Citation:
Peng Liu, Shengli Zhang, Tingting Zhang, Yu Si, Ziang Liu, Xiao Qian, Yingxu Wu, Yuan Liang, Wen Sun, Engin U. Akkaya, Lei Wang. Near-infrared light activatable nanoplatform for proteins labeling, enrichment and visualization in living systems[J]. Chinese Chemical Letters,
2026, 37(1): 110966.
doi:
10.1016/j.cclet.2025.110966
Near-infrared light activatable nanoplatform for proteins labeling, enrichment and visualization in living systems
English
Near-infrared light activatable nanoplatform for proteins labeling, enrichment and visualization in living systems
Received Date:
30 November 2024 Accepted Date:
17 February 2025 Revised Date:
15 February 2025 Available Online:
15 January 2026
Abstract:
The study of target proteins is crucial for understanding molecular interactions and developing analytical platforms, therapeutic agents and functional tools. Herein, we present a novel nanoplatform activated by near-infrared (NIR) light for triple-modal proteins study, which enabling target protein labeling, enrichment and visualization. Azido-naphthalimide-coated upconversion nanoparticles (UCNPs) serve as NIR light-responsive nanoplatforms, showing promising applications in studying interactions between various bioactive molecules and proteins in living systems. Under NIR light irradiation, azido-naphthalimides are activated by ultraviolet (UV) and blue light emitted from UCNPs and the resulting amino-naphthalimides intermediate not only crosslink nearby target proteins but also enable imaging performance. We demonstrate that this nanoplatform is capable of selective protein labeling and imaging in complex protein environments, achieving specific labeling and imaging of both intracellular and extracellular proteins in mammalian cells as well as bacteria. Furthermore, in vivo protein labeling has been achieved using this novel NIR light-activatable nanoplatform. This technique will open new avenues for discoveries and mechanistic interrogation in chemical biology.
The identification of biological proteins and study of the interactions of proteins and ligands are essential for comprehensively understanding the biological processes and design of new therapeutic agents [1]. Small molecules-based proximity labeling is a prevailing strategy which is purposefully crafted to resemble the structure of a ligand or substrate, enabling it to interact with biomolecular targets. Hamachi group developed ligand-directed chemistry which rely on the nucleophilic amino acid on proteins to react with the electrophilic group of small molecules-based probes [2]. However, this strategy mainly relies on the abundance of nucleophilic amino acid and their reactivity toward electrophilic group of probes. Photoaffinity-based approach has emerged as promising alternatives which enabling high spatiotemporal control and good labeling efficacy [3–6]. Typically, a bioactive molecule incorporates a photoreactive group capable of forming a covalent bond with the target molecule upon exposure to ultraviolet (UV) light. Until now, many efforts have been witnessed on efficient purification of the labeled targets, with different tags including biotin or a clickable tag as a post-labeling technique [7,8]. However, the purification process is often complicated by the tedious procedures and the inevitable loss of targets during the handling process. The combination with mass spectrometry (MS) [9] enables target identification on the proteome-wide scale and structural elucidation of the binding site but a successful and efficient isolation of small molecule-conjugated targets are precondition. While, post-labeling commonly requires distinct procedures for protein separation or visualization via bio-orthogonal chemistry to introduce a biotin or a fluorophore [10], which cannot be achieved simultaneously. Additionally, the efficiency of proteins labeling is often limited to specific targets, such as glycoproteins, typically exhibit low affinity and selectivity toward their ligands resulting in a lower labeling yield [11]. Very recently, Sakurai's group developed gold nanoparticle-based multivalent carbohydrate probes, demonstrating favorable affinity and efficient isolation of carbohydrate-binding proteins [12]. Toshima and colleagues devised a solid-phase affinity labeling technique for the target-selective isolation and modification of proteins [13]. Nanoplatforms were promising tools for proteins labeling and enrichment, but current strategies mainly rely on the irradiation by UV light which bring a risk of harm to biomolecular targets and cells [14,15]. Given the advantages of lower energy light, visible light-driven photoproximity labeling has emerged as a promising approach for protein study. MacMillan group developed a µMap photocatalytic labeling strategy using a photocatalyst excited by blue or red light to convert diazirines into reactive carbenes via energy transfer [16,17]. Meanwhile, Rovis and coworker reported photocatalytic activation of aryl(trifluoromethyl) diazos or aryl azide for protein labeling using red light [18]. These strategies commonly involved light conversion, from blue or red light to UV light, which are mediated by a metal photocatalyst. However, oxygen could inhibit intracellular photocatalytic reactions by quenching the excited-state photocatalysts, and singlet oxygen [19,20] commonly generated in the photosensitization process which is highly reactive and may cause protein degradation and cellular damage [21]. Upconverting nanoparticles (UCNPs) are remarkable for their ability to emit UV light when excited by continuous-wave near-infrared (NIR) laser diodes [22–27]. This property makes NIR-based photochemistry highly valuable in bioimaging, biocatalysis [28–33] and drug delivery because NIR light is advantageous due to its minimal photodamage to biomolecular targets and its superior tissue penetration compared to UV light. Therefore, we explored whether NIR light could activate photoreactive groups, especially those with imaging capabilities. If successful, this could enable the development of a NIR light-activated nanoplatform for protein labeling, enrichment, and visualization in living systems.
Keeping all these points in mind, in this work, we reported a triple-modal NIR light activatable nanoplatform enabling protein labeling, enrichment and visualization, simultaneously. The novel nanoplatform could be activated under lower power NIR light within short irradiation time, and it features easy preparation, good applicability, convenient targets enrichment. Meanwhile, azido-naphthalimide as photoreactive group can be converted amino-naphthalimide, a fluorescent moiety with emission at long wavelengths (λmax ~ 540 nm) enabling targets visualization simultaneously. Most importantly, we successfully demonstrated its applicability for targets labeling in bacteria, mammalian cells and living animals with good safety. Together, we envision that this nanoplatform can be developed as a novel photochemical tool for comprehensive understanding the biological processes and design of new therapeutic agents.
As Tm-loaded UCNPs emitted both blue and UV light, but the density of blue light is higher than the latter one [22]. Meanwhile, Wu reported that only blue light was emitted when the excitation intensity was below 2.2 W/cm2 [28], thus a higher excitation intensity was required to activate diazirine in previous work. Therefore, we hypothesized that a more sensitive photoreactive group, particularly one that can be activated under both UV and blue light, would be promising for achieving more effective proteins labeling. Azido-naphthalimide was recently utilized as an excellent photoreactive group, which demonstrates good photo-reactivity under UV or blue light irradiation [34,35], but not NIR light. Meanwhile, it owns good aqueous stability and its photoinduced product amino-naphthalimide is a fluorescent moiety with emission at long wavelengths [36], thus could enable the protein visualization after labeling process. Based on this, we assumed that azido-naphthalimide-loading upconverting nanoparticles could be a novel nanoplatform integrating protein labeling, isolation, and visualization under NIR light irradiation. To test our hypothesis, we firstly synthesized azido-naphthalimide and analyzed its photolysis reaction under various light irradiation (see Supporting information for syntheses). As expected, changes in absorption and fluorescence spectra under UV light indicated a fast photochemical transformation of azido-naphthalimide (Fig. S1 in Supporting information). In contrast, diazirine, a traditional photoreactive group displayed moderate transformation rate under the same irradiation condition (Fig. S2 in Supporting information). Comparable alterations were observed under blue light exposure, indicating its robust photoreactivity under dual-light conditions (Fig. S3 in Supporting information). Next, Tm-loaded UCNPs and silica shell-coated one (UCNPs-SiO2) were synthesized, and the transmission electron microscopy (TEM) analysis and dynamic light scattering (DLS) both revealed their diameters (Figs. S4–S6 in Supporting information). As expected, UCNPs-SiO2 successfully emitted UV and blue light when excited with a 980 nm laser, demonstrating its potential to activate azido-naphthalimide (Fig. S7 in Supporting information). With azido-naphthalimide and UCNPs in hand, we test their photoreaction potential under NIR light irradiation. As anticipated, significant changes in the fluorescence spectra of azido-naphthalimide were observed when UCNPs were present (Fig. S8 in Supporting information), while no changes were found in their absence (Fig. S9 in Supporting information), suggesting that blue and UV light emitted from UCNPs are able to activate azido-naphthalimide.
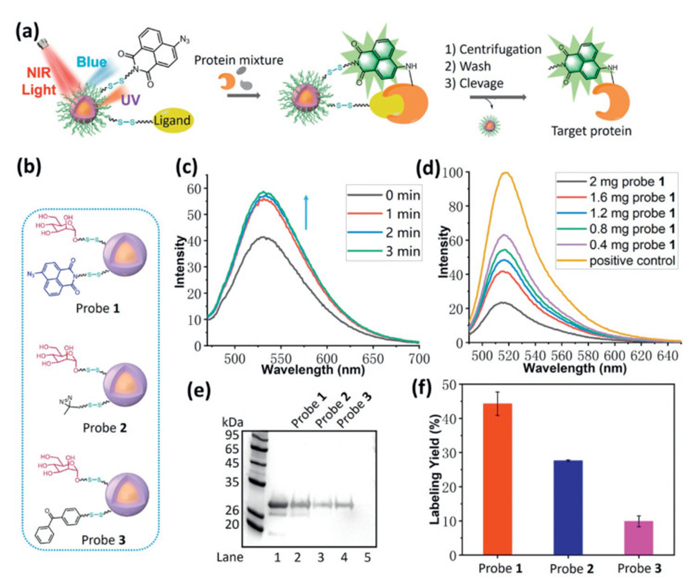
Next, we began testing the protein labeling potential of this NIR light-activatable nanoplatform. Carbohydrates serve as critical components in various biological processes, acting as recognition sites for cells and facilitating interactions with diverse binding partners, and carbohydrates-proteins interactions are involved in many biology processes including cell-cell interactions, tumor cell metastasis, bacteria and virus invasion, or immune response [37]. However, the exploration and understanding of these processes face significant challenges due to the typically weak affinities associated with carbohydrate-protein interactions (Kd, 10−3–10−6 mol/L) [38,39]. This limitation drives us to select carbohydrates as model ligands to test our novel nanoplatform (Fig. 1a). Concanavalin A (Con A) is an important lectin that binds primarily to α-D-mannopyranosides with high specificities but relatively low affinity [40], which was selected as the model target. Probe 1 was readily synthesized by a competing reaction of amino-modified azido-naphthalimide and mannose with NHS-functionalized UCNPs (Fig. 1b and Scheme S10 in Supporting information). Fluorescence spectra successfully demonstrated the azido-naphthalimide loading as well as its reactivity under NIR light irradiation (Fig. 1c). Using phenol/H2SO4 colorimetric reaction [41], the amount of mannose of probe 1 was determined as 26.7 nmol/mg (Fig. S10 in Supporting information). Next, fluorescein isothiocyanate (FITC)-labeled Con A was mixed with probe 1 for 4 h, and the supernatant was monitored by fluorescence after centrifugation. As shown in Fig. 1d, interaction of Con A and probe 1 was furnished in concentration dependent manner, which was strongly inhibited by competition with D-(+)-mannose (Fig. S11 in Supporting information), thus suggesting the specific interaction between probe 1 and target proteins. To determine the photoreactivity and labeling efficiency of probe 1, control probes with diazirine and benzophenone as photoreactive group were prepared (probe 2 and 3, Fig. 1b). Then, Con A were incubated with probe 1, 2 or 3 in phosphate buffered saline (PBS) at 4 ℃ for 2 h to form specific binding, and the incubating buffer was irradiated using NIR light (980 nm) on ice. The labeled proteins were enriched and purified through several cycles of centrifugation and washing PBS buffer. Subsequently, they were released from nanoparticles using mercaptoethanol and were subjected to sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) for analysis. As can be seen from Fig. 1e and Fig. S12 (Supporting information), all probes successfully labeled Con A, but probe 1 loaded with azido-naphthalimide showed the highest labeling yield (44%), indicating its higher reactivity and labeling efficiency (Fig. 1f). As shown in Figs. S13 and S14 (Supporting information), protein labeling followed both a power- and time-dependent manner, and 3 W of NIR light with 15 min irradiation was opted as the optimal condition due to the good labeling efficiency and moderate thermal effect (Fig. S15 in Supporting information). Competition experiment using excess D-(+)-mannose as competitor led to a significant decrease of labeling yield, suggesting the specific interaction of mannose with targets (Fig. 2a and Fig. S16 in Supporting information). Another control experiment using the probe lacking ligand but containing azido-naphthalimide displayed similar results (Fig. S17 in Supporting information). Obvious fluorescence was found in the in-gel fluorescence scanning, suggesting the protein visualization ability of probe 1 induced by NIR light. Meanwhile, labeling selectivity of probe 1 was further assessed with a mixture consisting of Con A and 10-fold excess nonspecific protein, bovine serum albumin (BSA). As expected, the enriched proteins in SDS-PAGE indicated that probe 1 showed good labeling selectivity towards Con A over BSA (Fig. 2b and Fig. S18 in Supporting information). The labeling of Con A from complicated environment was study, and a mixture of Con A (2 µg) and E. coli lysate (30 µg) were incubated with probe 1 for 2 h (Fig. S19 in Supporting information). Through NIR light irradiation, centrifugation, wash and cleavage by mercaptoethanol, the enriched protein was subjected to SDS-PAGE. As anticipated, probe 1 successfully labeled and enriched Con A in complicated cell lysate, shown in both regular staining and fluorescence gel.
Figure 1
Figure 1.
(a) Schematic illustration of protein labeling and visualization under NIR light irradiation. (b) Structures of probes 1–3. (c) Fluorescence spectra of probe 1 (2 mg/mL) under NIR light irradiation (980 nm). Ex. slit: 5 nm, Em. slit: 5 nm. (d) Binding study of FITC-labeled Con A (16 µg) with probe 1, fluorescence of the supernatant after removal of Con A/probe 1 complex. Ex. slit: 10 nm, Em. slit: 10 nm. (e) Labeling of Con A using different probe. Lane 1: Con A input (2 µg), lanes 2–4: labeling of Con A (2 µg) using indicated probe (1 mg), lane 5: negative control without NIR irradiation using probe 2 (1 mg). (f) Labeling yield of Con A using different probe (error bar indicates SD, n = 4).
Figure 2.
(a) Labeling of Con A (2 µg) using probe 1 (1 mg) with or without competitor. Lane 1: Con A input, lane 2: labeling of Con A using probe 1, lane 3: labeling of Con A using probe 1 with competitor (D-(+)-mannose, 300 µmol/L), lane 4: negative control without NIR irradiation. (b) Labeling of Con A in mixed proteins using probe 1 (1 mg), data were shown in stained gel (left) and in-gel fluorescence (right). Lane 1: Con A input, lane 2: Con A (2 µg) and BSA (20 µg), lane 3: labeling of Con A from mixed proteins. (c) Labeling of HSA (2 µg) using probe 4 (1 mg). Lane 1: HSA input, lane 2: labeling of HSA under NIR light (980 nm), lane 3: labeling of HSA under NIR light (980 nm) through 3 mm of pork tissue, lane 4: negative control without light irradiation. (d) Labeling of HSA (2 µg) using probe 4 (1 mg) with or without competitor. Lane 1: HSA input, lane 2: labeling of HSA using probe 4, lane 3: labeling of HSA using probe 4 with competitor (ibuprofen, 300 µmol/L), lane 4: negative control without NIR irradiation, lane 5: labeling of HSA using probe 4 with competitor (camptothecin, 300 µmol/L), lane 6: labeling of HSA using probe 4 with competitor (oxaliplatin, 300 µmol/L). (e) TEM of E. coli treated with probe 1 under NIR light. (f) TEM of E. coli treated with probe 1 with competitor under NIR light.
Human serum albumin (HSA) is the most abundant protein in plasma and serves as a major carrier for various drugs. Ibuprofen and fatty acids are known to exhibit specific binding to HSA which has been extensively studied. To demonstrate the application potential of our platform for investigating drug delivery, we designed and tested ibuprofen- and fatty acid-based probes (probes 4 and 5). As shown in Fig. 2c, probe 4 labeled HSA (purchased from Shanghai Yuanye Bio-Technology Co., Ltd., China) with a yield of 38%, and successful labeling was also observed through pork muscle tissue (29%) indicating the platform's potential for drug discovery as well as in vivo studies. Meanwhile, competition experiments were conducted (Fig. 2d). When excess ibuprofen was introduced as competitor, the labeling yield decreased significantly to 3%, highlighting the effect of competitive inhibition. Interestingly, the labeling in the presence of other drugs competitor (camptothecin and oxaliplatin) which bind to HSA at sites different from ibuprofen [42,43] resulted in less inhibition (10% and 12% respectively), thus further supporting the binding specificity of HSA to ibuprofen. Additionally, probe 5 functionalized with fatty acids demonstrated a similar trend (Fig. S20 in Supporting information). Taken together, our platform shows strong promise for studying drug-carrier proteins interactions and facilitating targets investigation in vivo.
Nowadays, bacterial infections [44] pose a significant threat to global human health and the rise of antibiotic-resistant bacteria has exacerbated this issue, making it increasingly difficult to treat common infections. Study of carbohydrate-lectin interactions is crucial for microbial decontamination and rapid detection, which is of elevated significance for many applications including antibiotics screening assays, microbiology studies, and clinical examinations. It is well known that bacteria exploit cell surface oligosaccharides for adhesion to host cell which is the initial step in many infectious diseases [45]. The probe designed in this work featuring multivalent ligands on the surface could enhance the binding affinity to bacteria, which drive us to test its ability for bacteria detection. E. coli (DH-5α) was incubated with probe 1 in PBS buffer for 6 h, and a parallel experiment in the presence of competitor (D-(+)-mannose) was conducted as a control. After exposure with NIR light, probe 1/E. coli complex was then imaged with TEM. As shown in Fig. 2e, probe 1 aggregates could be easily observed on the surface of the E. coli after light irradiation. However, the complex was obviously inhibited in the presence of mannose competitor (Fig. 2f), suggesting the specific bacterial recognition of probe 1 led by the binding activity of mannose to the FimH lectin on the pili [46].
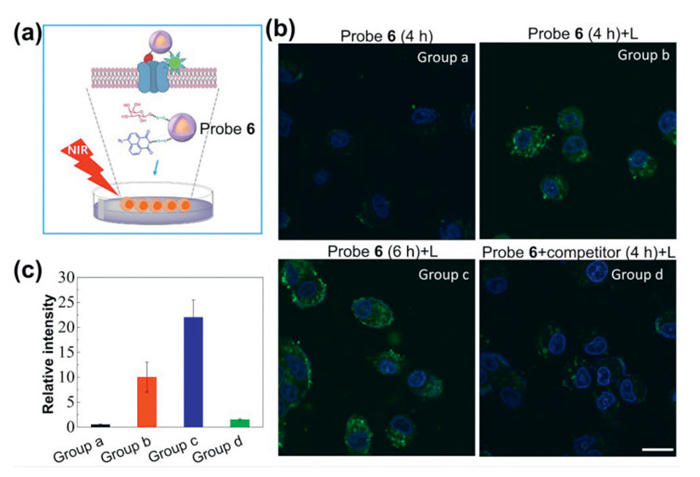
Inspired by the efficient protein labeling and imaging capability of our new strategy, we move our attention to another lectin, peanut agglutinin (PNA), which showed weak binding affinity to methyl β-D-galactose [47]. Probe 6 with D-galactose as ligand was prepared according to the above-mentioned strategy using galactose and azido-naphthalimide with a 1/3 mole ratio (Scheme S11 in Supporting information). Following previously described procedure, PNA were incubated with probe 6 in PBS buffer at 4 ℃ for 2 h to form specific binding and was purified by centrifugation and washing which was finally subjected to SDS-PAGE after cleavage by mercaptoethanol. Interestingly, PNA was purified and enriched in 43% yield, which is a great improvement compared to our previous work (Fig. S21 in Supporting information). Next, competition experiment was conducted using excess methyl-β-D-galactopyranoside as competitor and it was found that competitor led to a significant decrease of labeling yield, suggesting the specific interaction of D-galactose with targets (Fig. S22 in Supporting information). Cell surface-associated proteins play crucial roles in various biological processes, including intercellular communication, immune response, cell differentiation, development, and signal transduction. One particularly significant protein is the asialoglycoprotein receptor (ASGPR), which is highly expressed on hepatocytes as an extracellular protein [48]. The ASGPR functions by binding and clearing asialoglycoproteins from the bloodstream, facilitating receptor-mediated endocytosis [49]. Moreover, it plays essential roles in pathogen recognition and serves as a target for therapeutic interventions. D-Galactose residue serves as a commonly employed ligand for targeting liver cells via ASGPR recognition. Previous efforts mainly focus on imaging the cell surface-associated proteins in vitro [50], while one probe integrate the advantages of both fluorescent imaging and protein labeling and isolation have rarely been explored. Initially, the features of probe 6 for protein labeling and isolation were studied by incubating it with HepG2 liver cancer cell lysate, followed by NIR light irradiation, centrifugation and targets release by mercaptoethanol treatment. It has been reported that ASGPR levels in HepG2 cells are generally low but can be induced with biotin [51], thus we prepared the cell lysate from HepG2 under different treatments. As shown in the Western blot assay (Fig. S23 in Supporting information), probe 6 efficiently labeled and enriched ASGPR in biotin-induced HepG2 cells, whereas labeling was significantly inhibited in the presence of methyl-β-D-galactopyranoside or in the HepG2 lysate without biotin treatment, suggesting the specific binding between probe 6 and ASGPR of HepG2 cells. Next, we evaluated the extracellular protein recognition and imaging capabilities of probe 6 by incubating it with HepG2 cell (Fig. 3a). Figs. 3b and c and Fig. S24 (Supporting information) showed that no fluorescence was detected from cells treated with probe 6 without NIR light exposure. In contrast, strong fluorescence was detected upon NIR light irradiation, indicating the activation of azido-naphthalimide and formation of a fluorescent structure. Prolonged incubation time resulted in even stronger fluorescence, whereas fluorescence was markedly reduced in the presence of methyl β-D-galactose as a competitor, suggesting that the labeling is mediated by the interaction of D-galactose and ASGPR.
Figure 3
Figure 3.
(a) Schematic illustration of labeling extracellular protein using probe 6 in living cells. (b) Fluorescence imaging of HepG2 cells under different treatments (probe 6: 80 µg/mL; competitor: methyl β-D-galactose, 30 µmol/L; L: 3 W of 980 nm light, 5 min). Group a: HepG2 cells and probe 6 were incubated for 4 h, Group b: HepG2 cells and probe 6 were incubated for 4 h, then irradiated with NIR light, Group c: HepG2 cells and probe 6 were incubated for 6 h, then irradiated with NIR light, Group d: HepG2 cells, probe 6 and competitor were incubated for 4 h, then irradiated with NIR light. Scale bar: 20 µm. (c) Relative fluorescence intensity of HepG2 cells under different treatments (error bar indicates SD, n = 3).
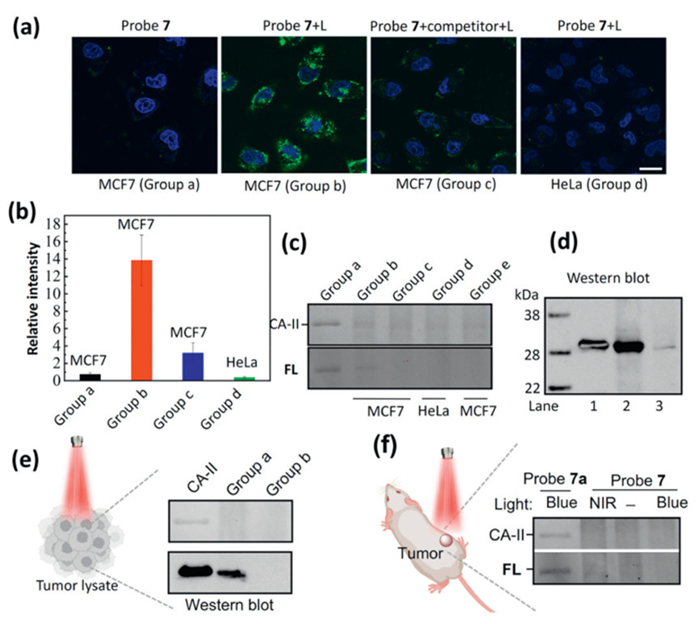
Based on the promising capabilities for labeling, enrichment, and imaging of extracellular proteins of our nanoplatform, we further evaluated its utilization potentiality for intracellular protein in living cells. Human carbonic anhydrase Ⅱ (CA-Ⅱ) is indispensable for maintaining acid-base balance, facilitating gas exchange, regulating ion transport, and influencing physiological processes crucial for human health and function [52]. Its multifaceted roles underscore its importance in both normal physiological functions and potential therapeutic interventions. As CA-Ⅱ can be reversibly inhibited by sulfonamide-carrying ligands, we prepared a probe 7 incorporated with benzenesulfonamide moiety for specifically binding of CA-Ⅱ (Scheme S11). Probe 7 was incubated with CA-Ⅱ in PBS buffer at 4 ℃ for 2 h before irradiation under NIR light. Following NIR-mediated protein labeling mentioned previously, the resultant mixture containing probe 7 and labeled proteins underwent a purification process involving several cycles of centrifugation and wash. Subsequently, the labeled proteins were liberated from probe 7 using mercaptoethanol and subjected to analysis via SDS-PAGE. As anticipated, the experimental results revealed that probe 7 effectively labeled and enriched CA-Ⅱ, achieving a yield of 40% compared to the control group that did not undergo NIR light irradiation (Fig. S25 in Supporting information, left). The fluorogenic property of amino-naphthalimide facilitated the visualization of proteins during in-gel fluorescence scanning (Fig. S25, right). Additional competition experiment using excess of BSA (10-fold) was designed and the result clear indicated the labeling efficiency and specificity of probe 7 toward CA-Ⅱ (Fig. S26 in Supporting information). Next, imaging of the intracellular CA-Ⅱ in living cell was attempted using MCF7 human breast cancer cells which endogenously express CA-Ⅱ (Figs. 4a and b, Fig. S27 in Supporting information) [53]. Upon incubation with probe 7, MCF7 cells without light irradiation showed no fluorescence, while strong green fluorescence was readily detected upon NIR light irradiation, suggesting the endogenously imaging of CA-Ⅱ. In the presence of 4-(2-aminoethyl)benzenesulfonamide as competitor, endogenously imaging fluorescence was significantly suppressed and hardly no fluorescence was shown in HeLa cell, which show less CA-Ⅱ expression. To detail the labeling of endogenous CA-Ⅱ, MCF7 and HeLa cell labeled with probe 7 were lysed respectively. The cell lysate was harvested and separated by SDS-PAGE and analyzed by western blot. As shown in Fig. 4c, Figs. S28 and S29 (Supporting information), obvious fluorescence band was found in the MCF7 cell treated with probe 7 under light irradiation. Meanwhile, Western blot data indicated that MCF7 express more CA-Ⅱ compared to HeLa (Fig. 4d, Fig. S30 in Supporting information), which is consistent to the previous report [54].
Figure 4
Figure 4.
(a) Fluorescence imaging of cells under different treatments (probe 7: 80 µg/mL, incubation time: 6 h, competitor: (4-(2-aminoethyl)benzenesulfonamide, 30 µmol/L, L: 3 W of 980 nm light for 5 min). Group a: MCF7 cells incubated with probe 7, Group b: MCF7 cells incubated with probe 7, then irradiated with NIR light, Group c: MCF7 cells incubated with probe 7 and competitor, then irradiated with NIR light, Group d: Hela cells incubated with probe 7, then irradiated with NIR light. Scale bar: 20 µm. (b) Relative fluorescence intensity of cells (error bar indicates SD, n = 3). (c) Labeling and visualization of CA-Ⅱ (group a: CA-Ⅱ treated with control probe 7a (see Supporting information) under 470 nm blue light; group b: MCF7 cells treated with probe 7 under NIR light; group c: MCF7 cells treated with probe 7 without NIR irradiation; group d: HeLa cells treated with probe 7 under NIR light; group e: MCF7 cells treated with probe 7 and competitor) (d) Western blot for CA-Ⅱ (from left to right: CA-Ⅱ, MCF7 cells treated with probe 7 under irradiation, HeLa cells treated with probe 7 under NIR light. (e) Western blot for labeling of CA-Ⅱ using probe 7 (1 mg) from tumor lysate (60 µg). Lane 1: CA-Ⅱ input; group a: labeling of CA-Ⅱ from tumor lysate using probe 7 with NIR light; group b: labeling of CA-Ⅱ using probe 7 without NIR light. (f) Labeling and visualization of CA-Ⅱ in vivo (from left to right: CA-Ⅱ treated with probe 7a under 470 nm light, the tumor was irradiated with 980 nm laser, the tumor was not irradiated, the tumor was irradiated with 470 nm light).
Until now, photoaffinity-based approach has been primarily limited to cell-based models because conventional UV light struggles to penetrate the skin. One of the most important advantages of our nanoplatform is the potential for protein labeling in vivo as NIR light penetrate deeper than UV light [55]. Inspired by the exciting result in pork tissue model, we evaluated the in vivo protein labeling potential using probe 7. MCF7 xenograft tumor model was developed using BALB/c mice and all experiments were approved by the Animal Ethics Committee of Dalian University of Technology (ethics approval No. DUTSCE240626-02). Before in vivo proteins labeling, we conducted the CA-Ⅱ labeling from MCF7 tumor lysate using probe 7. As shown in Fig. 4e and Fig. S31 (Supporting information), probe 7 successfully labeled and enriched CA-Ⅱ, indicating its potential for protein labeling in complex environment. Encouraged by this result, in vivo protein labeling was carried out and probe 7 in PBS solution was injected into tumor via intratumoral injection. Upon NIR light irradiation, the tumor was harvested, lysed and the supernatant was subjected to SDS-PAGE for staining analysis. As can be seen from Fig. 4f and Fig. S32 (Supporting information), In-gel fluorescence demonstrating the protein labeling potential of probe 7in vivo. Meanwhile, the safety evaluation of the NIR light activatable nanoplatform in vivo were performed and no adverse physiological effects and organ damages were found during the treatment of probe 7, consistent with the cell viability assay (Figs. S33–S36 in Supporting information).
In conclusion, we have developed a novel NIR light-activated nanoplatform and evaluated its potential applications for protein labeling, enrichment and visualization. Azido-naphthalimides can be easily activated by UV and blue light emitted from upconversion nanoparticles under NIR light irradiation. The resulting amino-naphthalimides not only crosslink nearby target proteins but also facilitate their imaging. Benefitting from the nanocharacteristics of this platform, we successfully labeled, enriched lectins and drugs carrier proteins with excellent efficiency, selectivity and convenience. Due to its high ligand loading capacity, this nanoplatform has proven valuable for bacterium detection. Furthermore, we demonstrated the versatility of this NIR light-activated nanoplatform in affinity labeling and imaging of both extracellular and intracellular proteins within living cells. Moreover, in vivo protein labeling was achieved using this novel NIR light-activatable nanoplatform, leveraging the superior tissue penetration of NIR light. We believe this platform will expand the application of protein modification strategies, especially photoaffinity-based approach in living systems, thereby facilitating biological discoveries and mechanistic interrogation.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Peng Liu: Methodology, Investigation, Data curation. Shengli Zhang: Data curation. Tingting Zhang: Data curation. Yu Si: Data curation. Ziang Liu: Data curation. Xiao Qian: Data curation. Yingxu Wu: Data curation. Yuan Liang: Methodology, Investigation. Wen Sun: Investigation. Engin U. Akkaya: Investigation. Lei Wang: Writing – original draft, Supervision, Methodology, Funding acquisition.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 22007008), the LiaoNing Revitalization Talents Program (No. XLYC1907021) and the Fundamental Research Funds for the Central Universities (Nos. DUT23YG120, DUT19RC(3)009).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2025.110966.
[1]
D.E. Scott, A.R. Bayly, C. Abell, J. Skidmore, J. Nat. Rev. Drug Discov. 15 (2016) 533–550. doi: 10.1038/nrd.2016.29
[2]
I. Hamachi, S. Tsukiji, M. Miyagawa, Y. Takaoka, T. Tamura, Nat. Chem. Biol. 5 (2009) 341–343.
[3]
M. Hashimoto, Y. Hatanaka, Eur. J. Org. Chem. 2008 (2008) 2513–2523. doi: 10.1002/ejoc.200701069
[4]
A.V. West, Y. Amako, C.M. Woo, J. Am. Chem. Soc. 144 (2022) 21174–21183. doi: 10.1021/jacs.2c08257
[5]
L. Wang, A. Ishida, Y. Hashidoko, M. Hashimoto, Angew. Chem. Int. Ed. 56 (2017) 870–873. doi: 10.1002/anie.201610371
[6]
X. Ma, Y. Han, K. Liu, et al., Chin. Chem. Lett. 34 (2023) 107595.
[7]
P. Thirumurugan, D. Matosiuk, K. Jozwiak, Chem. Rev. 113 (2013) 4905–4979. doi: 10.1021/cr200409f
[8]
D. Saimi, Z. Chen, Smart Mol. 1 (2023) e20230002.
[9]
H.A. Flaxman, C. Chang, H. Wu, C.H. Nakamoto, C.M. Woo, J. Am. Chem. Soc. 141 (2019) 11759–11764. doi: 10.1021/jacs.9b03764
[10]
W. Chi, Q. Qiao, C. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 20215–20223. doi: 10.1002/anie.202010169
[11]
J.C. Paulson, O. Blixt, B.E. Collins, Nat. Chem. Biol. 2 (2006) 238–248. doi: 10.1038/nchembio785
[12]
K. Sakurai, Y. Hatai, A. Okada, Chem. Sci. 7 (2016) 702–706.
[13]
D. Kuwahara, T. Hasumi, H. Kaneko, et al., Chem. Commun. 50 (2014) 15601–15604.
M.H. Lee, J.H. Han, P. Kwon, et al., J. Am. Chem. Soc. 134 (2012) 1316–1322. doi: 10.1021/ja210065g
[51]
A.M. Pujol, M. Cuillel, O. Renaudet, et al., J. Am. Chem. Soc. 133 (2011) 286–296. doi: 10.1021/ja106206z
[52]
V. Alterio, A. Di Fiore, K.D. Ambrosio, C.T. Supuran, G. De Simone, Chem. Rev. 112 (2012) 4421–4468. doi: 10.1021/cr200176r
[53]
J.C. Mallory, G. Crudden, A. Oliva, et al., Mol. Pharmacol. 68 (2005) 1747–1756. doi: 10.1124/mol.105.016519
[54]
S. Dai, D. Yang, J. Am. Chem. Soc. 142 (2020) 17156–17166. doi: 10.1021/jacs.0c08068
[55]
Y. Cai, Z. Wei, C. Song, et al., Chem. Soc. Rev. 48 (2019) 22–37. doi: 10.1039/c8cs00494c
Figure 1
(a) Schematic illustration of protein labeling and visualization under NIR light irradiation. (b) Structures of probes 1–3. (c) Fluorescence spectra of probe 1 (2 mg/mL) under NIR light irradiation (980 nm). Ex. slit: 5 nm, Em. slit: 5 nm. (d) Binding study of FITC-labeled Con A (16 µg) with probe 1, fluorescence of the supernatant after removal of Con A/probe 1 complex. Ex. slit: 10 nm, Em. slit: 10 nm. (e) Labeling of Con A using different probe. Lane 1: Con A input (2 µg), lanes 2–4: labeling of Con A (2 µg) using indicated probe (1 mg), lane 5: negative control without NIR irradiation using probe 2 (1 mg). (f) Labeling yield of Con A using different probe (error bar indicates SD, n = 4).
Figure 2
(a) Labeling of Con A (2 µg) using probe 1 (1 mg) with or without competitor. Lane 1: Con A input, lane 2: labeling of Con A using probe 1, lane 3: labeling of Con A using probe 1 with competitor (D-(+)-mannose, 300 µmol/L), lane 4: negative control without NIR irradiation. (b) Labeling of Con A in mixed proteins using probe 1 (1 mg), data were shown in stained gel (left) and in-gel fluorescence (right). Lane 1: Con A input, lane 2: Con A (2 µg) and BSA (20 µg), lane 3: labeling of Con A from mixed proteins. (c) Labeling of HSA (2 µg) using probe 4 (1 mg). Lane 1: HSA input, lane 2: labeling of HSA under NIR light (980 nm), lane 3: labeling of HSA under NIR light (980 nm) through 3 mm of pork tissue, lane 4: negative control without light irradiation. (d) Labeling of HSA (2 µg) using probe 4 (1 mg) with or without competitor. Lane 1: HSA input, lane 2: labeling of HSA using probe 4, lane 3: labeling of HSA using probe 4 with competitor (ibuprofen, 300 µmol/L), lane 4: negative control without NIR irradiation, lane 5: labeling of HSA using probe 4 with competitor (camptothecin, 300 µmol/L), lane 6: labeling of HSA using probe 4 with competitor (oxaliplatin, 300 µmol/L). (e) TEM of E. coli treated with probe 1 under NIR light. (f) TEM of E. coli treated with probe 1 with competitor under NIR light.
Figure 3
(a) Schematic illustration of labeling extracellular protein using probe 6 in living cells. (b) Fluorescence imaging of HepG2 cells under different treatments (probe 6: 80 µg/mL; competitor: methyl β-D-galactose, 30 µmol/L; L: 3 W of 980 nm light, 5 min). Group a: HepG2 cells and probe 6 were incubated for 4 h, Group b: HepG2 cells and probe 6 were incubated for 4 h, then irradiated with NIR light, Group c: HepG2 cells and probe 6 were incubated for 6 h, then irradiated with NIR light, Group d: HepG2 cells, probe 6 and competitor were incubated for 4 h, then irradiated with NIR light. Scale bar: 20 µm. (c) Relative fluorescence intensity of HepG2 cells under different treatments (error bar indicates SD, n = 3).
Figure 4
(a) Fluorescence imaging of cells under different treatments (probe 7: 80 µg/mL, incubation time: 6 h, competitor: (4-(2-aminoethyl)benzenesulfonamide, 30 µmol/L, L: 3 W of 980 nm light for 5 min). Group a: MCF7 cells incubated with probe 7, Group b: MCF7 cells incubated with probe 7, then irradiated with NIR light, Group c: MCF7 cells incubated with probe 7 and competitor, then irradiated with NIR light, Group d: Hela cells incubated with probe 7, then irradiated with NIR light. Scale bar: 20 µm. (b) Relative fluorescence intensity of cells (error bar indicates SD, n = 3). (c) Labeling and visualization of CA-Ⅱ (group a: CA-Ⅱ treated with control probe 7a (see Supporting information) under 470 nm blue light; group b: MCF7 cells treated with probe 7 under NIR light; group c: MCF7 cells treated with probe 7 without NIR irradiation; group d: HeLa cells treated with probe 7 under NIR light; group e: MCF7 cells treated with probe 7 and competitor) (d) Western blot for CA-Ⅱ (from left to right: CA-Ⅱ, MCF7 cells treated with probe 7 under irradiation, HeLa cells treated with probe 7 under NIR light. (e) Western blot for labeling of CA-Ⅱ using probe 7 (1 mg) from tumor lysate (60 µg). Lane 1: CA-Ⅱ input; group a: labeling of CA-Ⅱ from tumor lysate using probe 7 with NIR light; group b: labeling of CA-Ⅱ using probe 7 without NIR light. (f) Labeling and visualization of CA-Ⅱ in vivo (from left to right: CA-Ⅱ treated with probe 7a under 470 nm light, the tumor was irradiated with 980 nm laser, the tumor was not irradiated, the tumor was irradiated with 470 nm light).

 DownLoad:
DownLoad:


 下载:
下载:





 下载:
下载:
