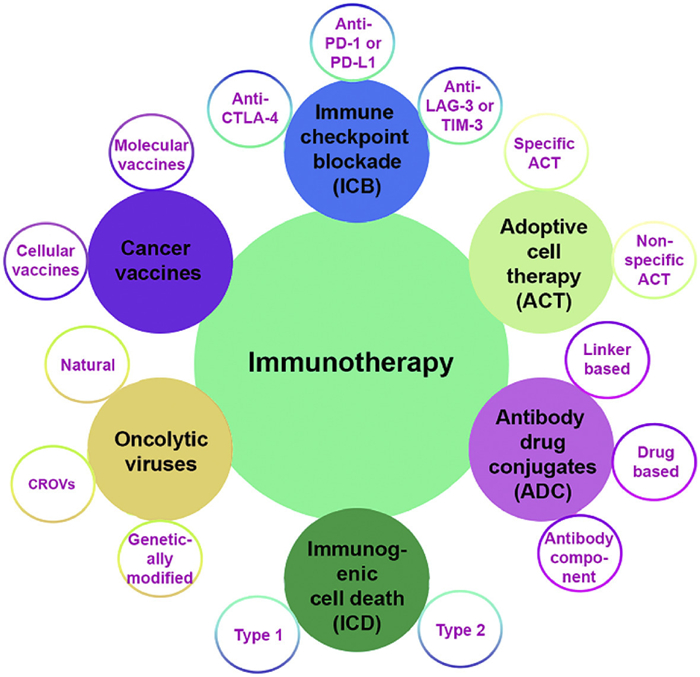
Figure 1.
The current treatment modalities of cancer immunotherapy and their associated sub-classification.

Immunotherapy can revolutionize cancer treatment by harnessing the body's own immune system to identify and eradicate cancer cells [1,2]. Recent advances in tumor biology have greatly enhanced our understanding of the immunotherapeutic mechanism. Cancer immunotherapies utilize different approaches to augment the immune responses (Fig. 1). These include adoptive cell therapy (ACT), antibody-drug conjugates (ADC), tumor vaccines, oncolytic viruses, immunogenic cell death (ICD) therapy, and immune checkpoint blockade (ICB) therapy [3–5]. In ACT, immune effector cells, typically T cells, are collected from a patient, adapted or expanded ex vivo to boost their anticancer efficacy, and then infused back into the patient. This enhanced the body's innate capacity to combat cancer by fortifying or amplifying the immune system with a substantial quantity of highly activated immune cells [6]. ACT therapy is divided into non-specific and specific cell therapy. In non-specific cell therapy, ex vivo primed immune cells such as tumor-infiltrating lymphocytes (TILs), γ/δ T cells, and natural killer (NK) cells are directly infused into the cancer. In contrast, in specific cell therapy, chimeric antigen receptors are genetically engineered onto T cells or NK cells to selectively identify antigens on cancer cells, resulting in the elimination of tumors [7,8].
Similar to ACT, tumor vaccines were designed to provoke a specific immune response by targeting antigens present on tumor cells [9]. Cellular and molecular vaccines are the main categories of tumor vaccines. Cellular vaccines use dendritic cells (DCs) or tumor cells that have been genetically engineered ex vivo with tumor-specific antigens. These are then reintroduced into patients to activate T lymphocytes that target and eliminate cancer cells with the same antigen. In molecular vaccines, tumor-specific antigens are administered as proteins, peptides, or nucleic acid to antigen-presenting cells to elicit an immune response [10–12].
In contrast, ADC therapy adopts a distinct approach to stimulate immune response by utilizing the particular binding site of monoclonal antibodies (mAbs) to conjugate cytotoxic drugs via a linker, thus mitigating the adverse effects of tethered drugs. Mechanistically, the mAb specifically binds to an antigen on tumor cells, initiating endocytosis, which facilitates the internalization of the ADC inside the tumor cell, followed by destruction or cleavage of the linker and the release of the cytotoxic payload. The delivered cytotoxic drug then triggers apoptosis and eliminates adjacent cancer cells via bystander effects. ADC therapy is often categorized based on the selection of antibodies, cytotoxic drugs, and linker types [13,14].
Oncolytic virus-based immunotherapy aligns with the aforementioned approaches by directly targeting tumors and indirectly stimulating the immune system. Oncolytic virus-based immunotherapy has been categorized into three primary types: Natural, engineered, and chimeric recombinant oncolytic viruses (CROVs) [15,16]. Generally, it employs replication-competent viruses that generate more viral particles upon internalization into tumor cells. This replication ultimately causes the lysis of the tumor cells. The dying cells further discharge viruses that can subsequently destroy adjacent tumor cells, along with tumor-specific antigens and other danger signals that activate the immune system [17,18]. Conversely, ICD-based therapy promotes release of danger-associated molecular patterns (DAMPs), tumor-associated antigens, and immune-stimulating molecules from dying cancer cells, thereby initiating a robust antitumor immune response [19,20]. Mechanistically, DAMPs such as calreticulin (CRT), ATP, high mobility group protein B1 (HMGB1), and heat-shock proteins (HSPs) are released from dying cancer cells, which are subsequently captured by antigen-presenting cells, which facilitates the differentiation of immature T cells into effector T cells (often referred as cytotoxic T lymphocytes (CTLs)) [21]. These CTLs identify particular antigens displayed on the surface of cancer cells and subsequently secrete granules (perforin and granzymes). These proteins form pores in cancer cell membranes, causing apoptosis and tumor cell death [22,23]. ICD is often classified into intrinsic (type 1) and extrinsic (type 2) categories, depending on the pathways engaged and the manner in which the immune system is triggered during the process of cell death [24–26]. Providing more comprehensive details on all forms of immunotherapy exceeds the scope of this article. Therefore, for an in-depth understanding of the discussed forms of immunotherapy, we will direct our readers to the aforementioned cited articles.
Targeting immune checkpoints (commonly called gatekeepers of immune responses) and regulatory pathways in the immune system that maintain self-tolerance and modulate the duration and amplitude of immune responses is a crucial component of immunotherapy [27,28]. The key checkpoints encompass cytotoxic T-lymphocyte antigen 4 (CTLA-4), lymphocyte activation gene-3 (LAG-3), T-cell immunoglobulin and mucin domain 3 (TIM-3), and programmed death protein 1 (PD-1) or its ligand PD-L1, which tumors utilize to circumvent immune recognition and suppression [29,30]. PD-1/PD-L1 checkpoint interaction transpires peripherally compared to CTLA-4/B7 contact, which is mostly located in the lymphoid organs [31]. Therefore, anti-PD-1/PD-L1 treatment may cause elevated response rates and reduced immune-related adverse events. Both PD-1 and its ligand PD-L1 are classified as type Ⅰ transmembrane proteins, characterized by their classical immunoglobulin-like extracellular domains [32]. PD-1 protein is overexpressed on the surface of T cells, B cells, NK cells, and macrophages engaged in recognizing and eliminating cancer cells and infected cells. Upon binding to its ligand, PD-L1, which is upregulated in various types of cancer cells and immune cells, PD-1 transmits an inhibitory signal to the T-cell. In essence, this signal instructs the T-cell to dampen its activity, thereby causing a "break" in the immunological response [33–35]. This immune evasion mechanism is intricately connected to the advancement of tumors since it enables uncontrolled tumor proliferation and spread by compromising one of the body's main defense mechanisms against tumor growth.
Since 2014, immune checkpoint inhibitors, including mAbs like pembrolizumab (anti-PD-1), durvalumab (anti-PD-L1), as well as small molecules, have shown significant potential in obstructing the interaction between PD-1 and PD-L1 (Fig. 2) [36,37]. This blockade enhances immune system functionality, allowing it to recognize and eliminate cancer cells, hence extending the life of cancer patients, with some attaining complete remission. It is worth highlighting a recent clinical study of nivolumab, an immune checkpoint (PD-1) inhibitor. In conjunction with chemotherapy, nivolumab demonstrates progression-free survival in patients with advanced classical Hodgkin lymphoma. Investigators saw a response rate of 100% with the combination of nivolumab and AVD (adriamycin/doxorubicin, vinblastine, and dacarbazine), and a 96% response rate with nivolumab monotherapy, indicating the potential for future use of nivolumab alone [38]. However, subsequent healthcare records showed that not all patients were sensitive to mAbs treatment. For example, in skin cancer therapy, PD-1 antibodies demonstrate 50% effectiveness; however, the overall efficacy in treating solid tumors was only about 15%–20% [39–41]. This is mainly caused by the impairment or lack of T cell infiltration in the tumor tissue and microenvironment [42]. Despite their high specificity, mAbs frequently encounter challenges with inadequate tumor penetration and elevated production costs, hence restricting their availability [43]. Conversely, small organic molecules lack the stability and functional versatility necessary to tackle the intricacies of the tumor microenvironment (TME). Furthermore, both modalities face challenges such as the development of resistance mechanisms and the immunosuppressive characteristics of the TME, which obstruct optimal immune activation [44,45]. These challenges have stimulated interest in alternate treatment approaches, including the utilization of metal complexes for tumor immune modulation.
Metal-based drugs such as cisplatin, oxaliplatin, and carboplatin continue to be the mainstay of treatment for various cancers (Fig. 3) [46,47]. In contrast to mAbs, metal-based drugs provide notable benefits like the potential for oral administration, enhanced stability, and membrane permeability. Combining metal-based drugs with immune checkpoint inhibitors can boost their efficacy and mitigate adverse effects [48]. Recent clinical trial data showed that several studies are ongoing, concentrating on expanding the use of PD-L1 inhibitors while discovering beneficial combination therapies and mitigating the side effects of metallodrugs (Table 1). For example, a study is underway examining the efficacy of mAbs (adebrelimab) in conjunction with chemotherapeutic agents capecitabine and oxaliplatin for the treatment of resectable adenocarcinoma of the esophagogastric junction (NCT06482788). In another study, immunotherapeutic (PD-L1/CTLA-4 bispecific antibody) was coupled with FLORINOX (fluorouracil, irinotecan, oxaliplatin) for the treatment of locally advanced and metastatic pancreatic cancer (NCT04324307). Apart from this, preclinical findings indicate that the combination strategies involving PD-1/PD-L1 inhibitors with metal-based drugs result in superior tumor shrinkage, prolonged resistance delay, and increased patient outcomes relative to monotherapy [49,50]. Fournel et al. examined the clinicopathological features of non-small-cell lung cancer (NSCLC) patients and found that cisplatin therapy elevated PD-L1 expression. These findings are expected to establish a robust foundation for future investigations and support multiple clinical studies involving metal-based drugs like cisplatin [51]. Considering the limitations of mAbs, including exorbitant costs and challenges in storage and transportation, there is a clear need for developing new metal-based compounds that not only impede cancer cell proliferation but also target immune checkpoints, thereby reducing the risk of tumor relapse and metastasis [52,53]. Because of their unique modes of action, these metal-based agents offer considerable potential to enhance the efficacy of immunotherapy when used either alone or in combination with other agents.
 DownLoad:
CSV
DownLoad:
CSV
| PD-1/PD-L1 antibody | Chemotherapeutics | Clinical phase and status | Tumor stage and type | Clinical trial |
| Atezolizumab | Carboplatin and nab-paclitaxel | Phase 3, completed | Stage Ⅳ, non-squamous NSCLC | NCT02367781 |
| Atezolizumab | Carboplatin and etoposide | Phase 1/3, completed | Extensive-stage, NSCLC | NCT02763579 |
| Atezolizumab | Bevacizumab plus carboplatin and paclitaxel | Phase 3, completed | Stage Ⅳ, non-squamous NSCLC | NCT02366143 |
| Camrelizumab | Carboplatin and pemetrexed | Phase 3, completed | Stage ⅢB/Ⅳ non-squamous NSCLC | NCT03134872 |
| Durvalumab | Etoposide and cisplatin/carboplatin | Phase 3, active | Non-squamous NSCLC | NCT03043872 |
| Nivolumab | Oxaliplatin Plus fluoropyrimidine | Phase 3, completed | Metastatic gastric or gastro-esophageal (GE) junction cancer | NCT02872116 |
| Pembrolizumab | Carboplatin and pemetrexed | Phase 1/2, completed | Metastatic non-squamous NSCLC | NCT02039674 |
| Pembrolizumab | Carboplatin and paclitaxel/nab-paclitaxel | Phase 3, completed | Metastatic squamous NSCLC | NCT02775435 |
| Pembrolizumab | Cisplatin and 5-fluorouracil | Phase 3, completed | Metastatic esophageal or GE junction cancer | NCT03189719 |
| Pembrolizumab | Trastuzumab plus cisplatin + 5-fluorouracil or oxaliplatin + capecitabine | Phase 3, active | HER2 + advanced gastric or GE junction adenocarcinoma | NCT03615326 |
| Sintilimab | Platinum complex and pemetrexed | Phase 3, completed | Metastatic non-squamous NSCLC | NCT03607539 |
| Sintilimab | Platinum complex and gemcitabine | Phase 3, completed | Metastatic squamous NSCLC | NCT03629925 |
| Tislelizumab | Platinum complex and pemetrexed | Phase 3, completed | Metastatic squamous NSCLC | NCT03663205 |
Exploring metal-based complexes as immune checkpoint inhibitors is a relatively new research area. This concise review covers the most recent strategies and approaches in rationally designed metallodrugs for ICB. We provide mechanistic insights into metallodrugs and their nanoformulation, which not only exhibited unique cytotoxic mechanisms but also obstructed PD-1/PD-L1 interactions. We also discussed challenges and potential opportunities in the advancement of metallodrugs aimed at targeting the PD-1/PD-L1 axis. The overall aim of this review is to highlight the potential of metallodrugs in targeting immune checkpoints and facilitating the recruitment of immune cells to initiate a robust antitumor response in cold tumors. We anticipate this short perspective will stimulate further discussions and research in this fledgling research topic.
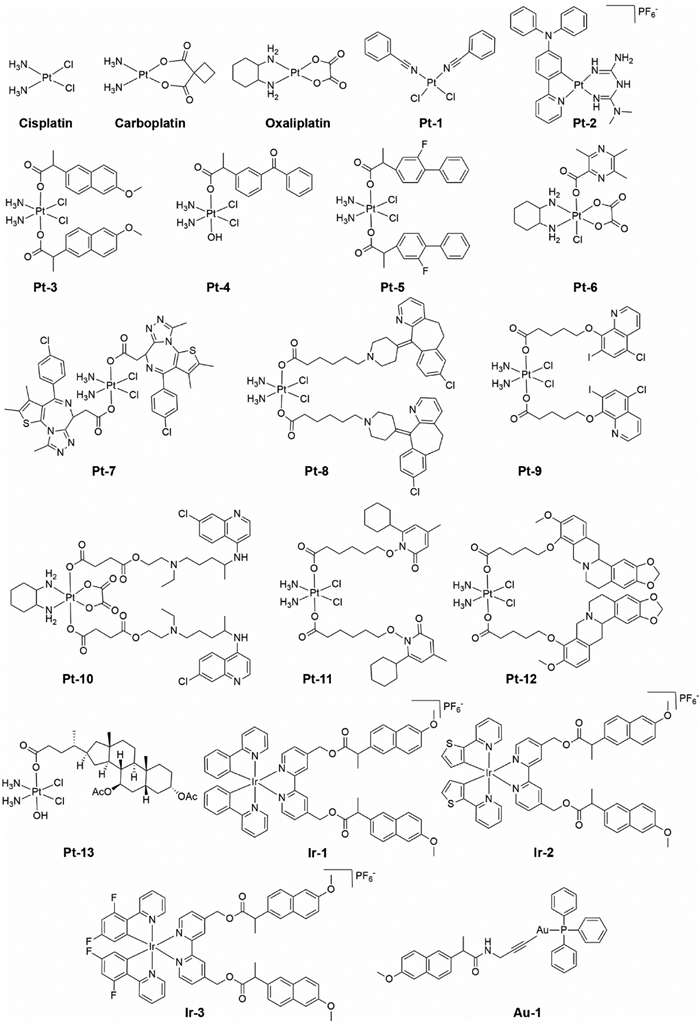
Beyond suppressing tumor cell proliferation, current advancements in metallodrug design strengthen the immune response by obstructing PD-1/PD-L1 interactions and minimizing immunosuppressive factors. This combined impact enhances the efficacy of cancer treatment and underscores the possibility of integrating chemotherapy with ICB to attain more robust antitumor responses. Wang and his colleagues discovered the first PtⅡ complex, (bis(benzonitrile)dichloroplatinum(Ⅱ)), which can bind to PD-Ⅰ but not to PD-L1, thereby inhibiting the interaction between them (Pt-1, Fig. 3) [54]. The complex demonstrated the ability to suppress tumor growth in murine models by promoting the proliferation and activity of tumor-infiltrating T cells. Metformin, an antidiabetic agent, is now well recognized for its anticancer effects [55,56]. Cellular studies revealed that metformin promoted AMP-activated protein kinase (AMPK), which could reduce PD-L1 expression in several cancer cell types, thereby amplifying antitumor immune responses [57,58]. By taking into account, Liu et al. synthesized metformin tethered cyclometalated PtⅡ complex (Pt-2, Fig. 3) which demonstrated superior therapeutic efficacy compared to cisplatin in suppressing tumor cell proliferation [59]. Experimental investigations reveal that Pt-2 mainly resides in lysosomes, inhibits PD-L1 expression as well as facilitates lysosomal-dependent PD-L1 degradation through the AMPK-transcription factor EB pathway. Moreover, the complex not only boosted antitumor efficacy but also effectively stimulated antitumor immunity by improving DCs maturation and increasing TILs, ultimately achieving highly effective immune-chemotherapy.
Apart from PtⅡ complexes, numerous octahedral PtⅣ complexes have been developed recently. These complexes, upon internalization, release their axial ligand and cytotoxic PtⅡ species, which can synergistically act together to eliminate cancer cells effectively [60]. A multitargeted PtⅣ complex (Pt-3, Fig. 3) has been developed by tethering non-steroidal anti-inflammatory drug (NSAID) naproxen at the axial positions of the PtⅣ precursor [61]. Pt-3 was synthesized by combining naproxen acid with oxoplatin in the presence of a catalyst and a base to promote an efficient coupling reaction. In vitro and in vivo investigations showed that Pt-3 inhibited the growth of breast cancer by downregulating the expression of cyclooxygenase 2 (COX-2) and PD-L1. This multispecific nature of Pt-3 provides a unique advantage in breast cancer treatment by integrating chemotherapy and immunotherapy into a single molecule. Inspired by the antimetastatic and COX-2 inhibitory properties of NSAID-PtⅣ conjugate, Li et al. designed and developed ketoprofen and loxoprofen tethered PtⅣ complexes [62]. The lead complex (Pt-4, Fig. 3), was shown to exhibit significant anticancer activity in both in vitro and in vivo models. Pt-4 causes DNA damage, induces mitochondrial injury, regulates inflammation, and enhances the immune response in the TME by decreasing the expression of PD-L1. The same group developed PtⅣ complexes with flurbiprofen and zaltoprofen-based NSAIDs, incorporating cisplatin and oxaliplatin cores [63]. They discovered that PtⅣ complexes containing cisplatin core exhibited potent anticancer activity and superior ability to overcome cisplatin associated resistance. Specifically, the disubstituted PtⅣ complex of flurbiprofen (Pt-5, Fig. 3) worked better than the monosubstituted complexes. Pt-5 induces DNA damage, fosters mitochondrial-mediated apoptosis, reduces inflammation, and suppresses PD-L1 expression by enhancing T cell immunity. The strong immune modulation drastically affects tumor growth and metastasis.
Ligustrazine, a bioactive component of Chinese medicine, has been extensively studied as an anticancer agent for many types of tumors [64,65]. A PtⅣ complex (Pt-6, Fig. 3), has been prepared by tethering ligustrazine at one of the axial positions of the oxaliplatin-based PtⅣ precursor [66]. In vivo antitumor studies demonstrated that Pt-6 suppressed tumor growth and exhibited promising antimetastatic effectiveness in pulmonary metastasis models. Pt-6 activates mitochondrial apoptotic pathways and reduces PD-L1 expression while simultaneously increasing the number of T cells. JQ-1, a nuclear protein inhibitor, plays an important role in suppressing tumor growth and lowering interferon gamma triggered PD-L1 expression. However, it underwent rapid metabolism and was secreted from the body because of its short half-life [67,68]. To overcome this drawback and to induce a synergistic effect, Fan et al. developed JQ-1 tethered PtⅣ complexes [69]. A disubstituted PtⅣ complex (Pt-7, Fig. 3) that consists of a cisplatin core was found to be the most potent one among the developed compounds. Pt-7 blocks PD-L1 synthesis and reverses the immunosuppression microenvironment by increasing T cell infiltration. In addition, the complex inhibits DNA replication and halts the proteins responsible for DNA repair damage, thus overcoming resistance to cisplatin. Various biochemical analyses demonstrate that tethering JQ-1 to the PtⅣ center improves the antitumoral efficacy by reshaping the TME.
Desloratadine is an antihistamine drug that has been approved by the FDA. Recent studies have demonstrated its potential in reversing immunity and antitumor efficacy against various solid tumors [70,71]. A PtⅣ complex (Pt-8, Fig. 3) with axially bound two desloratadine molecules shows potent antimetastatic potential by inhibiting epithelial−mesenchymal transition [72]. The complex displays excellent antitumor efficacy, similar to cisplatin but superior to oxaliplatin. The key cellular response to Pt-8 treatment involves DNA damage, stimulation of the mitochondrial apoptotic pathway, and activation of hypoxia-inducible factor (HIF), thus overcoming the resistance associated with clinically approved Pt drugs. Furthermore, Pt-8 provokes antitumor immunity by inhibiting PD-L1 while concurrently inducing macrophages to transition from M2 to M1. Autophagy is critical to cellular homeostasis because it promotes tumor cell survival and adaptability in a hostile environment [73]. In clinics, chloroquine and its derivatives have been investigated as possible treatment approaches to inhibit autophagy and delay metastases [74]. Zhang, Li, and co-workers developed chloroquine tethered PtⅣ complex (Pt-9, Fig. 3) as a potential prodeath autophagy inducer and immune activator [75]. Pt-9 suppresses DNA synthesis, induces mitochondrial enlargement, and stimulates autophagy. Furthermore, the complex suppresses PD-L1 expression and consequently enhances CD3+ and CD8+ T cell production, reversing the suppressed immune response. This work highlights the significant therapeutic potential of PtⅣ complexes against highly aggressive tumors. The same group developed hydroxychloroquine-tethered PtⅣ complex (Pt-10, Fig. 3) bearing oxaliplatin core [76]. Pt-10 displays superior antitumor efficacy and less toxicity in comparison with oxaliplatin. Cellular studies reveal that treatment with Pt-10 interferes with DNA synthesis, impairs autophagy, and triggers mitochondrial mediated apoptosis. Furthermore, the complex attenuates hypoxia and counteracts the adverse immune response in tumors by inhibiting the production of PD-L1. In a similar effort ciclopirox, a versatile fungicide, has been attached to the PtⅣ metal center in order to stimulate prodeath autophagy and immunological response (Pt-11, Fig. 3) [77]. Pt-11 complex shows enhanced anticancer characteristics and successfully overcomes drug resistance compared to its parent complex, cisplatin. Furthermore, Pt-11 suppresses the process of angiogenesis, obstructs DNA synthesis, and triggers mitochondrial mediated apoptosis. In addition, treatment with Pt-11 stimulates the immune response by suppressing PD-L1 expression, achieving a comparable rate of tumor growth suppression to cisplatin but with less toxicity.
Canadine, also referred to as tetrahydroberberine, is an alkaloid that showed significant promise as an antiproliferative and antimetastatic agent. Molecular investigations demonstrate that canadine suppresses inflammation and stimulates the immune response [78,79]. Chen et al. developed a series of canadine-tethered PtⅣ complexes [80]. One of the complex (Pt-12, Fig. 3) which contains a cisplatin core proved to be a promising candidate due to its superior antiproliferative effects in both in vitro and in vivo models. Mechanistically, Pt-12 damages nuclear DNA, generates reactive oxygen species (ROS), and strongly inhibits epithelial−mesenchymal transition. Furthermore, the complex boosts the immune response by inhibiting the expression of PD-L1 and CD47, while also inducing TILs accumulation and initiating macrophage polarization. This study demonstrates that the development of such hybrid systems warrants consideration when treating metastatic malignancies in clinics. Ursodeoxycholic acid (UDCA), the predominant secondary bile acid, is extensively used in clinics for the treatment of liver diseases [81]. Recent studies indicated that UDCA had significant promise as an anticancer agent [82,83]. Chen et al. synthesized UDCA conjugated PtⅣ complex (Pt-13, Fig. 3) that inhibited the Janus kinase (JAK)/STAT oncogenic signaling pathways and exhibited potent anticancer activity in both cells and murine models [84]. Pt-13 shows activity in hypoxic TME, severely damages DNA, and downregulates COX-2 and HIF-1, leading to inhibition of tumor angiogenesis. Moreover, the complex reverses the tumor suppressive microenvironment by increasing TILs density and inhibiting PD-L1 expression, thereby augmenting the antitumor immune response.
Iridium-based compounds have garnered significant interest in recent years owing to their broad range of therapeutic effects and activation mechanisms [85,86]. Motivated by the findings regarding NSAID-PtⅣ conjugates, Xie, Lu, and colleagues synthesized naproxen-linked IrⅢ complexes (Ir-1 to Ir-3, Fig. 3) [87]. These complexes have a relatively similar cytotoxicity profile when evaluated against various cancer cells. The complexes primarily localize in the mitochondria, where they initiate mitochondrial-mediated apoptosis. Furthermore, they induce ICD by releasing associated signaling molecules and downregulating the expression of COX-2 and PD-L1, hence facilitating a synergistic effect of chemotherapy and immunotherapy.
Gold complexes have attracted considerable attention due to their usually reduced toxicity, ability to circumvent drug resistance, and superior antitumor efficacy compared to traditional chemotherapies [88,89]. Liu et al. developed a series of Au1 complexes with NSAIDs or their equivalents and investigated their antiproliferative potential against cisplatin-resistant ovarian cancer cells [90]. Among them, Au-1 (Fig. 3) had superior potency relative to other investigated compounds, with IC50 values in the low micromolar range. Cellular studies revealed that Au-1 complex caused oxidative stress and inhibited thioredoxin reductase activity. The complex triggered the release of DAMP-related signals, which are vital for inducing the antitumor immune response. Moreover, Au-1 showed downregulation of the COX-2 and PD-L1 expression, activated DCs maturation, and elevated the expression of immunostimulatory cytokines.
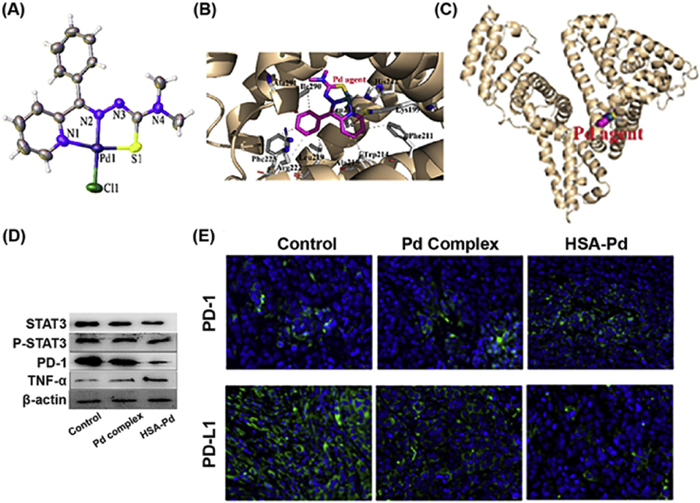
Palladium (Pd) derivatives were preferentially selected for cancer therapy among non-platinum metal complexes owing to their structural similarity to Pt(Ⅱ) complexes [91,92]. Li et al. constructed a multifunctional Pd complex (Fig. 4A), and its delivery was done using human serum albumin (HSA) [93]. Structural analysis reveals that Pd complex is bound to the big hydrophobic pocket of the HSA ⅡA subdomain, where His-242 of HSA substitutes a chlorido ligand coordinating to the Pd core (Figs. 4B and C). This prevents direct exposure of Pd complex with the blood. In vivo studies reveal that the HSA-Pd complex effectively inhibits tumor development and induces less toxicities compared to free Pd. Mechanistic studies demonstrate that HSA-Pd complex triggers DNA damage, inhibits angiogenesis, and reshapes the tumor immune microenvironment by raising the density of T cells and NK cells, which is subsequently associated with a reduction in PD-L1 expression and elevation in PD-1 expression (Figs. 4D and E). This dual ability of Pd compound tackles critical issues, providing an innovative and tailored strategy that may substantially enhance outcomes for patients with immune-resistant cancers.
Patients who showed favorable responses to mAbs often possess a "hot" immunological profile, marked by the presence of TILs and PD-L1 on tumor-associated immune cells. In contrast, non-responsive solid tumors may have a "cold" phenotype, which differs from the characteristics of "hot" tumors [94,95]. Accurate classification of "cold" and "hot" cancers is essential for predicting tumor response and treatment outcomes [96]. ICB is most efficacious against "hot" tumors and less so against "cold" tumors, highlighting the need for novel therapeutics that improve immune cell infiltration and transform "cold" tumors to immune responsive.
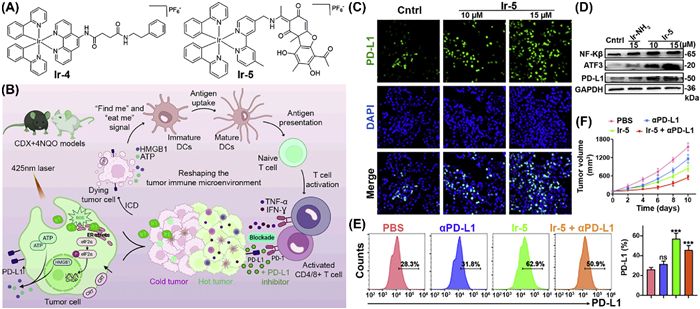
Photodynamic therapy (PDT) is a modern therapeutic technique that employs light-activated photosensitizers to produce ROS that eradicate cancer cells [97,98]. In addition to its cytotoxic properties, PDT functions as an in situ tumor vaccine by augmenting tumor immunogenicity and eliciting antitumor immune responses [99]. Zhou et al. develop light activated IrⅢ complex with 2-phenylpyridine (C˄N) and N1-(1,10-phenanthrolin-5-yl)-N4-phenethylsuccinamide (N˄N) ligands [100]. Upon light activation (425 nm), Ir-4 (Fig. 5A) demonstrated equivalent efficacy against murine squamous sarcoma cells in both hypoxia and normoxic environments. Moreover, Ir-4 induced ICD and enhanced PD-L1 expression, establishing a foundation for synergistic therapy. Subsequent mechanistic investigations demonstrate that the complex induced endoplasmic reticulum stress, stimulated ROS generation, and facilitated the maturation of DCs and T cells infiltration (Fig. 5B). The progression of oral cancer transpires sequentially, starting with hyperplasia and moving to invasive malignancy, presenting an imperative opportunity for the treatment [101,102]. In view of that, the authors establish the cell-derived xenograft (CDX) + 4-nitroquinoline 1-oxide (4NQO)-induced mouse tumor model, which offers a complex and clinically relevant framework for assessing the efficacy of Ir-4 against oral cancer. Upon administration with PD-L1 inhibitor, Ir-4 induces a synergistic impact, by shifting immune-suppressive tumors into immune-responsive tumors and significantly inhibiting the tumor development. An usnic acid-conjugated IrⅢ complex (Ir-5, Fig. 5A), has been engineered to transform cold tumors into hot ones [103]. Ir-5 entered into the cells through an energy-dependent pathway and mainly resided into the mitochondria. Subsequent analysis revealed that the complex substantially damaged mitochondria and disrupted energy production pathways, leading to cell death. Furthermore, Ir-5 treatment modulated the transcription factors of PD-L1 (nuclear factor kappa-light-chain-enhancer of activated B cells (NF-kβ), activating transcription factor 3 (ATF3)), thereby augmenting PD-L1 levels in both in vitro and in vivo models (Figs. 5C–E). Additionally, the concurrent administration of Ir-5 with an anti-PD-L1 antibody provoked a vigorous immune response and suppressed tumor development without damaging the key organs or tissues in murine models (Fig. 5F). In short, transforming cold tumors to hot tumors with metal-based complexes signifies a crucial advancement in immunotherapy, therefore enhancing outcomes for a wider range of cancer patients and facilitating the development of more tailored and effective treatment approaches.
Integrating metal complexes with ICB, particularly mAbs, offers potential for enhanced and more resilient response compared to monotherapy [104,105]. Oxaliplatin, a second-generation Pt metal-based drug, turned out to be as efficient as cisplatin in the treatment of gastric cancers with a better safety profile [106]. Recent research showed that the enhanced therapeutic effect of oxaliplatin depends at least in part on the induction of ICD, which in turn triggers strong immune responses against tumors [107,108]. Preclinical studies showed that oxaliplatin-treated tumors are sensitive to PD-1 immune checkpoint inhibitors, while cisplatin does not induce any such sensitizing effect. This finding appears to be supported by phase Ⅲ clinical trials that specifically target unresectable stomach and gastro-esophageal junction carcinomas [109]. Apart from oxaliplatin, another PtⅡ complex (PT-112, Fig. 6), stimulates ICD in metastatic solid tumors and exhibited therapeutic efficacy. When combined with PD-1 or PD-L1 blockade, Pt-112 triggers a robust immune response that has systemic outreach and limits the growth of untreated, distant lesions [110]. Based on the clinical and preclinical data, the European Medicines Agency and the FDA approved using platinum-based drugs in combination with PD-Ⅰ inhibitors [111].
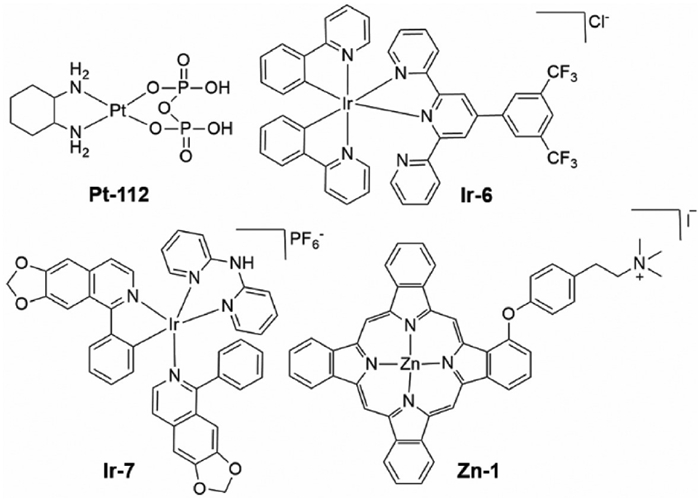
Rong et al. developed an estrogen (ER)-targeted IrⅢ complex, Ir-6 (Fig. 6), that markedly inhibits the growth of melanoma compared to cisplatin [112]. Cellular studies demonstrate that Ir-6 induces ROS production and initiates ICD by simultaneously enhancing the expression of phosphorylated eIF2 α and CHOP proteins, alongside the release of critical ICD markers, such as DAMPs, so facilitating enhanced cellular death. In vivo studies demonstrate that administration of Ir-6 markedly inhibits tumor growth. Histological analysis of Ir-6 treated tumors revealed substantial alterations in tumor tissues, including nuclear fragmentation and cytoplasmic structural disintegration, indicative of tumor cell necrosis or apoptosis. Moreover, the combination of Ir-6 with a PD-1 inhibitor transformed the TME by converting a "cold" tumor into a "hot" one by triggering the generation of proinflammatory cytokines such as interferon gamma and tumor necrosis factor-alpha. An isoquinoline and di(pyridin-2-yl)amine ligand-coordinated cyclometalated IrⅢ complex (Ir-7, Fig. 6), has been synthesized and investigated for its potential cytotoxic effects on triple-negative breast cancer [113]. In vitro studies demonstrated that Ir-7 was more potent than cisplatin and its potential cytotoxicity is associated with the generation of ROS. The complex triggers ER stress, induces ferroptosis, and inhibits the expression of indoleamine 2,3-dioxygenase, an enzyme responsible for the immunosuppressive environment. The complex markedly inhibited tumor development in murine models compared to oxaliplatin and anti-PD1, whereas the combination of Ir-7 and anti-PD1 synergistically enhanced the therapeutic impact.
Zinc phthalocyanines are promising candidates for PDT due to their strong red light absorption, high 1O2 quantum yield, little dark toxicity, and excellent efficacy in generating ROS [114,115]. Zhang, Li, and colleagues developed a cationic zinc phthalocyanine photosensitizer (Zn-1, Fig. 6) that has remarkable anticancer efficacy against both primary and metastatic cancers [116]. The presence of a positive charge on Zn-1 enhances the cooperative effectiveness of photosensitizers. The complex elicited a vigorous antitumor immune response when administered in conjunction with the PD-L1 antibody, resulting in an 89% tumor inhibition rate.
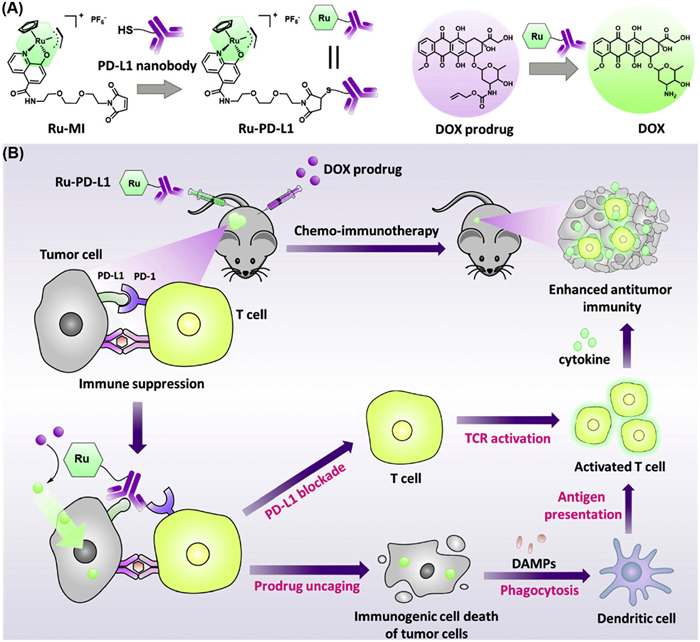
A hybrid strategy has recently been used to develop PD-L1 nanobody−Ru conjugate (Ru-Pd-L1, Fig. 7A) via the thiol maleimide reaction to enhance treatment efficacy while mitigating the adverse immunologic effects of chemotherapeutics [117]. Upon administration, Ru-Pd-L1 triggered the activation of spatially caged doxorubicin prodrug (DPD) via a ruthenium-catalyzed uncaging reaction. This in situ bioorthogonal reaction reduced the toxicity of the established anticancer drug DOX to normal cells. Biochemical analysis revealed that the combination of Ru-PD-L1 and DPD induced a reduction in HMGB1 levels and an elevation in CRT levels, confirming the ICD potential of combinational approach. Furthermore, this combination therapy obstructed the PD1/PDL1 interactions and enhanced TILs in both in vitro and in vivo models (Fig. 7B). The administration of Ru-Pd-L1 and DPD in a mouse cancer model resulted in a substantial reduction in tumor volume. This approach showed promise as a flexible platform for designing precise combination medicines for ICB therapy.
In contrast to conventional chemotherapeutic agents, nanoscale drug carriers such as nanoparticles (NPs) enhance therapeutic effectiveness and mitigate general toxicity [118–120]. This is because of their enhanced permeability and retention effects, high cellular uptake, and their ability to on-demand release of chemotherapeutics at the target tumor site [121]. The effectiveness of immunotherapy is now well acknowledged to be enhanced by the concurrent activation of ICD and suppression of PD-L1 [122,123]. Growing research data demonstrate that encapsulating antibodies and immune checkpoint inhibitors into NP-based delivery systems could boost immunotherapeutic responses while reducing off-target effects [124]. By keeping this in view, Liu et al. synthesized nanoscale coordination polymer NPs that concurrently encapsulate oxaliplatin and 2-bromopalmitic acid (2BP) to target both cancer cells and DCs for chemo-immunotherapy [125]. Cellular studies showed that oxaliplatin/BP encapsulated NPs markedly induce ICD with enhanced production of all DAMPs. Moreover, oxaliplatin/BP NPs treatment not only facilitated the maturation of DCs, but also suppressed PD-L1 expression and enhanced T cell priming in cancer cells, therefore synergistically eliciting chemotherapeutic and immunotherapeutic effects. Further experiments demonstrated that oxaliplatin/BP NPs inhibited tumor progression and metastasis in mice models with colorectal cancer by markedly reducing PD-L1 expression.
The inadequate anti-metastasis therapy and the restricted response rate of immunotherapy in breast cancer make effective treatment a critical concern [126]. Engineered macrophages loaded with a nanomedicine comprising an oxaliplatin prodrug (Oxa(Ⅳ)) and a photosensitizer (Zn phthalocyanine (ZnPc)) have been designed and developed to enhance synergistic effects in both primary and bone metastatic malignancies [127]. Following NIR treatment, the engineering macrophage (Oxa(Ⅳ)@ZnPc@M) undergoes a stimulated release of Oxa(Ⅳ) and ZnPc, which effectively eradicate primary tumors by a synergistic approach of chemotherapy and PDT, while simultaneously triggering ICD. When combined with anti-PD-L1, the engineered macrophages eliminate primary and distinct tumors, stimulate a tumor-specific antitumor immune response, and improve overall survival with little systemic toxicity. Thus, this multifunctional macrophage serves as a therapeutic platform for the successful treatment of primary and bone metastatic cancers.
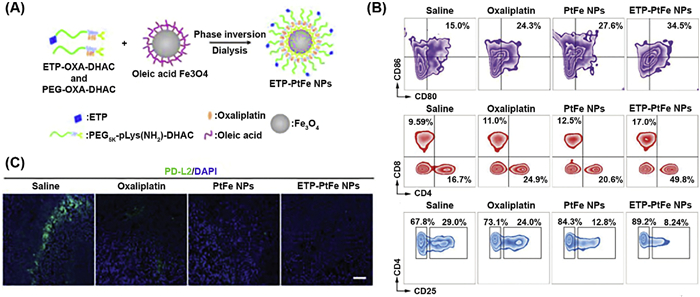
Iron oxide NPs are recognized as potent inducers of ferroptosis and function as adaptable carriers for the administration of ICD [128,129]. Upon destruction in the cellular lysosomes or endosomes, they enable the metabolized ferric/ferrous ions to participate in the intracellular Fenton reaction, leading to an enhanced antitumor immune response [130]. By keeping this in view, Chen et al. developed targeted and non-targeted core-shell magnetic NPs encapsulating the oxaliplatinⅣ prodrug (ETP-PtFe NPs) (Fig. 8A) [131]. Extracellular release studies revealed that in the presence of vitamin C (used to mimic in vivo reductive environment), around 80% of oxaliplatin is released from the NPs during the first 4 h, whereas 25% of ferric ions were liberated after 48 h. Upon cellular accumulation, encapsulated oxaliplatinⅣ prodrug escapes and reduces to its parental PtⅡ complex, which induces DNA lesions and generates ROS. Simultaneously, liberated ferric ions elicit highly toxic ROS species and trigger ER stress. When administrated in murine models, ETP-PtFe NPs significantly altered TME by stimulating DCs and releasing all DAMPs (Fig. 8B). Furthermore, ETP-PtFe NPs, counteracted immunosuppression induced by PD-L2, thereby provoking a robust antitumor immune response (Fig. 8C).
Shen et al. developed polymer NPs incorporating cisplatin (CNPs) to enhance the antitumor efficacy of PD1/PD-L1 inhibitors [132]. In comparison with cisplatin, CNPs generated prolonged PD-L1 overexpression over a period of time, facilitating the shift from low to high PD-L1 expression. The combination of CNPs with PD1/PD-L1 inhibitors resulted in a markedly enhanced tumor inhibition. In addition, when mice received a single administration of CNPs in conjunction with anti-PD-1, the resultant synergistic antitumor impact was much more potent than that seen with a single dosage of cisplatin combined with anti PD-1. The persistent overexpression of tumor PD-L1 underscored the importance of the PD-1/PD-L1 pathway in tumor cell proliferation and the attenuation of CD8+ T cells, enhancing the potential of PD1/PD-L1 inhibitors to obstruct this pathway. This study discovered a prospective therapeutic therapy with CNPs in conjunction with PD1/PD-L1 inhibitors.
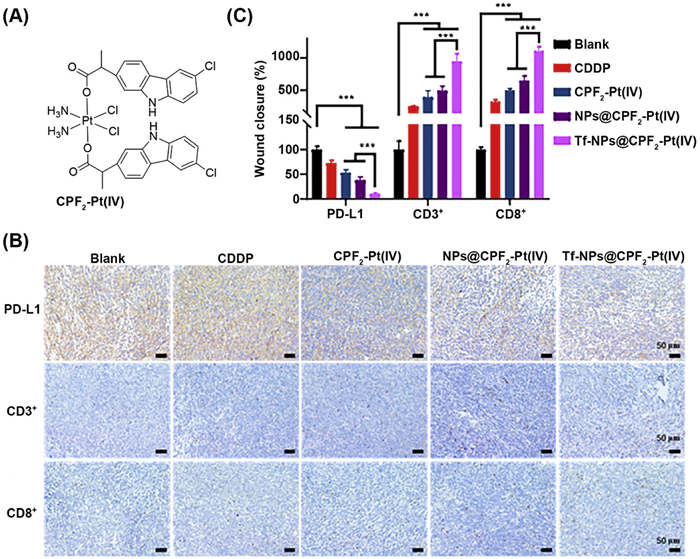
Recently, transferrin modified tumor-targeted NPs (Tf-NPs@CPF2−Pt(Ⅳ)) encapsulating carprofen (CPF2) tethered PtⅣ complex have been developed and investigated for their anticancer and antimetastatic properties (Fig. 9A) [133]. The NPs possess good stability in an aqueous medium and display prolonged release characteristics of CPF2−PtⅣ, which is favorably associated with the robust anticancer efficacy of the nanodrug. Tf-NPs@CPF2−Pt(Ⅳ) promotes apoptosis by concurrently impairing mitochondrial function and causing DNA injury via the upregulation of γ-H2AX and P53 in tumor cells. In murine models, the developed NPs enhance antitumor immunity by elevating the T cell density and inhibiting PD-L1 expression (Figs. 9B and C). Furthermore, compared with free drugs, Tf-NPs@CPF2−Pt(Ⅳ) demonstrated strong antigrowth and antimetastatic efficacy while exhibiting less toxicity, thus presenting considerable promise for treating metastatic malignancies. Likewise, Yuan et al. developed cisplatin-loaded polymeric micelles (CPMs), and assessed their capacity to modify the TME in conjunction with anti PD-L1 [134]. In vivo antitumor assay demonstrated that when CPMs were combined with a higher dose of anti PD-L1, 93.16% tumor growth inhibition rate was achieved. This therapeutic approach enhances the synergy between chemotherapy and immunotherapy, demonstrating significant potential to improve safety and antitumor effectiveness. Recently, a hierarchical pH/enzyme-responsive nanodrug has been developed by concurrently integrating Zn phthalocyanine and PD-L1 antibody to induce an immune-photodynamic synergistic effect [135]. The acidic TME disintegrated the polymer corona, resulting in the release of Zn phthalocyanine and PD-L1. Upon cellular internalization, the NPs strongly generate ROS and induce both apoptotic and necrotic mode of cell death. Intravenous administration in murine models with NIR light treatment promotes ICD and increases T cell density, therefore successfully inhibiting tumor development and demonstrating a synergistic effect.
Cancer immunotherapy harnesses the host's immune system to selectively eradicate malignant cells. Central to cancer immunotherapy is modulating the immune checkpoints such as PD-1 and its ligand PD-L1, which tumor cells exploit to evade immune surveillance. While mAbs inhibited the interaction between PD-1 and PD-L1 and showed remarkable effectiveness in treating certain cancers, but they have limited efficacy against other solid tumors such as glioblastoma likely because of immunosuppressive tumor microenvironment [136–138]. This necessitates developing alternative modalities that disrupt the interaction between PD-1 and PD-L1 and improve treatment outcomes for diverse patient populations. Metal-based compounds offer a significant therapeutic potential of combination therapy using metallodrugs with anti-PD-1/PD-L1 mAbs [139]. This also prompts considerable interest in the synthesis of metallodrugs with immune checkpoint targeting abilities. Specifically, PtⅣ and IrⅢ metal-based compounds have been significantly structurally modified to integrate various functionalities, including tethering or incorporation of biologically active ligands as well as their site-specific activation. In this regard, the most prevalent drugs tethered with metal centers are NSAIDs, which facilitate the inhibition of COX-2. COX-2 inhibition decreases the synthesis of prostaglandin E2, a crucial mediator that activates multiple oncogenic signaling pathways, which are involved in the elevation of PD-L1 expression. Furthermore, COX-2 inhibition can alter the tumor immune microenvironment by decreasing the presence of immunosuppressive cells, including regulatory T cells and myeloid-derived suppressor cells, thereby further diminishing PD-L1 expression. It is intriguing to note that such immunotherapeutic effects of NSAIDs were restricted when administered alone or used as a mixture with metal-based compounds, hence affirming the significance and benefit of linking with metal centers that improve the delivery and targeting of these bioactive compounds to the tumor site.
Additionally, newly developed IrⅢ complexes efficiently stimulated immune cell infiltration and PD-L1 expression, transforming cold tumors into hot ones, thus overcoming the resistance and enhancing the efficacy of mAbs. This strategy can potentially improve the effectiveness of cancer therapies across a broader range of patients. Besides, engineered NPs by incorporating metallodrugs with mAbs provided a robust and multipurpose drug delivery platform, improving the therapeutic effectiveness against primary and distinct tumors.
While the initial findings of metal-based PD-1/PD-L1 inhibitors are encouraging, however the field is in its infancy. Several unsolved challenges exist for improving ICB treatment in clinics. For instance, the inherent toxicity of metal complexes may result in off-target effects, damage to healthy tissues, and organ malfunction. Moreover, adjusting dosing regimens is challenging due to the intricate pharmacokinetics of metal complexes, necessitating a meticulous balance to attain therapeutic efficacy while avoiding side effects. Secondly, tumor heterogeneity hinders the efficacy of metal-based compounds in precisely targeting immune checkpoints due to the varied genetic and phenotypic attributes within the tumor. This is associated with the variations in immune cell infiltration and PD-1/PD-L1 expression which can lead to differing responses to treatment, with some tumor subpopulations remaining resistant to immune modulation. This variability reduces the consistency of outcomes and makes it challenging to develop novel compounds that can effectively target immune checkpoints across all cancer types and stages. Another major challenge is that the contact interfaces between PD-1 and PD-L1 proteins are notably flat and hydrophobic, leading to limited binding sites for metal-based compounds [140,141]. Therefore, comprehensive structural optimizations and an extensive library of metal-based compounds are necessary to overcome these challenges and to establish a structure-activity relationship.
Despite the limitation in structural analysis of metallotherapeutics that target PD-1/PD-L1, examining the structures of organic drugs currently in clinical trials can yield significant insights and establish a basis for the development of novel ligands that, once bound with metal centers, enhance the overall efficacy and safety of the metal complex. Additionally, a computationally driven methodology for designing these compounds may provide significant insights and improve the effectiveness of metal complexes. This is also coupled with the need for comprehensive research on preclinical animal models that adequately represent human physiology.
Moreover, the benefits of NPs-based delivery systems for ICB therapy are undoubtedly obvious; for example, Kim et al. recently engineered ferritin nanocages encapsulated with PD-L1 blocking peptide, which facilitates efficient tumor targeting and blood-brain barrier penetration [142]. Mechanistic investigations demonstrated that these nanocages efficiently interrupted PD-1/PD-L1 interactions and reinstated T-cell activation. This study underscores the significance of nanomaterials in ICB therapy, illustrating their capacity to improve delivery and circumvent immune evasion. However, further research is required on the biomarkers and intrinsic biology of tumors to optimize the targeted administration of these NPs. Moreover, the consistent and reproducible fabrication techniques for NPs are essential for scaling up production and ensuring batch consistency, which remains a significant barrier to their clinical translation [143]. In addition, rigorous preclinical and clinical studies are required to understand the safety profiles of these metallodrugs and their nanoformulations.
In summary, the studies reviewed here suggest that metal-based complexes have the potential to complement and even surpass existing antibody therapies for ICB. The versatility of metal-based agents, their unique cytotoxic mechanisms, and their ability to modulate immune responses through ICD or checkpoint blockade opens up new avenues for cancer treatment. This review aims to inspire future investigations and development of novel strategies that integrate metallodrugs for ICB in cancer treatment.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Muhammad Nafees: Writing – original draft, Conceptualization. Muhammad Hanif: Writing – review & editing. Piaoping Yang: Writing – review & editing.
This work was supported by the Fundamental Research Funds for the Central Universities and the National Natural Science Foundation of China (No. U22A20347). We would like to thank the Health Research Council of New Zealand for a Sir Charles Hercus Fellowship (M.H.).
P. Mahalingam, T. Newsom-Davis, Clin. Med. 23 (2023) 56–60. doi: 10.7861/clinmed.2022-0589
M. Darvishi, F. Tosan, P. Nakhaei, et al., Pathol. Res. Pract. 241 (2023) 154241. doi: 10.1016/j.prp.2022.154241
P. Dobosz, T. Dzieciątkowski, Front. Immunol. 10 (2019) 2965. doi: 10.3389/fimmu.2019.02965
Y. Zhang, Z. Zhang, Cell. Mol. Immunol. 17 (2020) 807–821. doi: 10.1038/s41423-020-0488-6
X. Meng, Y. Lei, X. Zhang, et al., Appl. Mater. Today 24 (2021) 101149. doi: 10.1016/j.apmt.2021.101149
S. Du, J. Yan, Y. Xue, et al., Exploration 3 (2023) 20210058. doi: 10.1002/EXP.20210058
E. Leon, R. Ranganathan, B. Savoldo, Semin. Immunol. 49 (2020) 101437. doi: 10.1016/j.smim.2020.101437
A.D. Waldman, J.M. Fritz, M.J. Lenardo, Nat. Rev. Immunol. 20 (2020) 651–668. doi: 10.1038/s41577-020-0306-5
M.J. Lin, J. Svensson-Arvelund, G.S. Lubitz, et al., Nat. Cancer 3 (2022) 911–926. doi: 10.1038/s43018-022-00418-6
Y. Igarashi, T. Sasada, J. Immunol. Res. 2020 (2020) 5825401. doi: 10.1155/2020/5825401
T. Fan, M. Zhang, J. Yang, et al., Sig. Transduct. Target. Ther. 8 (2023) 450. doi: 10.1038/s41392-023-01674-3
J. Wang, J. Ma, F. Xie, et al., Front. Immunol. 15 (2024) 1389173. doi: 10.3389/fimmu.2024.1389173
Z. Fu, S. Li, S. Han, et al., Sig. Transduct. Target. Ther. 7 (2022) 93. doi: 10.1038/s41392-022-00947-7
K. Tsuchikama, Y. Anami, S.Y.Y. Ha, et al., Nat. Rev. Clin. Oncol. 21 (2024) 203–223. doi: 10.1038/s41571-023-00850-2
R. Ma, Z. Li, E.A. Chiocca, et al., Trends Cancer 9 (2023) 122–139. doi: 10.1016/j.trecan.2022.10.003
Y. Chen, X. Chen, W. Bao, et al., Nat. Biotechnol. 42 (2024) 1876–1887. doi: 10.1038/s41587-023-02118-7
D. Lin, Y. Shen, T. Liang, Sig. Transduct. Target. Ther. 8 (2023) 156. doi: 10.1038/s41392-023-01407-6
X. Li, Z. Cheng, Adv. Ther. 7 (2024) 2300445. doi: 10.1002/adtp.202300445
D. Ding, X. Jiang, Small Methods 7 (2023) 2300354. doi: 10.1002/smtd.202300354
L. Galluzzi, E. Guilbaud, D. Schmidt, et al., Nat. Rev. Drug Discov. 23 (2024) 445–460. doi: 10.1038/s41573-024-00920-9
X. Chang, M. Bian, L. Liu, et al., Pharmacol. Res. 187 (2023) 106556. doi: 10.1016/j.phrs.2022.106556
K. Arimoto, S. Miyauchi, M. Liu, et al., Front. Immunol. 15 (2024) 1390263. doi: 10.3389/fimmu.2024.1390263
Z. Zhou, Y. Mai, G. Zhang, et al., Cancer Lett. 598 (2024) 217079. doi: 10.1016/j.canlet.2024.217079
F. Radogna, M. Diederich, Biochem. Pharmacol. 153 (2018) 12–23. doi: 10.1016/j.bcp.2018.02.006
J. Fucikova, O. Kepp, L. Kasikova, et al., Cell Death Dis. 11 (2020) 1013. doi: 10.1038/s41419-020-03221-2
L. Zhu, Y. Wu, H. Zhao, et al., Sci. Rep. 14 (2024) 2025. doi: 10.1038/s41598-024-52353-4
P. Sharma, S. Goswami, D. Raychaudhuri, et al., Cell 186 (2023) 1652–1669. doi: 10.1016/j.cell.2023.03.006
A.C. Huang, R. Zappasodi, Nat. Immunol. 23 (2022) 660–670. doi: 10.1038/s41590-022-01141-1
T. Wieder, T. Eigentler, E. Brenner, et al., J. Allergy Clin. Immunol. 142 (2018) 1403–1414. doi: 10.1016/j.jaci.2018.02.042
A.J. Korman, S.C. Garrett-Thomson, N. Lonberg, Nat. Rev. Drug Discov. 21 (2022) 509–528. doi: 10.1038/s41573-021-00345-8
M.M. Hossen, Y. Ma, Z. Yin, et al., Front. Immunol. 14 (2023) 1198365. doi: 10.3389/fimmu.2023.1198365
M. Gao, J. Shi, X. Xiao, et al., Cancer Lett. 588 (2024) 216726. doi: 10.1016/j.canlet.2024.216726
C. Donini, F. Galvagno, R. Rotolo, et al., Br. J. Cancer 129 (2023) 1409–1416. doi: 10.1038/s41416-023-02363-2
A.V.R. Kornepati, R.K. Vadlamudi, T.J. Curiel, Nat. Rev. Cancer 22 (2022) 174–189. doi: 10.1038/s41568-021-00431-4
S.S. Kirthiga Devi, S. Singh, R. Joga, et al., Eur. J. Pharm. Biopharm. 200 (2024) 114323. doi: 10.1016/j.ejpb.2024.114323
C. Bailly, G. Vergoten, Biochem. Pharmacol. 178 (2020) 114042. doi: 10.1016/j.bcp.2020.114042
P. Jiao, Q. Geng, P. Jin, et al., Curr. Pharm. Des. 24 (2018) 4911–4920.
P. Torka, T. Feldman, K. Savage, et al., Hematol. Oncol. 41 (2023) 161–162. doi: 10.1002/hon.3163_107
Y. Jiang, M. Chen, H. Nie, et al., Hum. Vaccin. Immunother. 15 (2019) 1111–1122. doi: 10.1080/21645515.2019.1571892
J. Bai, P. Liang, Q. Li, et al., Recent Pat. Anticancer Drug Discov. 16 (2021) 239–248. doi: 10.2174/1574892816666210212145107
Q. Wu, L. Jiang, S.C. Li, et al., Acta Pharmacol. Sin. 42 (2021) 1–9.
P. Ouyang, L. Wang, J. Wu, et al., Front. Immunol. 15 (2024) 1344272. doi: 10.3389/fimmu.2024.1344272
E. Cruz, V. Kayser, Biologics 13 (2019) 33–51. doi: 10.2147/btt.s166310
M. de Miguel, E. Calvo, Cancer Cell 38 (2020) 326–333. doi: 10.1016/j.ccell.2020.07.004
P. Dobosz, M. Stępień, A. Golke, et al., Int. J. Mol. Sci. 23 (2022) 2847. doi: 10.3390/ijms23052847
N. Muhammad, M. Hanif, P. Yang, Coord. Chem. Rev. 499 (2024) 215507. doi: 10.1016/j.ccr.2023.215507
X. Wang, X. Wang, S. Jin, et al., Chem. Rev. 119 (2019) 1138–1192. doi: 10.1021/acs.chemrev.8b00209
C. Yu, Z. Wang, Z. Sun, et al., J. Med. Chem. 63 (2020) 13397–13412. doi: 10.1021/acs.jmedchem.0c00950
W. Song, L. Shen, Y. Wang, et al., Nat. Commun. 9 (2018) 2237. doi: 10.1038/s41467-018-04605-x
M. Yi, X. Zheng, M. Niu, et al., Mol. Cancer 21 (2022) 28. doi: 10.1186/s12943-021-01489-2
L. Fournel, Z. Wu, N. Stadler, et al., Cancer Lett. 464 (2019) 5–14. doi: 10.1016/j.canlet.2019.08.005
L. Zhang, N. Montesdeoca, J. Karges, et al., Angew. Chem. Int. Ed. 62 (2023) e202300662. doi: 10.1002/anie.202300662
S. Sen, M. Won, M.S. Levine, et al., Chem. Soc. Rev. 51 (2022) 1212–1233. doi: 10.1039/d1cs00417d
R.N. Wang, Q. Yu, X.B. Wang, et al., Acta Pharmacol. Sin. 44 (2023) 2103–2112. doi: 10.1038/s41401-023-01092-9
S.R. Lord, A.L. Harris, Br. J. Cancer 128 (2023) 958–966. doi: 10.1038/s41416-023-02204-2
L. Zhu, K. Yang, Z. Ren, et al., Transl. Oncol. 44 (2024) 101945. doi: 10.1016/j.tranon.2024.101945
M. Foretz, B. Guigas, B. Viollet, Nat. Rev. Endocrinol. 19 (2023) 460–476. doi: 10.1038/s41574-023-00833-4
Z. Wei, X. Zhang, T. Yong, et al., Nat. Commun. 12 (2021) 440. doi: 10.1038/s41467-020-20723-x
B. Liu, B.B. Liang, W.D. Cao, et al., Angew. Chem. Intl. Ed. 63 (2024) e202410586. doi: 10.1002/anie.202410586
K. Peng, B.B. Liang, W. Liu, et al., Coord. Chem. Rev. 449 (2021) 214210. doi: 10.1016/j.ccr.2021.214210
S. Jin, N. Muhammad, Y. Sun, et al., Angew. Chem. Int. Ed. 59 (2020) 23313–23321. doi: 10.1002/anie.202011273
Z. Li, Q. Wang, L. Li, et al., J. Med. Chem. 64 (2021) 17920–17935. doi: 10.1021/acs.jmedchem.1c01236
Z. Li, L. Li, W. Zhao, et al., Dalton Trans. 51 (2022) 12604–12619. doi: 10.1039/d2dt00944g
H. Shao, X. He, L. Zhang, et al., Front. Pharmacol. 12 (2021) 761722. doi: 10.3389/fphar.2021.761722
Y. Bian, L. Yang, W. Sheng, et al., Ann. Palliat. Med. 10 (2021) 1578–1588. doi: 10.21037/apm-20-288
Y. Chen, L. Li, Z. Liu, et al., Dalton Trans. 52 (2023) 13097–13109. doi: 10.1039/d3dt02358c
L. Feng, G. Wang, Y. Chen, et al., Med. Res. Rev. 42 (2022) 710–743. doi: 10.1002/med.21859
P. Filippakopoulos, J. Qi, S. Picaud, et al., Nature 468 (2010) 1067–1073. doi: 10.1038/nature09504
R. Fan, A. Deng, B. Qi, et al., J. Med. Chem. 66 (2023) 875–889. doi: 10.1021/acs.jmedchem.2c01719
I. Fritz, P. Wagner, H. Olsson, Transl. Oncol. 14 (2021) 101029. doi: 10.1016/j.tranon.2021.101029
X. Lin, J. Zhang, X. Wang, et al., Anticancer Drugs 31 (2020) 989–996. doi: 10.1097/cad.0000000000000972
M. Zhang, Y. Chen, Z. Liu, et al., J. Med. Chem. 67 (2024) 2031–2048. doi: 10.1021/acs.jmedchem.3c01845
J. Debnath, N. Gammoh, K.M. Ryan, Nat. Rev. Mol. Cell Biol. 24 (2023) 560–575. doi: 10.1038/s41580-023-00585-z
M. Mauthe, I. Orhon, C. Rocchi, et al., Autophagy 14 (2018) 1435–1455. doi: 10.1080/15548627.2018.1474314
M. Zhang, L. Li, S. Li, et al., J. Med. Chem. 66 (2023) 3393–3410. doi: 10.1021/acs.jmedchem.2c01895
L. Li, Y. Chen, M. Zhang, et al., Dalton Trans. 53 (2024) 13890–13905. doi: 10.1039/d4dt01794c
S. Li, S. Feng, Y. Chen, et al., J. Inorg. Biochem. 260 (2024) 112696. doi: 10.1016/j.jinorgbio.2024.112696
B. Xu, X. Jiang, J. Xiong, et al., J. Med. Chem. 63 (2020) 5841–5855. doi: 10.1021/acs.jmedchem.0c00088
Y. Ma, Q.Q. Yang, D.M. Gu, et al., Tissue Barriers 12 (2024) 2256641. doi: 10.1080/21688370.2023.2256641
Y. Chen, M. Zhang, Y. He, et al., J. Med. Chem. 67 (2024) 12868–12886. doi: 10.1021/acs.jmedchem.4c00843
X. Lin, X. Lu, G. Luo, et al., Eur. J. Med. Chem. 186 (2020) 111876. doi: 10.1016/j.ejmech.2019.111876
J.F. Goossens, C. Bailly, Pharmacol. Ther. 203 (2019) 107396. doi: 10.1016/j.pharmthera.2019.107396
J. Lee, E.M. Hong, J.H. Kim, et al., Oncol. Lett. 24 (2022) 448. doi: 10.3892/ol.2022.13568
Y. Chen, M. Zhang, Z. Liu, et al., J. Med. Chem. 67 (2024) 17551–17567. doi: 10.1021/acs.jmedchem.4c01549
T.C. Pham, V.N. Nguyen, Y. Choi, et al., Chem. Rev. 121 (2021) 13454–13619. doi: 10.1021/acs.chemrev.1c00381
Y. Chen, C. Liang, M. Kou, et al., Dalton Trans. 53 (2024) 11836–11849. doi: 10.1039/d4dt01345j
K. Xie, X.Y. Lu, H. Zhu, et al., Dalton Trans. 53 (2024) 8772–8780. doi: 10.1039/d4dt00575a
A. Casini, R.W. Sun, I. Ott, Met. Ions Life Sci. 18 (2018) 199–217.
G. Moreno-Alcántar, P. Picchetti, A. Casini, Angew. Chem. Int. Ed. 62 (2023) e202218000. doi: 10.1002/anie.202218000
Z. Xu, Q. Lu, M. Shan, et al., J. Med. Chem. 66 (2023) 7813–7833. doi: 10.1021/acs.jmedchem.3c00063
B.B. Zmejkovski, N. Đ. Pantelić, G.N. Kaluđerović, Inorg. Chim. Acta 534 (2022) 120797. doi: 10.1016/j.ica.2022.120797
R. Ouyang, S. Wang, K. Feng, et al., J. Inorg. Biochem. 244 (2023) 112205. doi: 10.1016/j.jinorgbio.2023.112205
W. Li, S. Li, Z. Zhang, et al., J. Med. Chem. 66 (2023) 8564–8579. doi: 10.1021/acs.jmedchem.3c00248
N.E. Donlon, R. Power, C. Hayes, et al., Cancer Lett. 502 (2021) 84–96. doi: 10.1016/j.canlet.2020.12.045
H. Yamaguchi, J.M. Hsu, W.H. Yang, et al., Nat. Rev. Clin. Oncol. 19 (2022) 287–305. doi: 10.1038/s41571-022-00601-9
P. Bonaventura, T. Shekarian, V. Alcazer, et al., Front. Immunol. 10 (2019) 168. doi: 10.3389/fimmu.2019.00168
D. Liu, Y. Li, H. Zhang, et al., Chin. Chem. Lett. 36 (2025) 109910. doi: 10.1016/j.cclet.2024.109910
Y. Li, X. Zhang, S. Liu, et al., Chin. Chem. Lett. 36 (2025) 110501. doi: 10.1016/j.cclet.2024.110501
L. Jia, Y. Hong, X. He, et al., Chin. Chem. Lett. 36 (2025) 109957. doi: 10.1016/j.cclet.2024.109957
J.Y. Zhou, Q.H. Shen, X.J. Hong, et al., Chem. Eng. J. 474 (2023) 145516. doi: 10.1016/j.cej.2023.145516
D.B. Nandini, R.S. Rao, J. Hosmani, et al., Dis. Mon. 66 (2020) 101036. doi: 10.1016/j.disamonth.2020.101036
I. Mohamad, M.D.E. Glaun, K. Prabhash, et al., Am. Soc. Clin. Oncol. Educ. Book 43 (2023) e389810.
D. Deng, M. Wang, Y. Su, et al., J. Med. Chem. 67 (2024) 6810–6821. doi: 10.1021/acs.jmedchem.4c00404
A.C. Jung, F. Moinard-Butot, C. Thibaudeau, et al., Pharmaceutics 13 (2021) 1788. doi: 10.3390/pharmaceutics13111788
Y. Shao, B. Liu, Z. Di, et al., J. Am. Chem. Soc. 142 (2020) 3939–3946. doi: 10.1021/jacs.9b12788
F. Zhang, Y. Zhang, Z. Jia, et al., J. Cancer 10 (2019) 1923–1929. doi: 10.7150/jca.28896
F. Montagnani, G. Turrisi, C. Marinozzi, et al., Gastric Cancer 14 (2011) 50–55. doi: 10.1007/s10120-011-0007-7
X. Wang, S. Li, T. Xie, et al., J. Clin. Oncol. 38 (2020) 280-280.
F. Sun, L. Cui, T. Li, et al., J. Recept. Sig. Transd. 39 (2019) 208–214. doi: 10.1080/10799893.2019.1655050
T. Yamazaki, A. Buqué, T.D. Ames, et al., Oncoimmunology 9 (2020) 1721810. doi: 10.1080/2162402X.2020.1721810
P. Liu, J. Chen, L. Zhao, et al., OncoImmunol. 11 (2022) 2093518. doi: 10.1080/2162402X.2022.2093518
Y. Rong, Z. Fan, Z. Yu, et al., Inorg. Chem. Front. 10 (2023) 5278–5291. doi: 10.1039/d3qi00841j
Y. Lu, S.S. Wang, M.Y. Li, et al., Acta Pharm. Sin. B 15 (2025) 424–437. doi: 10.1016/j.apsb.2024.06.017
M. Pellei, F. Del Bello, M. Porchia, et al., Coord. Chem. Rev. 445 (2021) 214088. doi: 10.1016/j.ccr.2021.214088
L.P. Roguin, N. Chiarante, M.C. García Vior, et al., Int. J. Biochem. Cell Biol. 114 (2019) 105575. doi: 10.1016/j.biocel.2019.105575
B.Y. Zheng, S.Z. Li, B.D. Zheng, et al., Dyes Pigm. 185 (2021) 108907. doi: 10.1016/j.dyepig.2020.108907
Z. Zhao, X. Wang, J. Wang, et al., J. Med. Chem. 66 (2023) 11951–11964. doi: 10.1021/acs.jmedchem.3c00557
M.J. Mitchell, M.M. Billingsley, R.M. Haley, et al., Nat. Rev. Drug Discov. 20 (2021) 101–124. doi: 10.1038/s41573-020-0090-8
V. Jadhav, A. Roy, K. Kaur, et al., Nano-Struct. Nano-Objects 37 (2024) 101103. doi: 10.1016/j.nanoso.2024.101103
Y. Du, X. Zhao, F. He, et al., Adv. Mater. 36 (2024) 2403253. doi: 10.1002/adma.202403253
J. Zhang, Y. Lin, Z. Lin, et al., Adv. Sci. 9 (2022) 2103444. doi: 10.1002/advs.202103444
J. Li, J. Dai, Z. Zhuang, et al., Biomaterials 291 (2022) 121899. doi: 10.1016/j.biomaterials.2022.121899
H. Xiao, X. Li, S. Liang, et al., ACS Nano 18 (2024) 11070–11083. doi: 10.1021/acsnano.3c11464
R. Toy, K. Roy, Bioeng. Transl. Med. 1 (2016) 47–62. doi: 10.1002/btm2.10005
J. Liu, X. Jiang, Y. Li, et al., ACS Nano 18 (2024) 5152–5166. doi: 10.1021/acsnano.3c12678
J. Yu, Q. Mu, M. Fung, et al., Pharmacol. Ther. 236 (2022) 108108. doi: 10.1016/j.pharmthera.2022.108108
Y. Huang, Z. Guan, X. Dai, et al., Nat. Commun. 12 (2021) 4310. doi: 10.1038/s41467-021-24564-0
C. Luo, X. Li, H. Yan, et al., Sci. Rep. 14 (2024) 31383. doi: 10.1038/s41598-024-82917-3
S. Zhan, Z. Cao, J. Li, et al., Bioconjug. Chem. 36 (2025) 80–91. doi: 10.1021/acs.bioconjchem.4c00488
J. Yang, J. Zhu, B. Ren, et al., Chem. Eng. J. 497 (2024) 154470. doi: 10.1016/j.cej.2024.154470
Q. Chen, L. Liu, Y. Lu, et al., Adv. Sci. 6 (2019) 1802134. doi: 10.1002/advs.201802134
N. Shen, C. Yang, X. Zhang, et al., Acta Biomater. 135 (2021) 543–555. doi: 10.1016/j.actbio.2021.08.013
M. Zhang, Y. Chen, S. Feng, et al., J. Med. Chem. 67 (2024) 16416–16434. doi: 10.1021/acs.jmedchem.4c01265
H. Yuan, Y. Xue, C. Guo, et al., Int. J. Pharm. 622 (2022) 121860. doi: 10.1016/j.ijpharm.2022.121860
Z. Su, Z. Xiao, J. Huang, et al., ACS Appl. Mater. Interfaces 13 (2021) 12845–12856. doi: 10.1021/acsami.0c20422
M. Mandalà, C. Tondini, B. Merelli, et al., Am. J. Clin. Dermatol. 18 (2017) 597–611. doi: 10.1007/s40257-017-0282-0
F. Martins, L. Sofiya, G.P. Sykiotis, et al., Nat. Rev. Clin. Oncol. 16 (2019) 563–580. doi: 10.1038/s41571-019-0218-0
W. Wu, J.L. Klockow, M. Zhang, et al., Pharmacol. Res. 171 (2021) 105780. doi: 10.1016/j.phrs.2021.105780
L. Paz-Ares, M. Dvorkin, Y. Chen, et al., Lancet 394 (2019) 1929–1939. doi: 10.1016/S0140-6736(19)32222-6
K.M. Zak, P. Grudnik, K. Magiera, et al., Structure 25 (2017) 1163–1174. doi: 10.1016/j.str.2017.06.011
R. Kitel, I. Rodríguez, X. del Corte, et al., ACS Chem. Biol. 17 (2022) 2655–2663. doi: 10.1021/acschembio.2c00583
M. Kim, H.J. Yoon, C. Lee, et al., ACS Biomater. Sci. Eng. 10 (2024) 575–587. doi: 10.1021/acsbiomaterials.3c01200
Z. Zhou, H. Wang, J. Li, et al., Int. J. Biol. Macromol. 254 (2024) 127911. doi: 10.1016/j.ijbiomac.2023.127911

Figure 1 The current treatment modalities of cancer immunotherapy and their associated sub-classification.

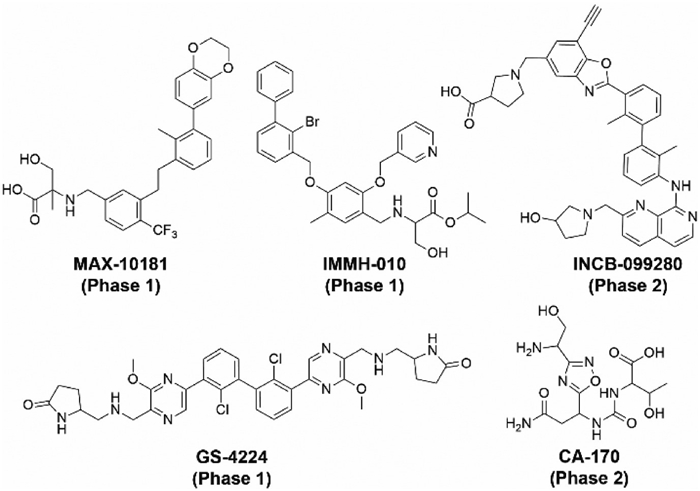
Figure 2 Small organic molecules as immune checkpoint inhibitors currently under clinical trials.

Figure 3 Examples of metal complexes that directly target PD-1/PD-L1 immune checkpoints for ICB therapy. Cisplatin, carboplatin, and oxaliplatin are the clinically approved Pt-based drugs. Novel metal complex Pt-1 decreases PD-1 expression, whereas the Pt-2 complex, which comprises metformin, inhibited PD-L1 expression. Other examples of metal-based compounds contain either anti-inflammatory drugs (complexes Pt-3 to Pt-5, Ir-1 to Ir-3, and Au-1) or biologically active ligands (complexes Pt-6 to Pt-13) that function synergistically with the metal complex to suppress PD-L1 expression and consequently improve the therapeutic response.

Figure 4 (A) Crystal structure of Pd complex. (B) The interaction between Pd complex and HSA was analyzed using a protein−ligand interaction profiler. (C) Structure of Pd-HSA complex. (D) Western blot images of different proteins after treatment with Pd complex and Pd-HSA. (E) PD-1 and PD-L1 protein expression in tumor tissues after treatment. Reproduced with permission [93]. Copyright 2023, American Chemical Society.

Figure 5 (A) Chemical structures of Ir-4 and Ir-5. (B) Mechanistic insights into the Ir-4 mediated PDT treatment which activates multiple cell death pathways and stimulate the immune response. (C, D) Confocal laser scanning microscopy and Western blot images show alterations in PD-L1, NF-k β, and ATF3 expressions after treatment with Ir-5 at designated concentrations. (E) Flow cytometry accompanied by statistical analysis of tumor tissues demonstrating changes in PD-L1 expression after Ir-5 treatment. (F) Time-dependent tumor growth curves of tumor-bearing mice after treatment. Reproduced with permission [100]. Copyright 2023, Elsevier Ltd. Reproduced with permission [103]. Copyright 2024, American Chemical Society.

Figure 6 Structure of metal complexes used in combination therapy to target PD-1/PD-L1 immune checkpoints.

Figure 7 (A) Schematic structure of Ru-Pd-L1 nanoconjugate and the doxorubicin prodrug (DPD) for combinational therapy. (B) Mechanistic insights into the combinational approach involving Ru-PD-L1 and DPD which alleviates the immunosuppressive TME and induces ICD and PD-L1 inhibition, culminating in synergistic T-cell activation and improved antitumor immunity. Reproduced with permission [117]. Copyright 2024, American Chemical Society.

Figure 8 (A) Schematic representation of the synthesis of ETP-PtFe NPs. (B) Illustrative graphs of flow cytometry analyses of DCs and T lymphocytes in treated tumors. (C) Representative image of immunofluorescence labelling of the tumor sections demonstrating reduced PD-L2 expression after treatment. Reproduced with permission [131]. Copyright 2019, Wiley-VCH.

Figure 9 (A) Chemical structure of CPF2−PtⅣ complex. (B, C) Immunohistochemical analysis shows alteration in PD-L1 and TILs in tumor tissue after treatment with mentioned formulations. Reproduced with permission [133]. Copyright 2024, American Chemical Society.
Table 1.
Metal complexes under clinical trials in conjunction with PD-1/PD-L1 antibodies. Data taken from
| PD-1/PD-L1 antibody | Chemotherapeutics | Clinical phase and status | Tumor stage and type | Clinical trial |
| Atezolizumab | Carboplatin and nab-paclitaxel | Phase 3, completed | Stage Ⅳ, non-squamous NSCLC | NCT02367781 |
| Atezolizumab | Carboplatin and etoposide | Phase 1/3, completed | Extensive-stage, NSCLC | NCT02763579 |
| Atezolizumab | Bevacizumab plus carboplatin and paclitaxel | Phase 3, completed | Stage Ⅳ, non-squamous NSCLC | NCT02366143 |
| Camrelizumab | Carboplatin and pemetrexed | Phase 3, completed | Stage ⅢB/Ⅳ non-squamous NSCLC | NCT03134872 |
| Durvalumab | Etoposide and cisplatin/carboplatin | Phase 3, active | Non-squamous NSCLC | NCT03043872 |
| Nivolumab | Oxaliplatin Plus fluoropyrimidine | Phase 3, completed | Metastatic gastric or gastro-esophageal (GE) junction cancer | NCT02872116 |
| Pembrolizumab | Carboplatin and pemetrexed | Phase 1/2, completed | Metastatic non-squamous NSCLC | NCT02039674 |
| Pembrolizumab | Carboplatin and paclitaxel/nab-paclitaxel | Phase 3, completed | Metastatic squamous NSCLC | NCT02775435 |
| Pembrolizumab | Cisplatin and 5-fluorouracil | Phase 3, completed | Metastatic esophageal or GE junction cancer | NCT03189719 |
| Pembrolizumab | Trastuzumab plus cisplatin + 5-fluorouracil or oxaliplatin + capecitabine | Phase 3, active | HER2 + advanced gastric or GE junction adenocarcinoma | NCT03615326 |
| Sintilimab | Platinum complex and pemetrexed | Phase 3, completed | Metastatic non-squamous NSCLC | NCT03607539 |
| Sintilimab | Platinum complex and gemcitabine | Phase 3, completed | Metastatic squamous NSCLC | NCT03629925 |
| Tislelizumab | Platinum complex and pemetrexed | Phase 3, completed | Metastatic squamous NSCLC | NCT03663205 |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: