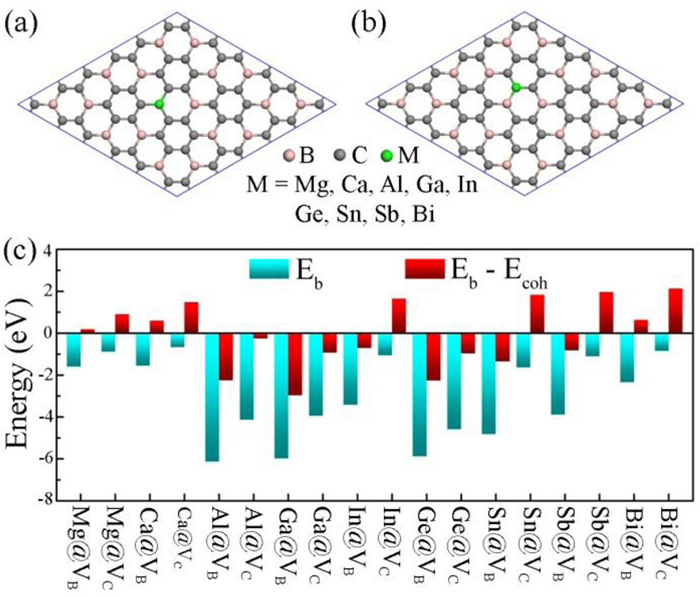
Figure 1.
Top view of (a) M@VB and (b) M@VC systems. (c) Computed binding energies (Eb) of main-group metal-embedded BC3 monolayer and the energy difference between Eb and cohesive energies (Ecoh) of main-group metals.
Unveiling the electrocatalytic potential of main-group metal-embedded BC3 monolayer for highly efficient NO reduction to NH3
Jiajun Wang , Chen Sun , Li Sheng , Zhiwen Zhuo , Shujuan Li , Jiayao Wang , Weiyi Wang , Jinbo Sun , Juqian Yang , Ke Xu , Shulai Lei

NH3 holds significant importance as a vital chemical compound within the industrial sector, playing a crucial role in modern economic development [1,2]. The electrochemical reduction of N2 (NRR) with minimal emissions of CO2 stands as a sustainable and environmentally friendly approach for N2 fixation under ambient conditions, facilitating the revolution for NH3 synthesis [3-7]. However, NRR encounters obstacles attributed to the concurrent hydrogen evolution reaction (HER) and the formidable N≡N bond [8]. In contrast, electrochemical reduction of NO to NH3 (NORR) offers a more energy-efficient and practical pathway for sustainable NH3 synthesis [9-14]. Additionally, NO itself serves as a prevalent atmospheric pollutant generated by vehicle exhaust and incomplete combustion of fossil fuels. NORR emerges as an efficient and eco-friendly technique for simultaneous NO treatment and NH3 electro-synthesis. Nevertheless, NORR presents a multi-step process involving intricate five-electron mechanism, resulting in slow reaction kinetics [15-17]. Consequently, the exploration of highly performant catalysts becomes imperative to enhance the NO-to-NH3 conversion efficiency.
Single-atom catalysts (SACs) have garnered increasing attention in heterogeneous catalysis, owing to the exceptional potential in delivering high activity and selectivity for a diverse range of chemical reactions [18-26]. To date, a variety of transition metal-based SACs are developed as NORR electrocatalysts, attributed to the ability of transition metal to augment NO adsorption and activation facilitated by the partially occupied d-orbitals [27-35]. Nevertheless, it has been noted that these d-orbitals also promote the binding of *H on catalyst surfaces, subsequently promoting the undesired HER and thus impeding NH3 synthesis [36]. Consequently, the exploration of alternative elements as active sites for SACs, beyond transition metal atoms, remains a topic of ongoing research. Promisingly, main-group metals (Al, Ga, In, Sn, Sb, and Bi, etc.) with partially occupied p-orbitals exhibit robust ability in restraining adsorption of *H, and can overcome the shortcoming of transition metal-based SACs [37-41]. Hence, main-group metals are expected to serve as promising SACs for NORR [42-45]. For instance, Chen et al. [42] experimentally reported that Sb single atoms confined in MoO3 were highly efficient for the NORR with the NH3 yield rate of 273.5 µmol h−1 cm−2 and Faradaic efficiency of 91.7%. Wu et al. [45] computationally predicted that single Mg/Al/Ga atom supported on N-doped graphene can act as active centers for direct NO-to-NH3 conversion. Despite the great potential, main-group metal-based SACs for NORR have not been explored extensively, and it is meaningful to extend the scope both from theoretical and experimental aspects.
Recently, graphene-like boron carbide (BC3) monolayer, a semiconductor with a bandgap spanning between 0.46 eV and 0.73 eV, garners enormous attention due to its superior mechanical strength, optical characteristics, and thermal stability [46,47]. These exceptional properties enable it to hold great potential in various fields, including hydrogen storage [48,49], sensor technology [50,51], lithium-ion batteries [52], and catalytic applications [53-57]. Particularly, the graphitic BC3 nanosheet has been successfully synthesized in experiment via the reaction of benzene with sodium borohydride at 800 ℃ and hold great potential to host SACs for achieving superior electrocatalytic performance [58]. Herein, we systematically investigate the potential of nine main-group metals (i.e., Mg, Ca, Al, Ga, In, Ge, Sn, Sb, and Bi) embedded BC3 monolayer as SACs for the NORR toward NH3 synthesis. Through first-principles calculations, we screen out Al@VB, Ga@VB, and Ge@VC systems with outstanding NORR activity and high selectivity for NH3 production from a pool of 18 candidate systems. Importantly, the NORR process is found to occur spontaneously in the three screened systems. Our research results provide a valuable guidance for further developing in NORR catalysts, as well as demonstrate the great potential of main-group metals as SACs.
All spin-polarized first-principles calculations are performed using the Vienna Ab initio Simulation Package (VASP) [59] with the projector augmented wave (PAW) approach [60] and the Perdew-Burke-Ernzerhof (PBE) exchange-correlation functional [61]. Additionally, the van der Waals weak interactions are further treated using Grimme’s semi-empirical correction scheme (DFT-D3) [62]. A plane-wave energy cutoff of 500 eV is applied in all calculations. To minimize interactions between two periodic images, a vacuum layer of 20.0 Å is set in the z-direction. For structure optimization and electronic property calculations of a 3 × 3 × 1 supercell, 2 × 2 × 1 and 4 × 4 × 1 k-point grids centered at the Γ point are used following the Monkhorst-Pack scheme [63], respectively. The convergence thresholds are set to be 10−5 eV for the total energy and 0.02 eV/Å for atomic forces, respectively. More other computational details are provided in Supporting information.
The pristine unit cell of BC3 monolayer composed of two boron (B) atoms and six carbon (C) atoms shows a graphene-like planar structure with the space group of P6/mmm (No. 191), in which each B atom is coordinated to three neighboring C atoms. The optimized lattice parameters are a = b = 5.17 Å, and the lengths of C–C and C-B bond are 1.42 and 1.57 Å, respectively, which are consistent with previous reports [64-66]. To simulate the main-group metal-embedded BC3 monolayer, we construct a 3 × 3 × 1 supercell and consider four kinds of vacancies, including B monovacancy (VB), C monovacancy (VC), BC divacancy (VBC), and CC divacancy (VCC). To evaluate the formation possibility of the four vacancies, we calculate their respective formation energies (Ef), which are 5.37 eV for VB, 4.44 eV for VC, 7.47 eV for VBC, and 6.05 eV for VCC, indicating the thermodynamically favorable formation of monovacancies (VB and VC) rather than divacancies (VBC and VCC). Compared to that of monovacancy in graphene (7.45 eV) [67-69], the lower Ef values of VB and VC monovacancies suggest the high feasibility for experimental synthesis. Consequently, we concentrate on VB and VC monovacancies for main-group metal-based SACs.
As shown in Figs. 1a and b, nine main-group metals (i.e., Mg, Ca, Al, Ga, In, Ge, Sn, Sb, and Bi) are chosen and embedded into VB and VC monovacancies, forming the structures of M@VB and M@VC, respectively. To quantitatively evaluate the stability of these M@VB and M@VC systems, we calculate the binding energy (Eb) as shown in Fig. 1c, and find that all systems exhibit negative Eb values ranging from −6.13 eV to −0.67 eV, indicating the strong binding interactions between main-group metals and BC3 monolayer. Additionally, considering the large cohesive energy (Ecoh) of metal bulk, the experimental feasibility of main-group metal-based SACs needs to be further assessed. Our results shown in Fig. 1c reveal that nine candidate systems (i.e., Al@VB, Al@VC, Ga@VB, Ga@VC, In@VB, Ge@VB, Ge@VC, Sn@VB, and Sb@VB) have smaller Eb in comparison with their corresponding Ecoh, and thus satisfy the requirement Eb - Ecoh < 0, indicating that the main-group metals in these systems prefer to be stably anchored on BC3 monolayer, without aggregating into metal clusters. To further examine the thermal stabilities of the nine systems, we also perform ab initio molecular dynamics (AIMD) simulations in the NVT ensemble, with a total time scale of 5 ps at 500 K. As shown in Fig. S1 (Supporting information), the total energies of the nine systems oscillate slightly, and their corresponding geometric structures remain intact without deformation at the end of the AIMD simulation, verifying the good thermodynamic stability. Therefore, Al@VB, Al@VC, Ga@VB, Ga@VC, In@VB, Ge@VB, Ge@VC, Sn@VB, and Sb@VB are selected as potential candidates for the subsequent NORR.
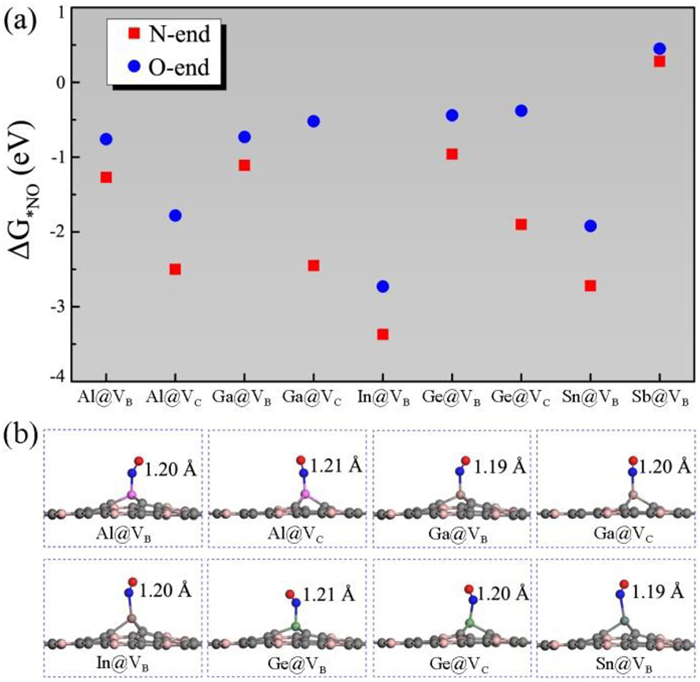
It is well-established that NO adsorption and activation, the first step of the NORR process, plays a crucial role in the following reaction pathways [70]. Therefore, we consider three initial NO adsorption configurations (i.e., N-end, O-end, and side-on) for the above screened nine systems, and calculate their adsorption free energies (ΔG*NO) as shown in Fig. 2a. For all candidate systems, we can find that NO molecule is favored to adsorb via the N-end configuration, while the side-on adsorption is unstable and will be transformed into the N-end configuration after fully structural relaxation. To ensure the effective activation of adsorbed NO molecule, spontaneous chemisorption (ΔG*NO < 0) is considered a necessary prerequisite for the NORR. Evidently, the Sb@VB system should be filter out from the NORR candidates because of the weak physisorption of NO (ΔG*NO = 0.28 eV). The remaining eight candidates present sufficient negative ΔG*NO values, ranging from −0.96 eV to −3.37 eV for the N-end configuration, which indicates that NO molecule would spontaneously chemisorb on these systems. Meanwhile, as shown in Fig. 2b, the chemisorbed NO molecules have considerable N–O bond elongation from 1.17 Å (free NO molecule) to 1.19 - 1.21 Å, indicating the strong activation of NO molecule. To evaluate the competitive effects of NO adsorption, we further calculate the adsorption free energies of N2 (ΔG*N2) and NH3 (ΔG*NH3), and compare with NO (ΔG*NO). As shown in Fig. S2 (Supporting information), ΔG*NO is more negative than ΔG*N2 and ΔG*NH3 for the M@VB and M@VC systems, indicating the NO molecule could be preferentially adsorbed on them.
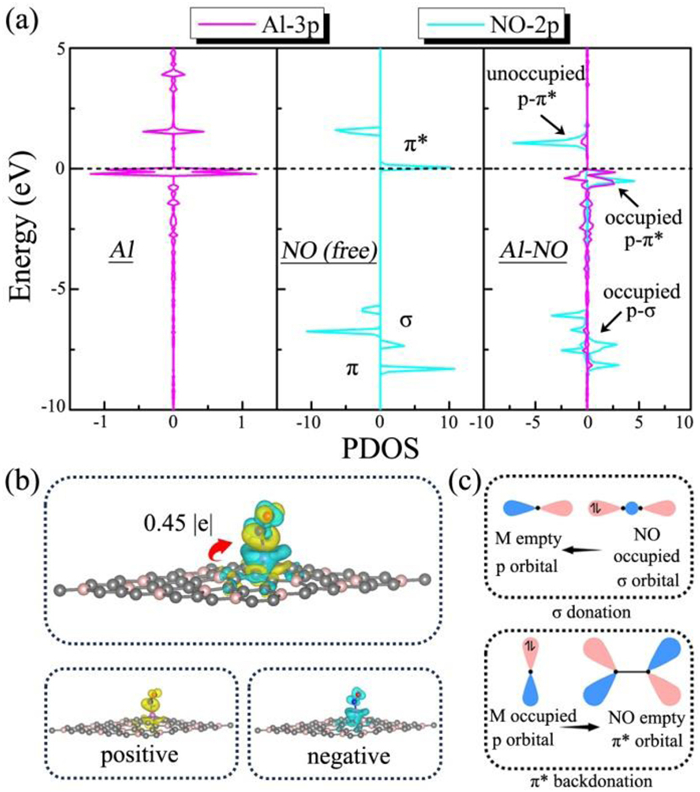
To elucidate the underlying mechanism of NO adsorption and activation on these main-group metal-embedded BC3 systems, we further calculate the partial density of states (PDOS) and charge density differences to investigate the interaction between main-group metal atom and NO molecule. Take for instance the Al@VB system illustrated in Fig. 3a. When a NO molecule chemisorbs on it, the π* states of NO would interact with the Al-3p states, resulting in the appearance of occupied and unoccupied p-π* states around the Fermi level (EF). Additionally, due to the hybridization between the occupied Al-3p states and NO-σ states below the EF, the occupied p-σ states can be observed. As displayed in Fig. 3b, we can see not only a significant charge transfer with 0.45 |e| but also bidirectional charge transfer, namely, both electron accumulation (yellow color) and depletion (cyan color) on the NO molecule and Al atom. Thus, as illustrated in Fig. 3c, the interaction nature between Al and NO can be attributed to the "acceptance-donation" of electrons analogous to that in NRR [71,72]. The Al atom can accept lone-pair electrons of NO and simultaneously donate electrons back to the empty π* orbitals of NO Similarly, as shown in Figs. S3-S9 (Supporting information), such electrons transfer mechanism can also be verified in other main-group metal-embedded BC3 systems.
Having confirmed the effective activation of NO on the eight main-group metal-embedded BC3 systems, we then move on to examine their catalytic performances of NORR toward NH3 synthesis. Depending on the different adsorption configuration of NO, three possible reaction pathways shown in Fig. 4a, named as N-end, O-end, and side-on, are taken into consideration based on previous studies [73,74]. Noted that we only consider the N-end pathway for these systems since the N-end adsorption configuration is energetically more favorable. Subsequently, we optimize the geometric configurations of all reaction intermediates during the NORR process, and calculate the Gibbs free energy changes of each elementary step. The obtained NORR free energy diagrams for the eight main-group metal-embedded BC3 systems are illustrated in Fig. 4 and Fig. S10 (Supporting information), and the corresponding intermediate structures are given in Figs. S11-S18 (Supporting information). There are two possible intermediates (i.e., *HNO and *NOH) for the first hydrogenation elementary step. For all of the eight main-group metal-embedded BC3 systems, *HNO is preferred with respect to *NOH. Subsequently, these systems exhibit distinct NORR paths and potential-determining steps (PDS). Taking Al@VB, Ga@VB, and Ge@VC as example, the optimum NORR pathway for Al@VB shown in Fig. 4b can be summarized as NO(g) → *NO → *HNO → *HNOH → *H2NOH → *NH2 → *NH3, with the PDS being the second hydrogenation step of *HNO → *HNOH. In terms of Ga@VB, Fig. 4c shows that the second hydrogenation step prefers to form *H2NO instead of *HNOH, and the PDS is identified as the hydrogenation of *HNO to *H2NO. While for Ge@VC shown in Fig. 4d, the reaction path NO(g) → *NO → *HNO → *HNOH → *NH → *NH2 → *NH3 is predicted to be the preferable one with the PDS being the hydrogenation of NO to *HNO.
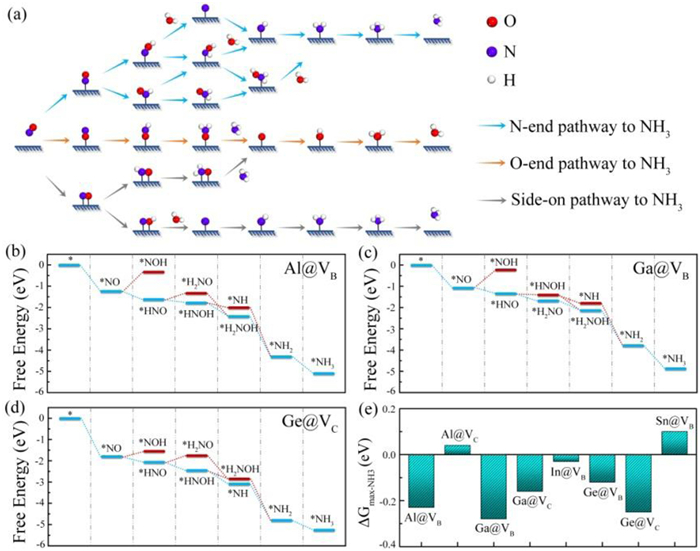
As an important index, the maximum free energy change (ΔGmax-NH3) along the most favorable reaction pathway, are usually used to estimate the NORR activity [45]. Here, ΔGmax-NH3 < 0 is selected as screening benchmark for targeting at finding potential NORR catalysts. As shown in Fig. 4e, the ΔGmax-NH3 values for Al@VC and Sn@VB are 0.04 and 0.10 eV, respectively. Thus, we rule out them from NORR candidates. It is quite interesting that the remaining six systems, including Al@VB (−0.23 eV), Ga@VB (−0.28 eV), Ga@VC (−0.16 eV), In@VB (−0.03 eV), Ge@VB (−0.12 eV), and Ge@VC (−0.25 eV), have significantly negative ΔGmax-NH3 values, indicating that the NORR process can proceed spontaneously on them without using any applied voltage. Furthermore, taking Al@VB as an example, we investigate the impact of solvation correction on the free energy during the NORR process by using an implicit solvation model as implemented in VASPsol [75]. As shown in Fig. S19 (Supporting information), the PDS and ΔGmax-NH3 of Al@VB are insensitive to the solvation effects. In this regard, the solvation effects are not considered in this work.
In addition to the catalytic activity, the NH3 selectivity is also an essential factor for achieving high catalytic performance [76,77]. At high NO coverage, N2O and N2 could be produced as main byproducts during the NORR process. To this end, we further investigate the electrochemical NO-to-N2O/N2 conversion following the reaction pathways illustrated in Fig. 5a, which starts with the 2O-side and 2N-side modes of NO dimer (labelled as *N2O2–O and *N2O2–N), respectively. The free energy diagrams of N2O and N2 formation on the remaining six systems (Al@VB, Ga@VB, Ga@VC, In@VB, Ge@VB, and Ge@VC) are shown in Figs. 5b–g, and the optimized atomic structures of intermediates are given in Figs. S20-S25 (Supporting information). One can see that converting *N2O2–O to *O + N2O is the PDS in the NORR process toward N2O synthesis, which demand the energy inputs of 0.83, 0.49, 0.58, 0.68, 0.47, and 0.27 eV for the Al@VB, Ga@VB, Ga@VC, In@VB, Ge@VB, and Ge@VC systems, respectively. On the other hand, the protonation of *N2O to *N2OH in the NORR process toward N2 synthesis have considerably large ΔG values of 0.17, 0.40, and 0.12 eV for the Al@VB, Ga@VB, and Ge@VC systems, respectively. Noted that the NO-to-N2 conversion on the Ga@VC, In@VB, and Ge@VB systems is an exothermic process without any extra energy, indicative of the spontaneous N2 formation. Hence, the Al@VB, Ga@VB, and Ge@VC systems can effectively prevent the byproduct reactions of N2O and N2 formation, indicating high selectivity of NORR toward NH3 synthesis.
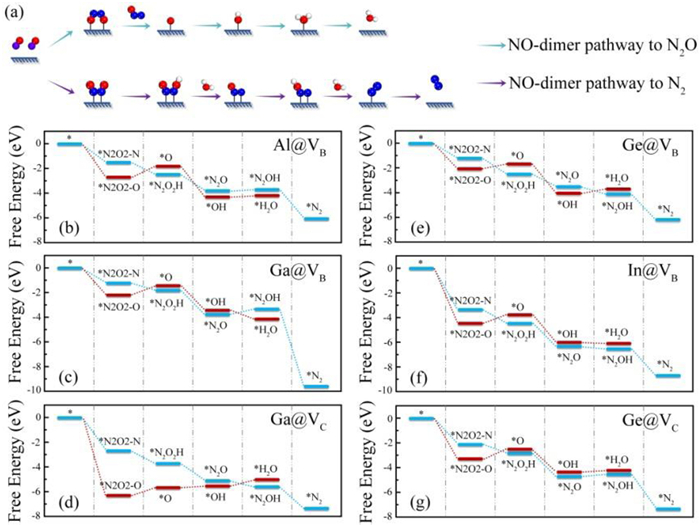
Another competing reaction to the NH3 selectivity is the H2 production through HER, which normally affects the Faradaic efficiency of NORR. Therefore, we compare the competition between H2 and NH3 production in the NORR process for the remaining three candidates. As shown in Fig. S26 (Supporting information), the calculated ΔG*H values are −0.25 eV for Al@VB, −0.39 eV for Ga@VB, and −1.65 eV for Ge@VC. Obviously, the free energy barriers for the HER are considerable larger than the PDS barriers for the NORR. This indicates that the HER process can be greatly suppressed on the three systems, ensuring the excellent NORR selectivity. According to these calculations, the Al@VB, Ga@VB, and Ge@VC systems are finally screened out as potential main-group metal-based SACs for NORR toward NH3 synthesis due to the superior catalytic activity and selectivity.
In conclusion, by performing first-principles calculations, we comprehensively investigate main-group metal-embedded BC3 monolayer as SACs for the NORR toward NH3 synthesis. Our calculations reveal that nine systems (i.e., Al@VB, Al@VC, Ga@VB, Ga@VC, In@VB, Ge@VB, Ge@VC, Sn@VB, and Sb@VB) out of 18 candidates satisfy the relationship Eb - Ecoh < 0, implying the high thermodynamic stability. Besides the Sb@VB, NO molecule can be stably adsorbed and effectively activated on the remaining eight systems via the "acceptance-donation" mechanism of electron. Further scrutiny of the catalytic activity, six candidate systems including Al@VB, Ga@VB, Ga@VC, In@VB, Ge@VB, and Ge@VC are considered as excellent NORR electrocatalysts with ΔGmax-NH3 < 0, since the electrochemical NO-to-NH3 conversion can proceed spontaneously without any limiting potential. In addition, after estimating the suppression of byproducts including N2O, N2, and H2, Al@VB, Ga@VB, and Ge@VC are expected to possess high NH3 selectivity, and finally screened out as promising main-group metal-based SACs for NORR. Consequently, these theoretical findings provide valuable insights for designing efficient NORR electrocatalysts and expanding SACs to main-group metal-based systems.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jiajun Wang: Writing – original draft, Project administration, Conceptualization. Chen Sun: Resources, Data curation. Li Sheng: Data curation. Zhiwen Zhuo: Investigation. Shujuan Li: Writing – review & editing, Supervision. Jiayao Wang: Validation, Investigation. Weiyi Wang: Writing – review & editing, Supervision, Funding acquisition. Jinbo Sun: Visualization. Juqian Yang: Writing – original draft. Ke Xu: Funding acquisition. Shulai Lei: Supervision, Funding acquisition.
This work is supported by the Postdoctoral Fellowship Program (No. GZC20232540), the General Program of China Postdoctoral Science Foundation (No. 2024M753088), Natural Science Foundation of Hubei Province (No. 2022CFC030), the Science and Technology Research Project of Hubei Provincial Department of Education (No. D20212603) and Hubei University of Arts and Science (No. 2020kypytd002). Hefei Advanced Computing Center is acknowledged for computational support.
Supplementary material associated with this article can be found, in the online version, at doi:
T.Y. Dai, T.H. Wang, Z. Wen, et al., Adv. Funct. Mater. 34 (2024) 2400773. doi: 10.1002/adfm.202400773
Y. Yang, J. Wang, Y. Shu, et al., Phys. Chem. Chem. Phys. 24 (2022) 8591–8603. doi: 10.1039/d1cp05442b
C. He, J. Wang, L. Fu, et al., Chin. Chem. Lett. 35 (2024) 109037. doi: 10.1016/j.cclet.2023.109037
G. Qing, R. Ghazfar, S.T. Jackowski, et al., Chem. Rev. 120 (2020) 5437–5516. doi: 10.1021/acs.chemrev.9b00659
J. Wang, M. Shi, G. Yi, et al., Mol. Catal. 511 (2021) 111726.
J. Wu, L. Yang, X. Liu, et al., Chin. Chem. Lett. 34 (2023) 107659. doi: 10.1016/j.cclet.2022.07.002
J. Wang, M. Shi, G. Yi, et al., Chin. Chem. Lett. 33 (2022) 4623–4627. doi: 10.1016/j.cclet.2021.12.040
Y. Ren, S. Li, C. Yu, et al., J. Am. Chem. Soc. 146 (2024) 6409–6421. doi: 10.1021/jacs.3c11676
D. Wang, X.F. Lu, D. Luan, et al., Adv. Mater. 36 (2024) 2312645. doi: 10.1002/adma.202312645
Y. Zang, Q. Wu, S. Wang, et al., Adv. Energy Mater. 14 (2024) 2303953. doi: 10.1002/aenm.202303953
X. Sun, Y. Dai, B. Huang, et al., J. Phys. Chem. Lett. 14 (2023) 11684–11690. doi: 10.1021/acs.jpclett.3c03105
C. He, J. Wang, L. Fu, et al., Chin. Chem. Lett. 33 (2022) 1051–1057. doi: 10.1016/j.cclet.2021.09.009
S. Zhao, Y. Li, Z. Guo, et al., J. Mater. Chem. A 10 (2022) 25201–25211. doi: 10.1039/d2ta06354a
C. He, R. Sun, L. Fu, et al., Chin. Chem. Lett. 33 (2022) 527–532. doi: 10.1016/j.cclet.2021.05.072
Z. Wu, Y. Liu, D. Wang, et al., Adv. Mater. 36 (2024) 2309470. doi: 10.1002/adma.202309470
L. Xiao, S. Mou, W. Dai, et al., Angew. Chem. Int. Ed. 63 (2024) e202319135. doi: 10.1002/anie.202319135
K. Chen, J. Xiang, Y. Guo, et al., Nano Lett. 24 (2024) 541–548. doi: 10.1021/acs.nanolett.3c02259
B. Qiao, A. Wang, X. Yang, et al., Nat. Chem. 3 (2011) 634–641. doi: 10.1038/nchem.1095
F. Kraushofer, G.S. Parkinson, Chem. Rev. 122 (2022) 14911–14939. doi: 10.1021/acs.chemrev.2c00259
S. Wang, X.T. Min, B. Qiao, et al., Chin. J. Catal. 52 (2023) 1–13.
J. Wang, G. Yi, S. Guo, et al., Chin. Chem. Lett. 35 (2024) 109050. doi: 10.1016/j.cclet.2023.109050
G. Xue, S. Ping, H. Ce, et al., Energy Mater. 3 (2023) 300016.
R. Cheng, C.N. Cui, Z.X. Luo, Rare Met. 43 (2024) 3810–3818. doi: 10.1007/s12598-024-02680-2
G. Song, R. Gao, Z. Zhao, et al., Appl. Catal. B: Environ. 301 (2022) 120809. doi: 10.1016/j.apcatb.2021.120809
W. Zhang, M. Xu, T. Wang, et al., Sep. Purif. Technol. 354 (2025) 129536. doi: 10.1016/j.seppur.2024.129536
S. Peng, Y. Rao, T. Li, et al., Chin. Chem. Lett. 35 (2024) 109219. doi: 10.1016/j.cclet.2023.109219
X. Yao, L. Huang, E. Halpren, et al., J. Am. Chem. Soc. 145 (2023) 26249–26256. doi: 10.1021/jacs.3c08936
L. Kong, X. Liang, M. Wang, et al., J. Colloid Interf. Sci. 647 (2023) 375–383. doi: 10.1016/j.jcis.2023.05.158
H. Niu, Z. Zhang, X. Wang, et al., Small 17 (2021) 2102396. doi: 10.1002/smll.202102396
L. Lin, L. Yan, L. Fu, et al., Fuel 308 (2022) 122068. doi: 10.1016/j.fuel.2021.122068
R. Zhao, Z. Ma, Y. Yu, et al., Nano Res. 16 (2023) 8533–8541. doi: 10.1007/s12274-023-5619-9
Y. Zang, Q. Wu, S. Wang, et al., Mater. Horiz. 10 (2023) 2160–2168. doi: 10.1039/d2mh01440h
Y. Wu, J. Lv, F. Xie, et al., J. Colloid Interf. Sci. 656 (2024) 155–167. doi: 10.3847/1538-4357/ad509f
L. Kong, M. Wang, C.M.L. Wu, ACS Mater. Lett. 6 (2024) 1711–1721. doi: 10.1021/acsmaterialslett.3c01604
H. Li, D. Wu, J. Wu, et al., Nanoscale 16 (2024) 7058–7067. doi: 10.1039/d4nr00028e
G.F. Chen, S. Ren, L. Zhang, et al., Small Methods 3 (2019) 1800337. doi: 10.1002/smtd.201800337
S. Liu, Z. Li, C. Wang, et al., Nat. Commun. 11 (2020) 938. doi: 10.1038/s41467-020-14565-w
T. Liu, X. Qu, Y. Zhang, et al., Chem. Eng. J. 457 (2023) 141187. doi: 10.1016/j.cej.2022.141187
F. Luo, A. Roy, L. Silvioli, et al., Nat. Mater. 19 (2020) 1215–1223. doi: 10.1038/s41563-020-0717-5
Q. Wang, M. Dai, H. Li, et al., Adv. Mater. 35 (2023) 2300695. doi: 10.1002/adma.202300695
L. Zhang, H. Zhou, X. Yang, et al., Angew. Chem. Int. Ed. 62 (2023) e202217473. doi: 10.1002/anie.202217473
K. Chen, Y. Zhang, J. Xiang, et al., ACS Energy Lett. 8 (2023) 1281–1288. doi: 10.1021/acsenergylett.2c02882
X. Li, G. Zhang, P. Shen, et al., Inorg. Chem. Front. 10 (2023) 280–287. doi: 10.1039/d2qi02118h
K. Chen, N. Zhang, F. Wang, et al., J. Mater. Chem. A 11 (2023) 6814–6819. doi: 10.1039/d3ta00606a
Q. Wu, B. Huang, Y. Dai, et al., npj 2D Mater. Appl. 6 (2022) 52. doi: 10.1038/s41699-022-00326-4
L. Zhao, Y. Li, G. Zhou, et al., Chin. Chem. Lett. 32 (2021) 900–905. doi: 10.1016/j.cclet.2020.07.016
H. Yanagisawa, T. Tanaka, Y. Ishida, et al., Phys. Rev. Lett. 93 (2004) 177003. doi: 10.1103/PhysRevLett.93.177003
Z. Yang, J. Ni, Appl. Phys. Lett. 100 (2012) 183109. doi: 10.1063/1.4711038
J. Li, Y. Guo, S. Ma, et al., Int. J. Hydrogen Energy 47 (2022) 24004–24013. doi: 10.1016/j.ijhydene.2022.05.154
H. Yang, C. He, L. Fu, et al., Chin. Chem. Lett. 32 (2021) 3202–3206. doi: 10.1016/j.cclet.2021.03.038
Y. Guo, X. Kang, S. Gao, et al., Phys. Chem. Chem. Phys. 25 (2023) 12420–12425. doi: 10.1039/d3cp00653k
R.P. Joshi, B. Ozdemir, V. Barone, et al., J. Phys. Chem. Lett. 6 (2015) 2728–2732. doi: 10.1021/acs.jpclett.5b01110
J. Ou, X. Kang, X. Duan, Nanoscale 14 (2022) 12823–12829. doi: 10.1039/d2nr02796h
W. Guo, S. Wang, H. Wang, et al., J. Energy Chem. 96 (2024) 336–344. doi: 10.1016/j.jechem.2024.05.002
C. Chen, B. Xiao, Z. Qin, et al., ACS Appl. Mater. Interfaces 15 (2023) 40538–40548. doi: 10.1021/acsami.3c07790
Y. Wang, N. Zhou, Y. Li, Chem. Eng. J. 425 (2021) 130631. doi: 10.1016/j.cej.2021.130631
Y. Zhou, G. Gao, W. Chu, et al., Nanoscale 13 (2021) 1331–1339. doi: 10.1039/d0nr07580a
D. McGlamery, C. McDaniel, D.M. Ladd, et al., Chem. Sci. 15 (2024) 4358–4363. doi: 10.1039/d3sc06837d
G. Kresse, J. Furthmuller, Phys. Rev. B 54 (1996) 11169. doi: 10.1103/PhysRevB.54.11169
P.E. Blöchl, Phys. Rev. B 50 (1994) 17953. doi: 10.1103/PhysRevB.50.17953
J.P. Perdew, K. Burke, M. Ernzerhof, Phys. Rev. Lett. 77 (1996) 3865. doi: 10.1103/PhysRevLett.77.3865
S. Grimme, J. Comput. Chem. 27 (2006) 1787–1799. doi: 10.1002/jcc.20495
H.J. Monkhorst, J.D. Pack, Phys. Rev. B 13 (1976) 5188. doi: 10.1103/PhysRevB.13.5188
L. Pan, X. Kang, S. Gao, et al., Phys. Chem. Chem. Phys. 26 (2024) 1011–1016. doi: 10.1039/d3cp04660e
G. Xing, S. Liu, J.Y. Liu, Mol. Catal. 553 (2024) 113771.
S. Li, M. Shi, J. Yu, et al., Chin. Chem. Lett. 32 (2021) 1977–1982. doi: 10.1016/j.cclet.2020.09.056
Y. Zhou, W. Chu, F. Jing, et al., Appl. Surf. Sci. 410 (2017) 166–176. doi: 10.1089/scd.2016.0162
D.H. Lim, J. Wilcox, J. Phys. Chem. C 115 (2011) 22742–22747. doi: 10.1021/jp205244m
L.J. Ma, J. Wang, J. Jia, et al., Int. J. Hydrogen Energy 47 (2022) 28423–28433. doi: 10.1016/j.ijhydene.2022.06.152
S.J. Qian, H. Cao, Y.G. Wang, et al., J. Am. Chem. Soc. 146 (2024) 12530–12537. doi: 10.1021/jacs.4c00827
M. He, X. Chen, Y. Zhou, et al., J. Phys. Chem. Lett. 14 (2023) 7100–7107. doi: 10.1021/acs.jpclett.3c01576
C. Ling, X. Niu, Q. Li, et al., J. Am. Chem. Soc. 140 (2018) 14161–14168. doi: 10.1021/jacs.8b07472
Y. Zang, Q. Wu, S. Wang, et al., J. Phys. Chem. Lett. 13 (2022) 527–535. doi: 10.1021/acs.jpclett.1c03938
Y. Sun, Z. Wang, Y. Liu, et al., Inorg. Chem. Front. 10 (2023) 2677–2688. doi: 10.1039/d3qi00225j
K. Mathew, R. Sundararaman, K. Letchworth-Weaver, et al., J. Chem. Phys. 140 (2014) 084106. doi: 10.1063/1.4865107
S. Liu, G. Xing, J.Y. Liu, Appl. Surf. Sci. 611 (2023) 155764. doi: 10.1016/j.apsusc.2022.155764
M. Tursun, C. Wu, Inorg. Chem. 61 (2022) 17448–17458. doi: 10.1021/acs.inorgchem.2c02247

Figure 1 Top view of (a) M@VB and (b) M@VC systems. (c) Computed binding energies (Eb) of main-group metal-embedded BC3 monolayer and the energy difference between Eb and cohesive energies (Ecoh) of main-group metals.

Figure 2 (a) Adsorption free energies (ΔG*NO) of NO on M@VB and M@VC systems with N-end and O-end configurations. (b) Optimized structures of NO adsorption on M@VB and M@VC systems through the N-end configuration, where the N—O bond lengths are labelled.

Figure 3 (a) Partial density of states (PDOS) of Al-3p orbitals for Al@VB, NO-2p orbitals for free NO molecule, and Al-3p and NO-2p orbitals for NO adsorbed on Al@VB. The Fermi level is set to be 0 eV. (b) Charge density difference of NO molecule adsorbed on Al@VB through the N-end configuration, where the positive and negative charges are shown in yellow and cyan, respectively. The isosurface value is set to be 0.005 e/Å3. (c) Schematic mechanism of the interaction between main-group metal and NO molecule.

Figure 4 (a) Schematic illustration of the NORR pathways toward NH3. Computed Gibbs free energy diagrams of the NORR on (b) Al@VB, (c) Ga@VB and (d) Ge@VC systems. (e) Maximum free energy change (ΔGmax-NH3) of the remaining eight systems via the respective most favorable NORR pathway.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: