeared below 900 ℃. Fortunately, BWO can be stabilized via partial replacement of W by Ta in Ba11W4-xTaxO23-x/2 (x ≤ 2), which shows pure oxide-ion conductivity over a wide oxygen partial pressure range and proton conductivity under wet-air below 800 ℃ [21]. The best performance is observed for the x = 0.5 case, showing comparable conductivity to the commercial YSZ, GDC, and LSGM around 400 ℃. The above discoveries suggest that, unlike conventional perovskite, the incorporation of relatively large cations into the perovskite B-site can drive considerable metal vacancies and oxygen disorder, encouraging further exploration for promising oxide-ion conductors. Moreover, accommodation of Sr in the B-site of Sr6–2xNb2+2xO11+3x (Sr2(Sr, Nb)BNbB'O6-δ) leaves oxygen vacancies in the original anionic sites and also drives some oxygen into interstitial sites, as reported in the first discovery of oxygen interstitials in double perovskite by neutron diffraction technique [18]. Thus, it is exciting and evokes researchers to synthesize new oxide-ion conductors via similar structural engineering, namely size-mismatched modification at the perovskite-related structures.
Chemically, Sr11Re4O24-type compounds [22–26], such as Ba11Os4O24 and BaLa10Ir4O24, seems similar to BWO but adopt tetragonal superstructure (I41/a, $a=2 \sqrt{2} a_{\mathrm{p}}$, c = 4ap, where ap symbols the cell dimension of simple perovskite). Their structural formula can be written as (A7/8□1/8)2ABBB'O6 with 1/8 A-site vacancy, in which the chemical valence variation of the B-site cations like Os+6/+7 or Ir+4/+5 should be largely responsible for the cubic-to-tetragonal distortion and absence of oxygen disordering/vacancies under charge balance. From the chemical and geometric point of view, Te6+ and W6+ possess comparable oxidation state and ionic size. Presumably, barium tellurate, saying Ba11Te(Ⅵ)4O23 if exists, could be a promising oxide-ion conductor as BWO. However, our attempts to make BWO-type Ba11Te4O23 was unsuccessful, and yielded a mixture of an uncertain superstructural phase and several barium tellurates [27]. Modification of the Ba-Te ratio finally resulted in a novel line phase in starting ratio of Ba/Te = 9.5/4 other than the expected Ba/Te = 11/4. In this work, we described the discovery of this nominal Ba9.50Te4O21.50 oxide-ion conductor, and solved its crystal structure via the combination of different diffraction techniques on polycrystalline specimen. The local crystal structure, electrical conductivity, and theoretical simulation were also studied to understand the ionic migration mechanism.
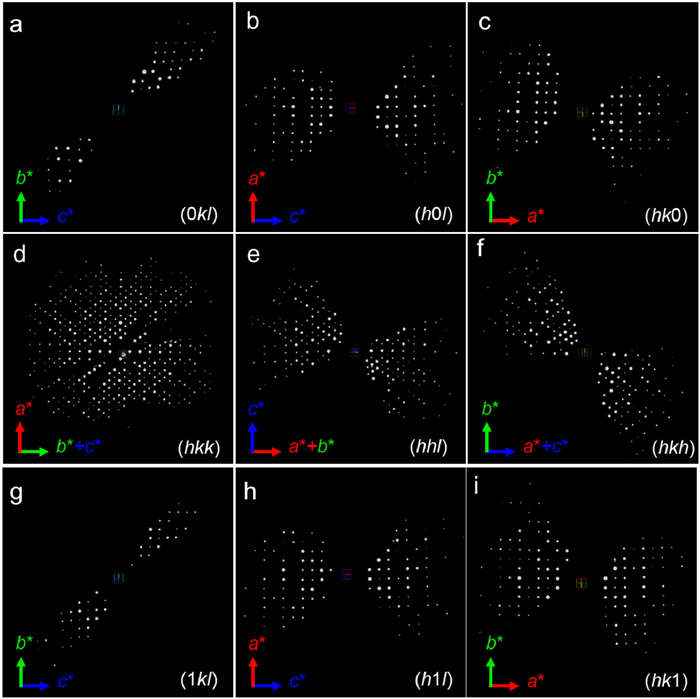
Our attempts to prepare Ba11Te4O23 were unsuccessful, and finally a single phase was obtained by varying the Ba/Te ratio from 2.75 to ~2.38. SEM-EDS and ICP-OES results of the as-made product (Fig. S1 in Supporting information) revealed an average Ba/Te ratio of 2.375 and 2.33(2), respectively, in line with the starting ingredient proportions. Considering the charge equilibrium, the nominal composition can be written as Ba9.50Te4O21.50. Initial structural phase analysis of the superfine PXD data does not match any known compounds in Ba-Te-O system [27–40], and chemically unstrained screening does not find any satisfying isostructural analogs. Thus, an ab initio indexing of the PXD data was carried out in TOPAS software [41], which yielded a cubic cell (a ~ 17.3 Å, Table S1 in Supporting information) with possible space group of F23 (No. 196), Fm-3 (No. 202), F432 (No. 209), F-43m (No. 216), or Fm-3m (No. 225) under the same extinction conditions. Therefore, 3D reciprocal lattice was reconstructed using the cRED technique for a clear image. As shown in Fig. 1 and Fig. S2 (Supporting information), the 3D-ED data illustrate the face-centered cubic unit cell parameters of Ba9.50Te4O21.50 of a = b = c ~ 17.3 Å and α = β = γ = 90°. The cubic lattice was further refined as a = 17.30612(2) Å through the PXD data. Based on the hkl file reduced from 3D-ED data, the crystal structure of this nominal Ba9.50Te4O21.50 oxide was solved in a quadruple perovskite structure with Fm-3m space group. Extraction of the structure factors from PXD data was carried out using the model-biased Le Bail method [42] based on the 3D-ED results, and direct procedures were then performed to get improved model for further model-biased extraction. Rietveld refinements were performed until no improvement was achieved after sequential iterations. The PXD data almost fit well, and positions of metal and three oxygen atoms were identified. However, Te2 atom was isolated, indicating the existence of low occupancy oxygen around, which cannot be captured by PXD diffraction. In succession, combined Rietveld refinements of the PXD and NPD data were evolved and the fitness of NPD data was totally unacceptable. As expected, disordered O4 was revealed around Te2 from Fourier and difference Fourier synthesis of NPD profiles (Fig. S3 in Supporting information). During refinements, the metal positions were recognized based on the metal-oxygen bond distances; occupancies of Ba1 and Te4 were restrained to maintain the metal ratio according to SEM-EDS results, and soft chemical restraint in light of electro-neutrality was applied to refine the O4 occupancy. The refinement parameters were histogram scale factors, background, unit cell parameters, peak profile coefficients, extinction coefficient (PXD only), atomic coordinates, atomic displacement parameters (ADPs), and occupancies for Te4, Ba1, and O4. The final refined profile fits are shown in Figs. 2a and b. The final refinement results and crystallographic data were presented in Tables S1-S4 (Supporting information).
Figure 1
Figure 1.
3D ED data of Ba10.55Te4.45O23.90. The diffraction patterns of (a) (0kl), (b) (h0l), (c) (hk0), (d) (hkk), (e) (hhl), (f) (hkh), (g) (1kl), (h) (h1l) and (i) (hk1) slices extracted from the reconstructed 3D reciprocal lattice. The reflection conditions are: hkl: h + k, h + l, k + l = 2n; 0kl: k, l = 2n; h0l: h, l = 2n; hk0: h, k = 2n; hkk: h + k = 2n; hhl: h + l = 2n; hkh: h + k = 2n.
Figure 2.
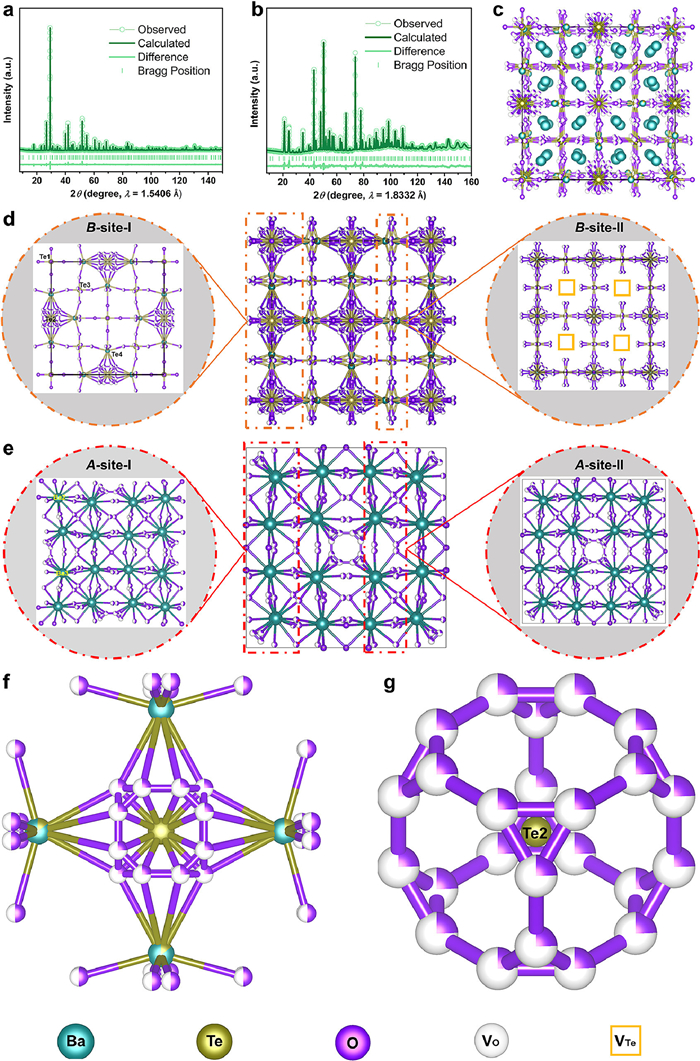
Rietveld refinements and crystal structure of Ba10.55Te4.45O23.90. The plots of (a) PXD and (b) NPD from joint refinements. (c) Perspective view of quadruple perovskite superstructure. (d) Crystal structure of the B-site sublattice in two different slabs, the left slab (B-site-Ⅰ) is comprised of Te1O6 and the average Te2O6, Te3O6, and (Te4/Ba1)O6, the right slab (B-site-Ⅱ) contains the average Te3O6, (Te4/Ba1)O6. (e) Crystal structure of the A-site sublattice in two different slabs comprised of Ba2-O and Ba3-O polyhedra. Local defect structure (f) face-sharing Te2(O4)24×0.242 and (Te4/Ba1)(O2)8×0.542(O4)8×0.242. (g) (O4)24 cage around Te2 viewed along the C3 direction in Ba10.55Te4.45O23.90. The colorful atomic spheres and vacancy (V) squares are labeled on the bottom.
The Ba/Te ratio from combined PXD and NPD refinements converges to 2.371(1) and gives Ba10.55Te4.45O23.90, which is in great agreement with the EDS result of 2.375. Original refinements with O2 and O3 at the 96j (y, z, 0) and 48g (1/4, 1/4, x) site, respectively, yielded unreasonably large atomic displacement parameters (ADPs) of O2 and O3, and too small ADPs for Te1. A close examination of the differential Fourier map based on the NPD data unveils somewhat density diffusion at O2 and O3 sites, indicating local site displacement. Therefore, each of them is set at a more general position with reduced occupancy. Decent results were obtained by placing O2 and O3 at doubled 192l (x, y, z) and 96k (x, x, z) site, respectively, with half occupancy as shown in Table S2. The three-fold symmetry axis leads to the splitting of the O4 atom at 96k site into three. Soft restraint of the occupancy refinement for O4 at (z, x, x) yielded 0.247(1), which is close to the expected value of 0.242 under charge balance giving Ba2+ and Te6+. The refined crystal structure of the titled compound comprises 3 crystallographically independent Te sites, 2 Ba sites, 1 mixed Ba/Te site, and 4 oxygen sites (Table S2 and Fig. 2c). For a better visualization of the atomic arrangement, separated sections for the perovskite-related A- and B-sites in the structure were extracted (Figs. 2d and e). Unlike the A-site defected crystal structure of BWO [20,21], all A sites are filled by Ba atoms (Fig. 2e), while only 7/8 of B sites accommodate Te/Ba or Te atoms in Ba10.55Te4.45O23.90. Approximately 1/8 B-site vacancies exist at the 8c (1/4, 1/4, 1/4) site (Fig. 2d, right). Consequently, the structure of Ba10.55Te4.45O23.90 can be regarded as a 4 × 4 × 4 supercell perovskite (ABO3)4 due to the positional order of Te, Ba/Te mixing, and cationic and anionic vacancies. According to the aforementioned analyses, the structural formula of Ba10.55Te4.45O23.90 with B-site and anionic vacancies can be written in quadruple-perovskite style Ba4(Ba1.28Te2.22□0.50)O11.94□0.06, namely (ABO3)4 in [(Ba)A (Ba0.320Te0.555□0.125)BO2.985□0.015]4, where □ symbols the vacancy. Notably, the Te/Ba mixing disrupts its local coordination environment from the octahedron. The Te/Ba atom at 24e site is surrounded by eight partially occupied O4, eight O2, and one O1 atom, forming a nearly statistical BO6 coordination (Fig. 2f). Likewise, the Te2 at 4b site, caged by 24 partially occupied O4 atoms (Fig. 2g), suggest its average coordination number ~6. Te3 at 24d position is connected to eight O2 and four O3 atoms with half occupancy, also yielding nominal Te3O6 octahedral environment. The above distinctive coordination polyhedron indicates the orientational disorder of the TeO6 octahedra (Table S3). The oxygen defect in the nominal Ba10.55Te4.45O23.90 suggests potential oxide ion conductivity as reported in other analogs.
In situ high-temperature PXD measurements were conducted to evaluate the thermal stability of the titled compound before any reliable temperature-dependent electrical conductivity evaluation. As shown in Fig. S4 (Supporting information), the phase evolution of Ba10.55Te4.45O23.90 behaved differently upon heating and cooling. In Fig. S4a, upon heating, the supercell cubic phase was stable up to 700 ℃ and transformed into Ba3TeO6 and small amount of other barium tellurates. Then, the pure cubic superstructural phase was recovered above 950 ℃. The precise recovering temperature must be around 950 ℃ and was not determined in this investigation. Stepwise cooling (Fig. S4b) resulted in almost no change, and the cubic phase was retained to room temperature. Here the overall trend of thermal stability of Ba10.55Te4.45O23.90 upon heating and cooling is similar to that of BWO [20], the difference is that, BWO is partially decomposed upon programmable cooling and can only be retained by temperature quenching, while Ba10.55Te4.45O23.90 is stable during the experimentally programmable cooling. More work is being undertaken to characterize the intermediate and slow cooled phases that will not be discussed in this paper. Apparently, the thermodynamic stability trend of Ba10.55Te4.45O23.90 is not equivalent at experimental condition, and the temperature dependent electrical conductivity measurements should be carried out upon cooling.
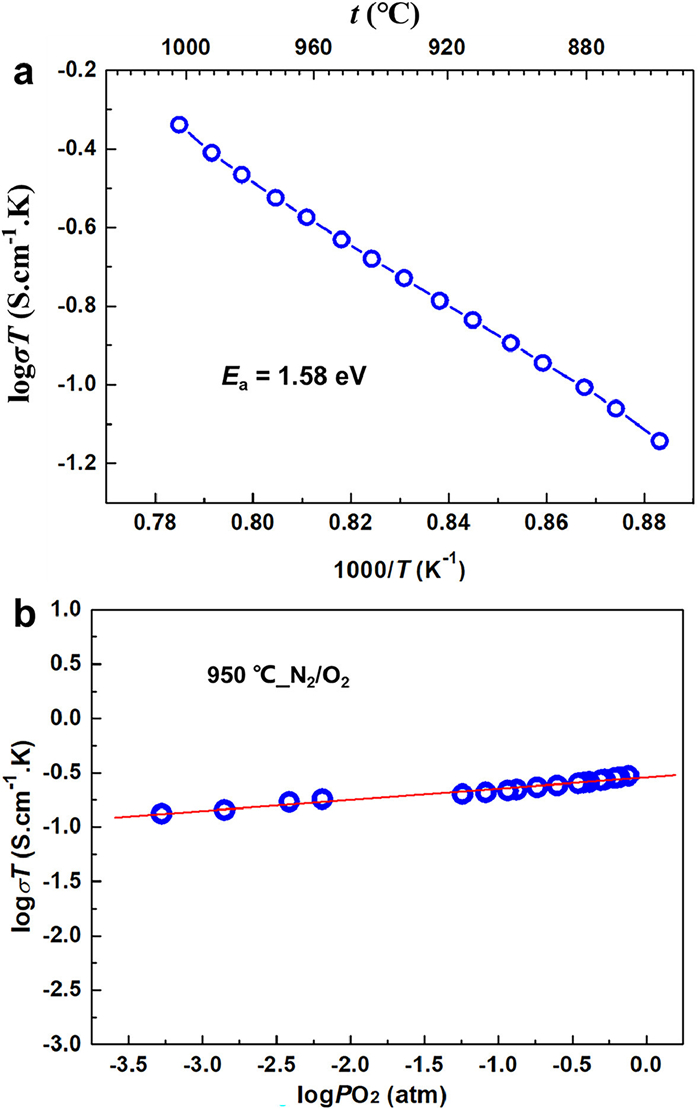
Thereby, the total electrical conductivity of Ba10.55Te4.45O23.90 was firstly measured on a sintered bar-pellet with relative density ~83% by four probe d.c. method in air, upon cooling in light of in situ high temperature PXD results. For the temperature range studied, the conductivity increases from 1.47 × 10–5 S/cm (850 ℃) to 6.60 × 10–4 S/cm (1000 ℃) with calculated activation energy of 1.58 eV (log σT based, Fig. 3a), which is somewhat too high compared with the Ta-doped BWO (0.78 eV) [21] and other conventional conductors (0.7–1.0 eV), such as YSZ (Y0.148Zr0.852O1.926) [8] and LSGM (La0.9Sr0.1Ga0.8Mg0.2O3-δ) [1,6]. Fig. 3b shows the plot of log(σT/S cm-1 K) versus log(PO2/atm) at 950 ℃ under N2/O2 gas pair for Ba10.55Te4.45O23.90. The conductivity appears to increase with increasing PO2 in the range of −4 ≤ log(PO2) ≤ 0, the rate(|∂ log σ /∂ log PO2|) is about 1/10 (0 < 1/10 < ¼). We may thus, conclude that the present conductivity is a mixture of dominated ionic conduction with some p-type ingredient. In contrast, the conductivities under further reducing atmospheres could not be measured reproducibly because the conductivity values fluctuated (Fig. S4). Although the crystal structure of Ba10.55Te4.45O23.90 contains considerable oxygen defect as in the BWO analogs, its electrical conductivity is rather poor. Thus, theoretical analysis was conducted to further understand the ionic migration path and energy barrier.
Figure 3
Figure 3.
Electrical conductivity of Ba10.55Te4.45O23.90. (a) Arrhenius plot showing the temperature dependence of electrical conductivity under air; (b) Oxygen partial pressure (PO2) dependent electrical conductivity at 950 ℃ under O2—N2 pair.
The BVSE method has been successfully applied to illustrate the ion migration process [43,44]. Based on the bond-valence sum (BVS) theory plus a morse-type potential, we calculated the entire energy landscape across the periodic structure. The most possible migration path can be determined by analyzing the connecting local minima and the relative energy barriers for each pathway segment. This method is particularly helpful for oxide-ion conductors with complex occupational and displacive disorder, which is usually computational expensive and difficult to cope with in ab initio calculations.
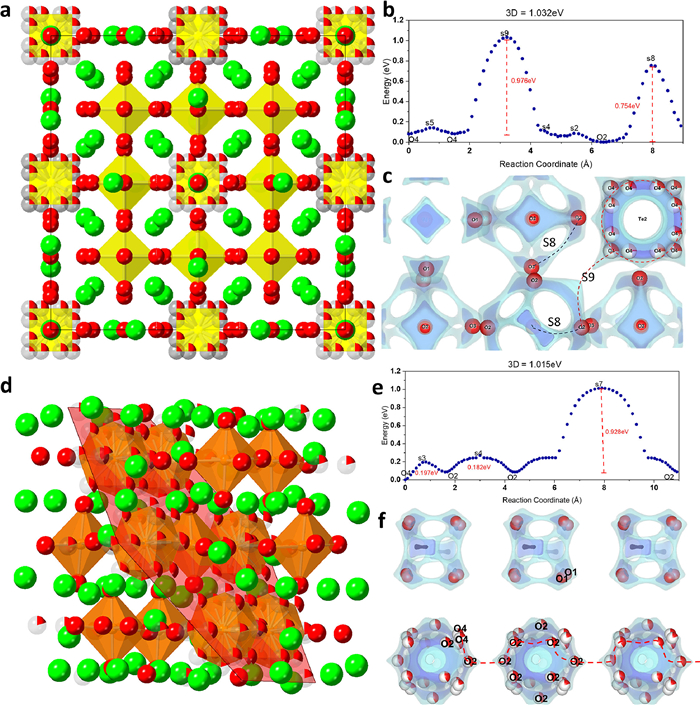
The average crystal structure of Ba10.55Te4.45O23.90 in Fig. 4a shows the occupational disorder is concentrated in the O4 sites, forming unusual defective Te-O cubes (Fig. 2g). This disorder creates a low oxygen migration barrier inside the Te-O cube as shown by the continuous stocky isosurface connection between O4 atoms in Fig. 4c, and a migration energy barrier as low as 0.058 eV in Fig. 4b. However, these disordered cubes are surrounded by oxygen fully localized and occupied Te-O octahedra (Fig. 4a). If an oxygen atom moves from the cube to the Te-O octahedral network, it has to overcome an energy barrier as high as 0.976 eV (to pass the saddle point s9 in Fig. 4c). The migration barrier for oxygen atoms to migrate within the Te-O octahedral network is 0.754 eV from BVSE calculation. The BVSE calculations suggest, although we introduced considerable oxygen vacancies through compositional design to a stable cubic superstructure, the uneven distribution of the oxygen occupational disorder within the material results in a moderate oxygen conductivity. This observation is further consolidated by another example BWO, with a similar composition (substituting W with Te) and superstructure [20,21].
Figure 4
Figure 4.
BVSE theoretical analysis. (a) The average crystal structure of Ba10.55Te4.45O23.90; Ba, green spheres; O, red spheres; anionic vacancy, white spheres; TeO6 polyhedra, yellow. (b) Calculated BVSE models of migration energy barriers for Ba10.55Te4.45O23.90. (c) Calculated BVSE isosurface map for Ba10.55Te4.45O23.90, here the dashed lines highlight the calculated migration path. (d) The average crystal structure of Ba11W4O23; Ba, green spheres; O, red spheres; anionic vacancy, white spheres; WO6 polyhedra, orange-red. (e) Calculated BVSE models of migration energy barriers for Ba11W4O23. (f) Calculated BVSE isosurface map for Ba11W4O23. The dashed lines highlight the calculated migration path.
As shown in Fig. 4d, the occupational disorder in BWO is also concentrated on an irregular W-O polyhedral. What's different from Ba10.55Te4.45O24 is, the oxygen fully occupied W-O octahedra are surrounded by 6 occupational disordered W-O irregular polyhedral on the (111) plane. Although the oxygen migration energy barrier across polyhedra to polyhedra (0.928 eV, Fig. 4e) in BWO is similar with that for Ba10.55Te4.45O23.90 (0.976 eV, Fig. 4b), the connected disordered polyhedra with occupational disorder is more likely for oxygen atoms to migrate along certain path. This configuration endows oxygen atoms to migrate along the continuous disordered W-O polyhedra as highlighted by the red dotted line in Fig. 4f. Hence, experimentally, the BWO analog has a higher oxide ion conduction than Ba10.55Te4.45O23.90. Obviously, the oxygen-ion conduction not only depends on the concentration of the vacancies, i.e., occupational disorder, but also relies on a continuous distribution of occupational disorder to lower the interpolyheral energy barrier along the ionic migration pathway.
The identified Ba10.55Te4.45O23.90 adopts a novel perovskite derived quadruple superstructure (ABO3)4 with ordered B-site vacancies and disordered oxygen vacancies in [(Ba)A (Ba0.320Te0.555□0.125)BO2.985□0.015]4, as solved via the combination of electron, X-ray, and neutron diffraction techniques from the polycrystalline sample. The existence of oxygen vacancies is further evidenced by the electrical conductivity measurements, which indicate nearly pure oxide ion conductivity above 800 ℃. Given the considerable oxygen vacancies, the oxide ion conductivity is rather poor compared with the parent superstructural materials and conventional oxide electrolytes. Theoretical simulation reveals that, although the energy barrier of intra-polyhedral oxide-ion migration is very small (0.058 eV), the value of inter-polyhedral oxide-ion migration is as high as 0.976 eV, which is responsible for the overall low electrical conductivity in Ba10.55Te4.45O23.90. Nevertheless, the discovery of Ba10.55Te4.45O23.90 proposes a new superstructure type to guide the design of new materials, and the physical properties like electrical conductivity, are expected to be significantly improved by chemical modification, such as chemical doping at the B-site by lower valence cations to increase oxygen vacancies at the neighbor polyhedron to reduce the energy barrier for oxide-on diffusion.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was financially supported by the Hainan Provincial Natural Science Foundation of China (No. 524RC473), the National Science Foundation of China (NSFC, Nos. 22090041, 22125102, U21A20285 and 12350710177), the Program for Guangdong Introducing Innovative and Entrepreneurial Teams (No. 2017ZT07C069), and the Guangdong Basic and Applied Basic Research Foundation (No. 2022B1515120014). The authors would like to thank Dr. Seung-Tae Hong at Daegu Gyeongbuk Insitute of Science and Technology for helpful discussion.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.111046.
N.A. Kabanova, Y.A. Morkhova, A.V. Antonyuk, et al., Solid State Ion. 391 (2023) 116142. doi: 10.1016/j.ssi.2022.116142
Figure 1
3D ED data of Ba10.55Te4.45O23.90. The diffraction patterns of (a) (0kl), (b) (h0l), (c) (hk0), (d) (hkk), (e) (hhl), (f) (hkh), (g) (1kl), (h) (h1l) and (i) (hk1) slices extracted from the reconstructed 3D reciprocal lattice. The reflection conditions are: hkl: h + k, h + l, k + l = 2n; 0kl: k, l = 2n; h0l: h, l = 2n; hk0: h, k = 2n; hkk: h + k = 2n; hhl: h + l = 2n; hkh: h + k = 2n.
Figure 2
Rietveld refinements and crystal structure of Ba10.55Te4.45O23.90. The plots of (a) PXD and (b) NPD from joint refinements. (c) Perspective view of quadruple perovskite superstructure. (d) Crystal structure of the B-site sublattice in two different slabs, the left slab (B-site-Ⅰ) is comprised of Te1O6 and the average Te2O6, Te3O6, and (Te4/Ba1)O6, the right slab (B-site-Ⅱ) contains the average Te3O6, (Te4/Ba1)O6. (e) Crystal structure of the A-site sublattice in two different slabs comprised of Ba2-O and Ba3-O polyhedra. Local defect structure (f) face-sharing Te2(O4)24×0.242 and (Te4/Ba1)(O2)8×0.542(O4)8×0.242. (g) (O4)24 cage around Te2 viewed along the C3 direction in Ba10.55Te4.45O23.90. The colorful atomic spheres and vacancy (V) squares are labeled on the bottom.
Figure 3
Electrical conductivity of Ba10.55Te4.45O23.90. (a) Arrhenius plot showing the temperature dependence of electrical conductivity under air; (b) Oxygen partial pressure (PO2) dependent electrical conductivity at 950 ℃ under O2—N2 pair.
Figure 4
BVSE theoretical analysis. (a) The average crystal structure of Ba10.55Te4.45O23.90; Ba, green spheres; O, red spheres; anionic vacancy, white spheres; TeO6 polyhedra, yellow. (b) Calculated BVSE models of migration energy barriers for Ba10.55Te4.45O23.90. (c) Calculated BVSE isosurface map for Ba10.55Te4.45O23.90, here the dashed lines highlight the calculated migration path. (d) The average crystal structure of Ba11W4O23; Ba, green spheres; O, red spheres; anionic vacancy, white spheres; WO6 polyhedra, orange-red. (e) Calculated BVSE models of migration energy barriers for Ba11W4O23. (f) Calculated BVSE isosurface map for Ba11W4O23. The dashed lines highlight the calculated migration path.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
