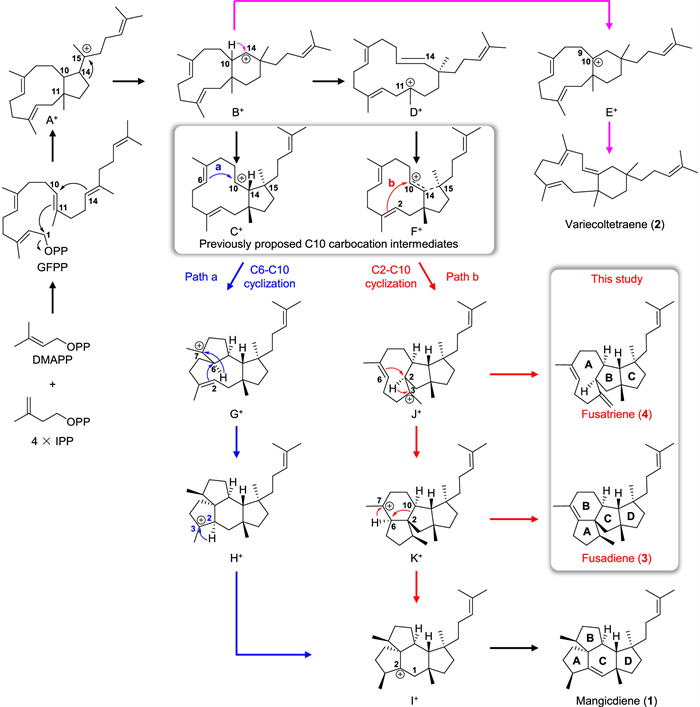
Figure 1.
Previously proposed and herein experimentally supported biosynthetic mechanisms of mangicdiene (1).
Identification of two novel sesterterpene skeletons offers the first experimental evidence for the cyclization mechanism of mangicdiene synthase
Pan Luo , Jian-Ming Lv , Hong-Ting Zhen , Ying-Qi Zhao , Jing-Yuan Liu , Jin-Yu Hong , Shao-Yang Li , Gao-Qian Wang , Guo-Dong Chen , Shui-Xing Zhang , Dan Hu , Hao Gao

Terpenoids are a remarkable class of natural products derived from linear polyprenyl diphosphate precursors. Impressively, they exhibit diverse structural complexity and a wide range of biological functions [1–3]. These fascinating molecules are formed through terpene synthases catalyzed cyclization cascade reactions, followed by tailoring enzymes mediated post-cyclization modifications [4–11], where terpene synthases play a pivotal role in constructing complicated polycyclic terpene skeletons. Therefore, there has been considerable interests in study on the terpene cyclization mechanism [12–14], which can set the stage for generation of novel terpene skeletons through protein engineering.
Mangicol-type sesterterpenoids are a small group of terpenoids, which are characterized with the 5–5–6–5 tetracyclic skeleton and exclusively found in fungi [15–17]. Notably, the tetracyclic skeleton of mangicols closely resembles that of the diterpene allokutznerene, except for the position of the double bond and an additional isoprene unit [18]. Thus far, 13 mangicol-type sesterterpenoids have been reported (Fig. S1 in Supporting information) [15–17], some of which possess anti-cancer and anti-inflammatory activities [15,17]. Among them, mangicol J showed comparable inhibitory activity to the pharmaceutical agent indomethacin in terms of treating phorbol myristate acetate-induced oedema in the mouse ear edema assay [17]. Therefore, biosynthesis of mangicols draws the extensive attention. Previously, the genetic basis for formation of the mangicol-type skeleton was unveiled. The chimeric sesterterpene synthase FgMS, which is composed of the prenyltransferase domain and terpene cyclase domain as other known fungal sesterterpene synthases [19], firstly elongates short prenyl chains into geranylfarnesyl diphosphate (GFPP), and then catalyzes cyclization of GFPP to mangicdiene [16]. In addition, several cytochrome P450 enzymes have also been identified to be responsible for biosynthesis of mangicol-type sesterterpenoids [17].
Regarding the formation of mangicdiene, it is well accepted that the cyclization begins with the departure of the pyrophosphate group of GFPP, followed by a concerted C1-C11/C10-C14 bicyclization to generate carbocation A+ (Fig. 1), which has been proposed to undergo a sequential 1,2-alkyl rearrangement to yield the C10 carbocation C+ via B+ [16]. However, the density functional theory (DFT) calculation revealed that A+ is likely to undergo two asynchronous events, including the 1,2-alkyl shift and C—C bond cleavage, to give D+ (Fig. 1), as the intermediate B+ is not a minimum on the potential energy surface [20]. This is in line with isolation of variecoltetraene, which is likely derived from E+ via B+. Subsequently, D+ is converted to the nonclassical C10 carbocation intermediate F+, wherein the single bond between C14 and C15, as well as the double bond between C10 and C14, is partially broken. After C10 carbocation formation, both C6-C10 cyclization (path a) and C2-C10 cyclization (path b) are able to lead to formation of mangicdiene (Fig. 1). The path a involves C6-C10 cyclization to G+, 1,2-hydride shift followed by C2-C6 cyclization to H+, 1,2-hydride shift to I+, and the final deprotonation of I+ (Fig. 1) [16]. Previously, the diterpene allokutznerene was also hypothesized to be formed in a similar manner [18]. In contrast, the path b seems a little prolix yet energetically reasonable, which requires C2-C10 cyclization to J+, 1,2-hydride shift followed by C2-C6 cyclization to K+, 1,2-hydride and alkyl shift to I+, and the final deprotonation of I+ (Fig. 1) [20]. Nevertheless, due to the lack of experimental evidence, it remains inconclusive whether mangicdiene is formed via the concise path a or the energetically favorable path b.
Due to being able to unequivocally determine the origin of carbons and hydrogens in the terpene skeleton, isotopic labelling experiments have been widely employed to unveil the terpene cyclization mechanism [21–28]. Alternatively, as carbocation intermediates are likely to be quenched via deprotonation or hydration to yield shunt products at any stage of the cascade cyclization reaction, identification of shunt products generated by the terpene cyclase or its variants can be used to confirm the presence of key carbocation intermediates, and then facilitate the elucidation of the cyclization mechanism [25,26,29–32]. For the case of biosynthesis of mangicdiene, as all the carbons and hydrogens in mangicdiene from the path a or path b share the same origin, isotopic labelling experiments cannot discern the specific pathway through which mangicdiene is formed. Therefore, the viable approach is to endeavor in the identification of shunt products related to mangicdiene. Previously, when FgMS was expressed in Escherichia coli (E. coli), the bicyclic shunt product variecoltetraene was concomitantly identified (Fig. 1) [16]. In addition, several trace shunt products were also detected, but none of them were isolated as a result of their rather low yields [16]. Considering the potential variability of the terpene cyclase product profile in different heterologous expression hosts, such as the notable difference observed when the amorpha-4,11-diene synthase (ADS) was expressed in Aspergillus nidulans and E. coli [33], the product profile of the mangicdiene synthase might also be altered in different hosts, thus resulting in isolation and identification of new shunt products.
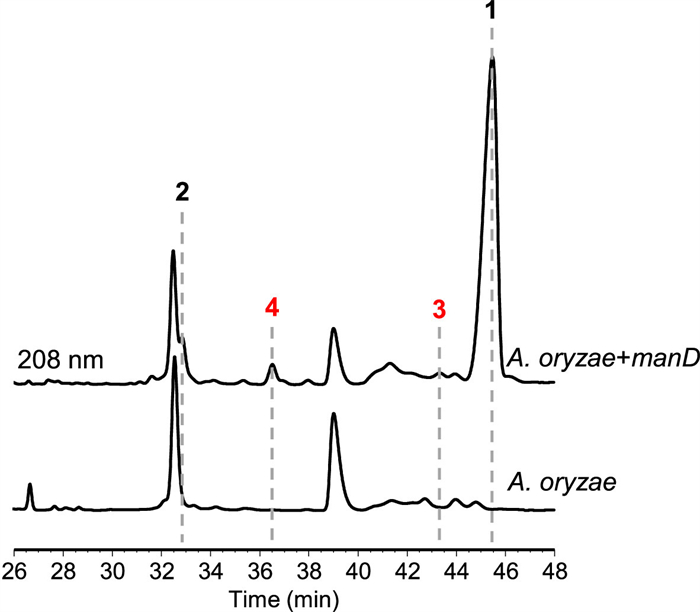
During our continuous mining of terpene cyclases [6,21,34–38], we located a candidate sesterterpene cyclase ManD from Fusarium sp. JNU-XJ070152–01, sharing the 93% amino acid identity with FgMS, also termed as MgcD [16,17]. Moreover, the tailoring enzymes encoded by genes in the flanking region of manD are also highly homologous (>90% identity) to those encoded by the mgc cluster (Fig. S2 in Supporting information), which are dedicated to biosynthesis of mangicols [17]. Based on these, we proposed that the man cluster (Genbank accession number: PQ623309) is functionally the same as the mgc cluster, and thus decided to utilize the mangicdiene synthase gene manD in hand to probe its product profile in Saccharomyces cerevisiae and A. oryzae, both of which have been widely used for heterologous expression of fungal sesterterpene synthases [17,25,27,29,31,35,39–45]. To this end, the intronless manD gene was prepared (Supplementary methods and Table S1 in Supporting information), and then incorporated into the engineered S. cerevisiae strain [34]. Upon gas chromatography-mass spectrometry (GC–MS) analysis, we found that the resulting transformant can only produce one sesterterpene hydrocarbon, the MS fragment pattern of which is completely consistent with that of mangicdiene (Fig. S3 in Supporting information) [16]. The result indicated that heterologous expression of manD in the engineered S. cerevisiae strain cannot accumulate any other related shunt products. We then turned to express manD in A. oryzae NSAR1 [46]. Intriguingly, high-performance liquid chromatography (HPLC) analysis showed that the A. oryzae NSAR1 transformant possessing manD could yield one major product 1 along with three minor products 2−4 (Fig. 2). Among them, 1 and 2 were individually identified as mangicdiene and variecoltetraene by comparing the NMR data and specific optical rotation with those reported values (Supplementary methods and Figs. S4−S7 in Supporting information) [16], while 3 and 4 were elucidated as two novel sesterterpene skeletons by exhaustive nuclear magnetic resonance (NMR) analysis, and named fusadiene and fusatriene, respectively (Tables S2−S4 and Figs. S8−S22 in Supporting information).
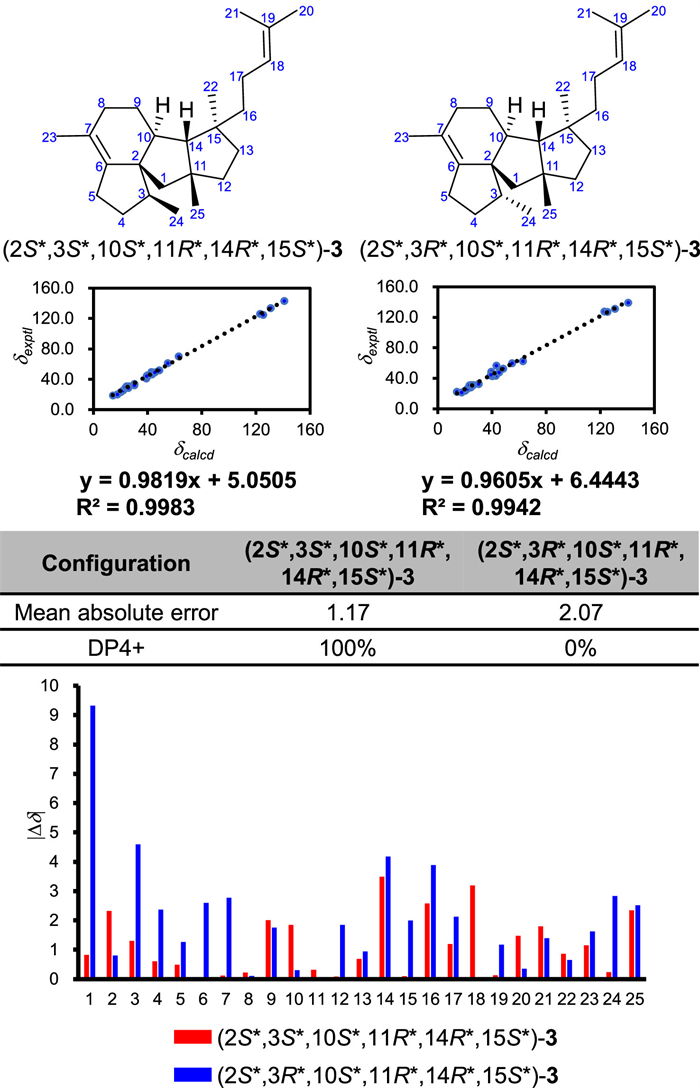
Fusadiene (3) was obtained as colorless oil, and its molecular formula was determined to be C25H40 (six degrees of unsaturation) based on the high resolution electron ionization mass spectrometry (HR-EI-MS) ion at m/z 340.31253 [M]•+ (calcd. for C25H40, 340.31245). In the 1H and 13C NMR spectra, one olefinic proton signal [δH 5.12 (1H, br t, J = 7.2 Hz)] and four olefinic carbon signals (δC 140.8, 130.8, 125.4, 123.1) were observed, implying that 3 possesses the tetracyclic skeleton with two double bonds. The 1H−1H homonuclear correlation spectroscopy (COSY) spectrum showed the presence of four spin coupling systems C-24–C-3–C-4–C-5, C-8–C-9–C-10–C-14, C-12–C-13, and C-16–C-17–C-18. And in the heteronuclear multiple-bond correlation (HMBC) spectrum, the correlations from H3–20 to C-18/C-19/C-21, from H3–21 to C-18/C-19/C-20, from H3–22 to C-13/C-14/C-15/C-16, from H3–23 to C-6/C-7/C-8, from H3–24 to C-2/C-3/C-4, from H3–25 to C-1/C-11/C-12/C-14, from Hb-1 to C-2/C-10, from Ha-5/Hb-5 to C-6, and from H-10 to C-2/C-3/C-6 were observed. The assignments of C-20 and C-21 were determined on the basis of the nuclear Overhauser effect (NOE) correlation between H-18 and H3–20. Accordingly, the planar structure of 3 was established (Fig. 3A). The NOE correlations between Ha-8 and H-14/H3–25 indicated that the rings B and C are fused in a cis configuration, and that H-10/C-3 and H-14/C-25 are located on the different sides of the ring C. The NOE correlations between H-10 and H3–22, and between H-14 and H2–17 revealed that in comparison with H-14 and C-25, C-22 is oriented in the opposite direction. However, the relative configuration of C-3 cannot be determined by NOE analysis. We then performed quantum chemical calculation of NMR chemical shifts. By comparison of the experimental 13C NMR data of 3 with those calculated values of two putative stereoisomers (Fig. 4), the relative configuration of 3 was determined to be 2S*,3S*,10S*,11R*,14R*,15S*. As both 1 and 3 are produced by ManD, and they should share the early-stage cyclization process, we thus assigned the absolute configuration of 3 as 2S,3S,10S,11R,14R,15S.
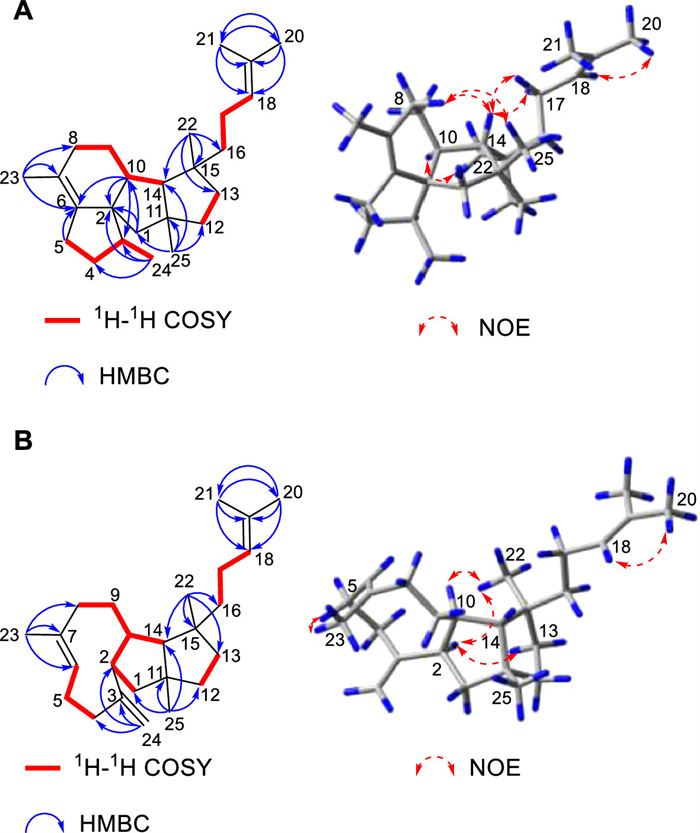
Fusatriene (4) was obtained as colorless oil, and the HR-EI-MS ion at m/z 340.31253 [M]•+ (calcd. for C25H40, 340.31245) revealed its molecular formula as C25H40 (six degrees of unsaturation). The presence of six olefinic carbon signals (δC 149.2, 136.3, 130.9, 125.3, 123.2, 112.0), as well as four olefinic proton signals [δH 5.30 (1H, br dd, J = 12.0, 3.2 Hz), 5.12 (1H, br t, J = 6.6 Hz), 4.83 (1H, br s), 4.77 (1H, br s)], in the 1D NMR spectra demonstrated that 4 is featured with the tricyclic skeleton and three double bonds. The 1H−1H COSY spectrum showed that there are four spin coupling systems C-1–C-2–C-10(–C-14)–C-9–C-8, C-4–C-5–C-6, C-12–C-13, and C-16–C-17–C-18. In combination with HMBC correlations from H3–20 to C-18/C-19/C-21, from H3–21 to C-18/C-19/C-20, from H3–22 to C-13/C-14/C-15/C-16, from H3–23 to C-6/C-7/C-8, from H3–25 to C-1/C-11/C-12/C-14, and from Ha-24/Hb-24 to C-2/C-3/C-4, and the NOE correlation between H-18 and H3–20, the planar structure of 4 was established (Fig. 3B). The NOE correlations between H-2 and Hb-13/H3–22 revealed that the rings B and C are cis fused, with H-2 and H-14/C-25 being positioned on the opposite sides of the ring B, and C-22 and H-14/C-25 being situated on the opposite sides of the ring C. And based on the NOE correlation between H-10 and H3–22, it can be determined that H-10 is oriented in a direction distinct from that of H-14. Taken together, the relative configuration of 4 was defined as 2R*,10R*,11R*,14R*,15S*. In addition, the geometrical configuration of the Δ6-double bond was assigned as E, due to the NOE correlation between Ha-5 and H3–23. Likewise, according to the biosynthetic relationship between 1 and 4, the absolute configuration of 4 was assigned as 2R,10R,11R,14R,15S.
Structural characterization of 3 and 4 proves the occurrence of J+ and K+ in the cyclization process from GFPP to mangicdiene, experimentally supporting the involvement of the path b for construction of mangicdiene (Fig. 1). Generally, isotopic labelling experiments serve as a powerful approach to unambiguously elucidate the cyclization mechanism of terpene synthases regardless of identification of shunt products [43,47,48]. Taking biosynthesis of the diterpene cattleyene as example, several carbons and hydrogens of cattleyene from two plausible pathways did not originate from the same source, and thus the cyclization mechanism for cattleyene was elucidated through extensive isotopic labelling experiments [47]. However, for some exceptional cases, where isotopic labelling experiments are not applicable, identification of shunt products is required to uncover the mystery [49,50]. For example, two plausible routes were presumed to be responsible for biosynthesis of the sesquiterpene β-terrecyclene, neither of which could be ruled out by isotopic labelling experiments. Therefore, to probe the mechanism involved in the formation of β-terrecyclene, several potential key amino acids in the active pocket of the β-terrecyclene synthase TerA was mutated, which led to detection of two additional products. Consequently, in combination with the isotopically sensitive branching experiments, the mechanism for β-terrecyclene biosynthesis was demonstrated [50]. Our work represents another rare example showing the significance of identifying shunt products, instead of isotopic labelling experiments, in elucidating the terpene cyclization mechanism. The other intriguing point is that heterologous expression of the mangicdiene synthase gene in different hosts can lead to alteration of its product profile, which might be attributed to the variation of intracellular conditions (such as pH, temperature, and metal ion species) in different hosts [38,51–53]. However, the similar phenomenon was few reported [33]. Notably, as ManD are not completely identical to FgMS, we could not rule out the possibility that alteration of the product profile was induced by the variance of their amino acid sequences.
In conclusion, we identified a candidate sesterterpene synthase ManD from Fusarium sp. JNU-XJ070152–01, which is highly homologous to the mangicdiene synthase FgMS. Through heterologous expression, we found that ManD is functionally the same as FgMS, but the product distribution of ManD in A. oryzae NSAR1 slightly differs from the previously reported product profile of FgMS in E. coli. As a result, we isolated and identified two novel sesterterpene skeletons fusadiene and fusatriene from the A. oryzae NSAR1 transformant expressing manD. The results, for the first time, experimentally confirmed that during the cyclization process from GFPP to mangicdiene, the C10 cation intermediate preferred to undergo C2-C10 cyclization (path b) rather than C6-C10 cyclization (path a).
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Pan Luo: Writing – original draft, Validation, Investigation. Jian-Ming Lv: Writing – original draft, Validation. Hong-Ting Zhen: Investigation. Ying-Qi Zhao: Investigation. Jing-Yuan Liu: Validation. Jin-Yu Hong: Formal analysis. Shao-Yang Li: Validation. Gao-Qian Wang: Validation. Guo-Dong Chen: Formal analysis. Shui-Xing Zhang: Writing – review & editing, Supervision, Conceptualization. Dan Hu: Writing – review & editing, Validation, Supervision, Conceptualization. Hao Gao: Writing – review & editing, Supervision, Conceptualization.
We thank Prof. K. Gomi (Tohoku University) and Prof. K. Kitamoto (The University of Tokyo) for providing the A. oryzae NSAR1 heterologous expression system. This work was financially supported by grants from the National Key Research and Development Program of China (No. 2024YFE0102000), the National Natural Science Foundation of China (Nos. 81925037, 82321004, U22A20371, U24A20782, 32170060, 22177037, 22207039, 22307045), the Guangdong Major Project of Basic and Applied Basic Research (No. 2023B0303000026), the Guangdong Natural Science Funds for Distinguished Young Scholars (No. 2022B1515020028, China), the Guangdong International Science and Technology Cooperation Base (No. 2021A0505020015, China), the Guangdong Basic and Applied Basic Research Foundation (Nos. 2023B1515040016, 2023A1515110388), the Innovative and Research Teams Project of Guangdong Higher Education Institution (No. 2021KCXTD001, China), the Guangzhou Science and Technology Project (Nos. 202206010020, 2024A04J6241, 2023A04J0080, China), the Fundamental Research Funds for the Central Universities (Nos. 21623105, 21624210).
Supplementary material associated with this article can be found, in the online version, at doi:
W. Du, Q. Yang, H. Xu, L. Dong, Chin. J. Nat. Med. 20 (2022) 737–748.
Z. Zhan, S. Li, W. Chu, S. Yin, Nat. Prod. Rep. 39 (2022) 2132–2174. doi: 10.1039/d2np00047d
K. Li, K.R. Gustafson, Nat. Prod. Rep. 38 (2021) 1251–1281. doi: 10.1039/d0np00070a
J.S. Dickschat, Nat. Prod. Rep. 33 (2016) 87–110. doi: 10.1039/C5NP00102A
P. Luo, J.H. Huang, J.M. Lv, et al., Nat. Prod. Rep. 41 (2024) 748–783. doi: 10.1039/d3np00052d
J.Y. Liu, F.L. Lin, J.M. Lv, D. Hu, H. Gao, Tetrahedron Lett. 112 (2022) 154224. doi: 10.1016/j.tetlet.2022.154224
Y. Li, J. Wang, L. Li, et al., Nat. Prod. Rep. 40 (2023) 1303–1353. doi: 10.1039/d2np00063f
Z. Hu, X. Liu, M. Tian, et al., Med. Res. Rev. 41 (2021) 2971–2997. doi: 10.1002/med.21816
J.D. Rudolf, T.A. Alsup, B. Xu, Z. Li, Nat. Prod. Rep. 38 (2021) 905–980. doi: 10.1039/d0np00066c
W. Yuan, F. Li, Z. Chen, et al., Chin. Chem. Lett. 35 (2024) 108788. doi: 10.1016/j.cclet.2023.108788
K. Wang, B. Liu, D. Yan, et al., Chin. Chem. Lett. 36 (2025) 109811. doi: 10.1016/j.cclet.2024.109811
D.J. Tantillo, Nat. Prod. Rep. 28 (2011) 1035–1053. doi: 10.1039/c1np00006c
J.S. Dickschat, Eur. J. Org. Chem. 2017 (2017) 4872–4882. doi: 10.1002/ejoc.201700482
D.W. Christianson, Chem. Rev. 117 (2017) 11570–11648. doi: 10.1021/acs.chemrev.7b00287
M.K. Renner, P.R. Jensen, W. Fenical, J. Org. Chem. 65 (2000) 4843–4852. doi: 10.1021/jo000081h
G. Bian, Y. Han, A. Hou, et al., Metab. Eng. 42 (2017) 1–8. doi: 10.1016/j.ymben.2017.04.006
Y. Yuan, S. Cheng, G. Bian, et al., Nat. Catal. 5 (2022) 277–287. doi: 10.1038/s41929-022-00762-x
L. Lauterbach, J. Rinkel, J.S. Dickschat, Angew. Chem. Int. Ed. 57 (2018) 8280–8283. doi: 10.1002/anie.201803800
A. Minami, T. Ozaki, C. Liu, H. Oikawa, Nat. Prod. Rep. 35 (2018) 1330–1346. doi: 10.1039/c8np00026c
H. Sato, B.X. Li, T. Takagi, et al., JACS Au 1 (2021) 1231–1239. doi: 10.1021/jacsau.1c00178
F.L. Lin, L. Lauterbach, J. Zou, et al., ACS Catal. 10 (2020) 4306–4312. doi: 10.1021/acscatal.0c00377
J.Y. Liu, F.L. Lin, K.A. Taizoumbe, et al., Angew. Chem. Int. Ed. 63 (2024) e202407895. doi: 10.1002/anie.202407895
Y.H. Wang, H. Xu, J. Zou, et al., Nat. Catal. 5 (2022) 128–135. doi: 10.1038/s41929-022-00735-0
H. Tao, L. Lauterbach, G. Bian, et al., Nature 606 (2022) 414–419. doi: 10.1038/s41586-022-04773-3
Z. Quan, A. Hou, B. Goldfuss, J.S. Dickschat, Angew. Chem. Int. Ed. 61 (2022) e202117273. doi: 10.1002/anie.202117273
T. Lou, A. Li, H. Xu, et al., J. Am. Chem. Soc. 145 (2023) 8474–8485. doi: 10.1021/jacs.3c00278
M. Okada, Y. Matsuda, T. Mitsuhashi, et al., J. Am. Chem. Soc. 138 (2016) 10011–10018. doi: 10.1021/jacs.6b05799
L. Zhang, B. Zhang, A. Zhu, et al., Angew. Chem. Int. Ed. 62 (2023) e202312996. doi: 10.1002/anie.202312996
J. Guo, Y.S. Cai, F. Cheng, et al., Org. Lett. 23 (2021) 1525–1529. doi: 10.1021/acs.orglett.0c03996
G. Bian, A. Hou, Y. Yuan, et al., Org. Lett. 20 (2018) 1626–1629. doi: 10.1021/acs.orglett.8b00366
L. Jiang, X. Zhang, Y. Sato, et al., Org. Lett. 23 (2021) 4645–4650. doi: 10.1021/acs.orglett.1c01361
X. Wang, Z. Wang, G. Zhu, et al., Chem. Commun. 58 (2022) 9476–9479. doi: 10.1039/d2cc03644d
D. Lubertozzi, J.D. Keasling, J. Ind. Microbiol. Biotechnol. 35 (2008) 1191–1198. doi: 10.1007/s10295-008-0400-3
P. Luo, J.M. Lv, Y.F. Xie, et al., Org. Chem. Front. 9 (2022) 3057–3060. doi: 10.1039/d2qo00408a
J.H. Huang, J.M. Lv, Q.Z. Wang, et al., Org. Biomol. Chem. 17 (2019) 248–251. doi: 10.1039/c8ob02832j
L. Dong, J.Y. Liu, G.Q. Wang, et al., Bioorg. Chem. 152 (2024) 107726. doi: 10.1016/j.bioorg.2024.107726
J.H. Huang, J.M. Lv, L.Y. Xiao, et al., Beilstein J. Org. Chem. 18 (2022) 1396–1402. doi: 10.3762/bjoc.18.144
F.L. Lin, K.A. Taizoumbe, Y.X. Wang, et al., Org. Biomol. Chem. 22 (2024) 7971–7975. doi: 10.1039/d4ob01348d
R. Chen, Q. Jia, X. Mu, et al., Proc. Natl. Acad. Sci. U. S. A. 118 (2021) e2023247118. doi: 10.1073/pnas.2023247118
D. Yan, J. Arakelyan, T. Wan, et al., Acta Pharm. Sin. B 14 (2024) 421–432. doi: 10.1016/j.apsb.2023.08.025
R. Chiba, A. Minami, K. Gomi, H. Oikawa, Org. Lett. 15 (2013) 594–597. doi: 10.1021/ol303408a
B. Qin, Y. Matsuda, T. Mori, et al., Angew. Chem. Int. Ed. 55 (2016) 1658–1661. doi: 10.1002/anie.201509263
Z. Quan, J.S. Dickschat, Org. Lett. 22 (2020) 7552–7555. doi: 10.1021/acs.orglett.0c02748
Y. Qiao, Q. Xu, Z. Huang, et al., Org. Chem. Front. 9 (2022) 5808–5818. doi: 10.1039/d2qo00983h
D. Li, M. Yang, R. Mu, et al., Chin. Chem. Lett. 34 (2023) 107469. doi: 10.1016/j.cclet.2022.04.067
F.J. Jin, J.i. Maruyama, P.R. Juvvadi, M. Arioka, K. Kitamoto, FEMS Microbiol. Lett. 239 (2010) 79–85. doi: 10.1016/j.femsle.2004.08.025
J. Rinkel, S.T. Steiner, J.S. Dickschat, Angew. Chem. Int. Ed. 58 (2019) 9230–9233. doi: 10.1002/anie.201902950
H. Xu, L. Lauterbach, B. Goldfuss, G. Schnakenburg, J.S. Dickschat, Nat. Chem. 15 (2023) 1164–1171. doi: 10.1038/s41557-023-01223-z
L. Zu, M. Xu, M.W. Lodewyk, et al., J. Am. Chem. Soc. 134 (2012) 11369–11371. doi: 10.1021/ja3043245
Y. Song, W. Wang, J. Yang, et al., Chem. Sci. 15 (2024) 8750–8755. doi: 10.1039/d4sc01208a
R.J. Peters, R.B. Croteau, Proc. Natl. Acad. Sci. U. S. A. 99 (2002) 580–584. doi: 10.1073/pnas.022627099
F. Lopez-Gallego, S.A. Agger, D. Abate-Pella, M.D. Distefano, C. Schmidt-Dannert, ChemBioChem 11 (2010) 1093–1106. doi: 10.1002/cbic.200900671
P.E. O’Maille, J. Chappell, J.P. Noel, Arch. Biochem. Biophys. 448 (2006) 73–82. doi: 10.1016/j.abb.2005.10.028

Figure 1 Previously proposed and herein experimentally supported biosynthetic mechanisms of mangicdiene (1).

Figure 2 HPLC analysis of mycelial metabolites from the A. oryzae NSAR1 transformant harboring manD and the parent strain.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:


 下载:
下载: