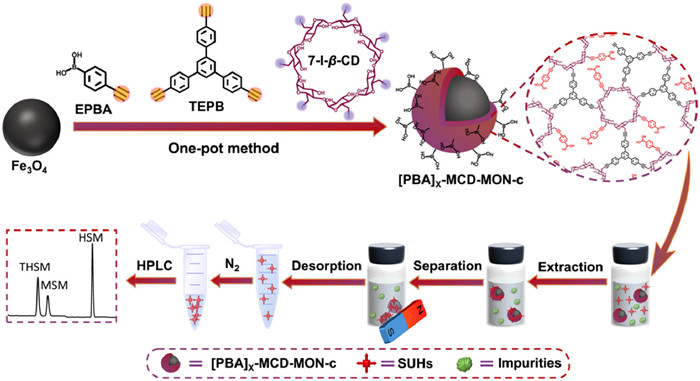
Figure 1.
Schematic of one-pot synthesis of [PBA]X−MCD-MON-c.
One-pot in situ doping synthesis of phenylboronic acid-functionalized magnetic-cyclodextrin microporous organic network for specific enrichment and detection of sulfonylurea herbicides
Chun-Ying Xu , Xiao-Lin Luan , Yuan-Yuan Cui , Cheng-Xiong Yang

Sulfonylureas herbicides (SUHs) are vital commercial herbicides, accounting to ~11% of global herbicide market [1]. Nearly 40 types of SUHs have been currently synthesized and extensively used for weed control owing to their ultra-high herbicidal activity, ultra-low application dosage, and high selectivity [2]. SUHs have strong polarity and good stability and are widely used; therefore, they are inevitably present in environmental water and cannot be easily degraded or removed [3]; this poses a potential risk on ecological, environmental, and human health. Therefore, the use of SUHs such as chlorsulfuron, metsulfuron-methyl (MSM) and ethametsulfuron is prohibited in China. The European Union, Council Directive regulations (80/778/EC) has recommended the maximum residue limit of SUHs in drinking water as 0.1 µg/L for single species and 0.5 µg/L for total SUH content. Thus, sensitive and reliable analytical methods must be urgently developed for monitoring trace amounts of SUHs in complex food and water samples.
However, SUHs have complex sample matrix and are present in low concentrations; therefore, their direct detection is difficult [4-8]. Introduction of effective sample pretreatment methods is essential before chromatographic determination of SUHs [9-14]. Magnetic solid-phase extraction (MSPE) has garnered considerable attention owing to its simple operation, time- and energy-saving processes, low solvent consumption, and easy recovery [15]. As adsorbents are crucial for MSPE, diverse magnetic adsorbents have been synthesized and used for enriching SUHs such as TpBD-(NH2)2@Fe3O4, Fe3O4@DODMAC@silica, Fe3O4@SiO2−MIP, AL-MoS2@Fe3O4, IL-MNPs, g-C3N4/Fe3O4@ZIF-8, and Fe3O4@PDA-DES [16-22]. However, these adsorbents more or less have the drawbacks of complex synthesis processes and insufficient selectivity. Therefore, the rational design and synthesis of highly efficient and selective magnetic adsorbents for SUHs remain a challenge.
Microporous organic networks (MONs) are new porous materials synthesized via Sonogashira coupling between aromatic alkynes and halides [23]. MONs have large surface areas, enhanced chemical and thermal stabilities, and controllable pore structures; therefore, they have been extensively used as advanced adsorbents in the sample pretreatment of diverse analytes from various matrices [24-29]. Cyclodextrin-based MON (CD-MON), a booming subclass of MONs, synthesized using unique CD monomers have garnered considerable attention recently. CD-MONs integrate the inherent cavity of CD and conjugated skeleton of MONs, which can provide additional host-guest and hydrogen bonding interaction sites. This improved the selectivity and performance of CD-MONs for pollutant removal and MSPE, making them ideal candidates for synthesizing efficient magnetic adsorbents [30,31].
Boronate affinity materials (BAMs) can covalently combine with cis-diol molecules to form five- or six-membered cyclic esters for the selective enrichment of cis-diol-containing compounds [32]. BAMs have been recently used to enhance the affinity of N-containing compounds via specific B-N coordination [33-37]. SUHs have characteristics of N-containing compounds (sulfonylurea and N-heterocycle rings) and CD-MONs have abundant hydrogen bonding, host-guest, hydrophobic, and π-π interaction sites; therefore, doping phenylboronic acid (PBA) molecules into CD-MONs for synthesis novel CD-MON-based magnetic BAMs with specific B-N coordination sites is a feasible approach for the efficient and selective enrichment of SUHs. However, such an approach has not been reported yet.
Magnetic MONs are currently synthesized via direct synthesis [26,28] and postsynthesis [27,29]. Direct synthesis uses different functional monomers for yielding magnetic MONs; however, the limited availability of such monomers restricts the applicability of this method. This limitation can be addressed via post-synthesis; however, it has multiple steps and low grafting rate and is time-consuming. Therefore, novel and simple synthesis methods have to be developed for synthesizing magnetic MONs.
Herein, we report a facile one-pot in situ doping strategy to prepare novel magnetic PBA-functionalized CD-MON for selective MSPE of trace SUHs from complex food and environmental water samples prior to HPLC determination. A three-components co-doping method was employed using heptakis-6-iodo-6-deoxy-beta-cyclodextrin (7-I-β-CD) as the knot, 1, 3, 5-tris(4-ethynylphenyl)benzene (TEPB) and (4-ethynylphenyl)boronic acid (EPBA) with different molar ratios (X=[EPBA]/[TEPB]) as the linkers, and Fe3O4 as the magnetic core to synthesize [PBA]X−MCD-MON-c (c denotes the molar amount of 7-I-β-CD) (Fig. 1). X and c were varied to regulate the SUHs extraction performance of [PBA]X−MCD-MON-c. [PBA]3/4−MCD-MON-0.04 had a large surface area, high saturation magnetism, and good reusability and stability. It also contained abundant B-N coordination, π-π interaction, hydrogen bonding, and host-guest interaction sites, which enabled efficient MSPE of SUHs. This study provides a new strategy to synthesize novel functionalized CD-MONs and reveals the prospects of [PBA]X−MCD-MON-c for enriching N-containing compounds.
For efficient SUH extraction, [PBA]X−MCD-MON-c with different EPBA/TEPB doping ratios (X=0/7, 1/6, 2/5, 3/4, and 4/3) was primarily optimized using 0.04 mmol of a 7-I-β-CD (c=0.04). The obtained adsorbents were characterized via Fourier transform infrared (FT-IR) spectroscopy, X-ray photoelectron spectroscopy (XPS), scanning electron microscopy (SEM), water contact angle measurements, and X-ray diffraction (XRD) (Figs. 2 and 3, Figs. S1 and S2 in Supporting information). The appearance of characteristic FT-IR spectra at 1490 and 1601 cm−1 corresponding to aromatic C=C [38], 2212 cm−1 corresponding to C≡C, 3425 cm−1 corresponding to -OH [39], and 1359 cm−1 corresponding to B-O on [PBA]X−MCD-MON-0.04 confirmed the successful incorporation of PBA molecules in these adsorbents via the proposed in situ doping strategy (Fig. 3a, Figs. S1a and b). B 1s peak in the XPS spectrum of [PBA]X−MCD-MON-0.04 further confirmed this result (Fig. S1c). SEM images revealed that Fe3O4 particles and [PBA]X−MCD-MON-0.04 exhibited uniform spherical morphology with a particle size of 250–300 nm, indicating that different doping ratios have insignificant effects on their morphologies (Figs. 2a-f). Similarly, the saturation magnetization of [PBA]X−MCD-MON-0.04 was ~50.0 emu/g (Table S1 in Supporting information), which was independent of the doping ratios (Fig. 3b). The water contact angles of [PBA]X−MCD-MON-c decreased from 137.7° to 108.6° as the doping ratios increased from 1:6 to 4:3 (Fig. S2), indicating that the doped EPBA effectively improved the hydrophilicity of [PBA]X−MCD-MON-0.04. The XRD peaks of [PBA]X−MCD-MON-c were observed at 30.2°, 35.5°, 43.8°, 53.4°, 57.1°, and 62.7°, corresponding to the (220), (311), (400), (422), (551), and (440) planes for Fe3O4, respectively (Fig. 3c, and Fig. S1d) [28]. These results indicate the successful synthesis of [PBA]X−MCD-MON-c.

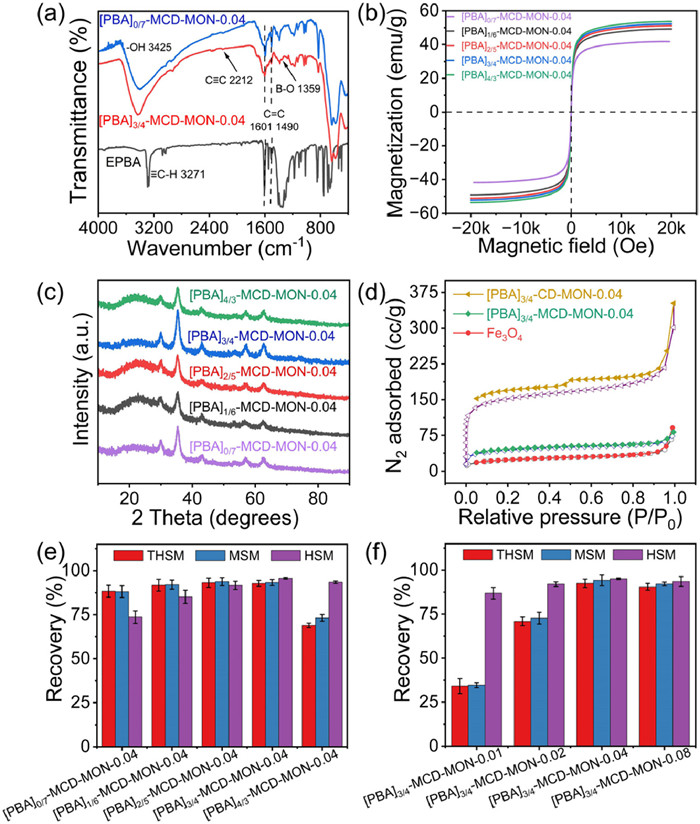
Three typical SUHs, namely thifensulfuron-methyl (THSM), MSM, and halosulfuron-methyl (HSM), were then used to evaluate the effect of doping ratios on their enrichment efficiencies (Fig. 3e, and Table S2 in Supporting information). The recoveries first increased and then decreased as the X doping ratios increased from 0:7 to 4:3. The highest recovery was achieved when X was 3:4.
The shell thickness of [PBA]3/4−MCD-MON-c was further regulated by tuning the monomer amount c (0.01, 0.02, 0.04, and 0.08 mmol) to determine its effect on the morphology, magnetism, and extraction performance for SUHs. The results revealed that the surface of [PBA]3/4−MCD-MON-c changed from rough to smooth as the c increased (Figs. 2e, g-i) and a uniform shell with thickness of ~15 nm was formed when c=0.04 (Fig. 2j). The XRD patterns of [PBA]3/4−MCD-MON-c were not affected by shell thickness (Fig. S1d) [31]. Moreover, magnetization decreased from 57.2 emu/g to 46.2 emu/g as c increased from 0.01 to 0.08 (Table S1), proving that thicker shells were obtained under higher monomer concentrations. Furthermore, [PBA]3/4−MCD-MON-0.04 was efficiently dispersed and rapidly recycled from aqueous solutions (insets in Fig. S1e). The effect of shell thickness on SUH recovery was also explored (Fig. 3f), which revealed that [PBA]3/4−MCD-MON-0.04 exhibited the best enrichment efficiency and was therefore used for subsequent experiments.
[PBA]3/4−MCD-MON-0.04, [PBA]3/4-CD-MON-0.04, and Fe3O4 were further characterized via N2 adsorption-desorption experiments (Fig. 3d). The Brunauer-Emmett-Teller (BET) surface areas of [PBA]3/4−MCD-MON-0.04, [PBA]3/4-CD-MON-0.04, and Fe3O4 were 160.6, 568.1, and 84.1 m2/g, respectively. [PBA]3/4−MCD-MON-0.04 had a lower BET surface area than [PBA]3/4-CD-MON-0.04, which indicated the successful integration of [PBA]3/4-CD-MON-0.04 with Fe3O4. [PBA]3/4−MCD-MON-0.04 also exhibited remarkable solvent and thermal stabilities after immersion in various solvents for seven days and heating up to 250 ℃ (Figs. S1f and S3 in Supporting information), promoting its practical application for sample pretreatment. Further, the extraction recoveries of SUHs on [PBA]3/4−MCD-MON-0.04 were higher than those on Fe3O4@CD-MON-PBA synthesized using the post-synthesis strategy (Fig. S4a in Supporting information), confirming the advantages of using one-pot in situ doping strategy.
The extraction parameters, including eluent type and volume, adsorbent dosage, extraction and elution time, solution pH, and ionic strength, of [PBA]3/4−MCD-MON-0.04 were optimized for the MSPE of SUHs (Fig. 4, and Fig. S4b in Supporting information). Based on the structural characteristics of SUHs, binary solvents of MeOH and formic acid (FA) were used as eluents. SUHs was efficiently desorbed from [PBA]3/4−MCD-MON-0.04 using 0.6 mL of 5% FA MeOH (5:95, v/v) with 4 min ultrasonication (Figs. 4a and b, and Fig. S4b). The effects of adsorbent dosage and extraction time of 2–6 mg and 3–15 min, respectively, on the SUH adsorption were investigated (Figs. 4c and d). The results showed that the SUHs were extracted using 4 mg of [PBA]3/4−MCD-MON-0.04 and 9 min of extraction time.
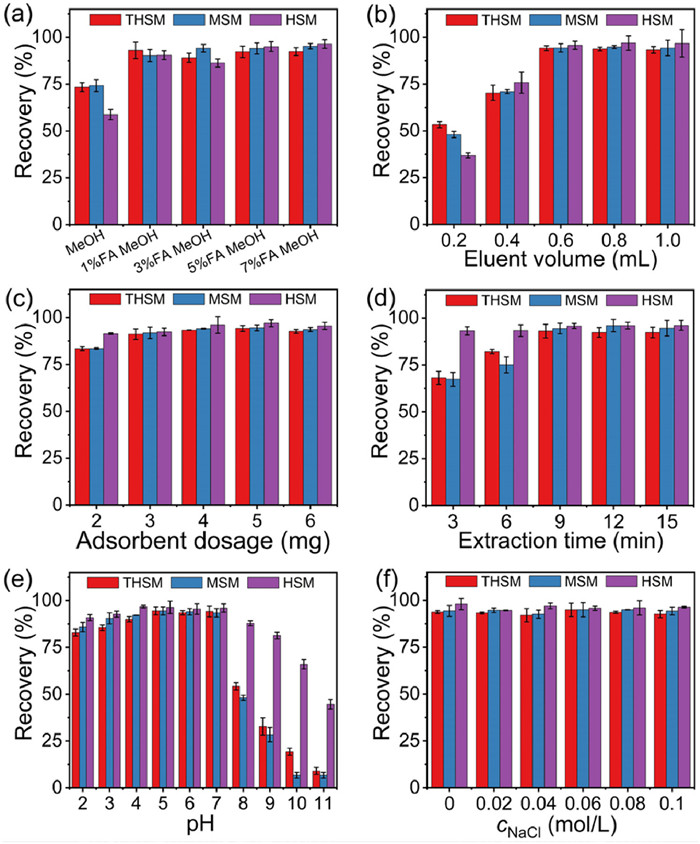
The effect of varying pH within 2.0–11.0 on the SUH recovery was investigated (Fig. 4e). The SUH recovery increased when pH increased from 2 to 5; it remained constant at pH 5–7 and obviously declined as the pH increased to 11. B-N coordination is generally formed when N atoms on SUHs are not protonated and PBA on [PBA]3/4−MCD-MON-0.04 is not dissociated [35]. However, the urea groups on SUHs appeared protonated at low pH range [37], which was not conducive to the formation of B-N coordination between SUHs and [PBA]3/4−MCD-MON-0.04 at pH 2–5. Therefore, π-π interaction was considered the dominant mechanism in this pH range. Similarly, PBA molecules (pKa=7.97) doped in [PBA]3/4−MCD-MON-0.04 dissociated under alkaline conditions; therefore, they are also not favorable for the generation of B-N coordination as the extraction efficiencies decreased at pH 8–11. Moreover, electrostatic repulsion occurred at pH 8–11 due to the deprotonation of SUHs and dissociation of PBA, decreasing the extraction efficiency. In contrast, B-N coordination and hydrogen bonding, hydrophobic, and π-π interactions between SUHs and [PBA]3/4−MCD-MON-0.04 synergistically yielded the maximum SUH recovery at pH 5–7 (Fig. 4e). These findings confirmed that the pH of sample solution need not be excessively adjusted during each MSPE.
SUH recovery was only slightly impacted within NaCl concentrations of 0–0.1 mol/L, indicating the good anti-interference ability of [PBA]3/4−MCD-MON-0.04 toward NaCl (Fig. 4f). In addition, the morphology, magnetization, hydrophobicity, structure, and thermostability of [PBA]3/4−MCD-MON-0.04 after eight extraction cycles were similar to those of fresh [PBA]3/4−MCD-MON-0.04, indicating its excellent stability during MSPE (Fig. S5 in Supporting information). Furthermore, the recovery rate of SUHs on regenerated adsorbents after eight extraction-desorption cycles remained above 90%, proving the outstanding reusability of [PBA]3/4−MCD-MON-0.04 for SUH adsorption (Fig. S6a in Supporting information). Moreover, four parallelly synthesized [PBA]3/4−MCD-MON-0.04 exhibited comparable recoveries for SUHs, indicating its good reproducibility (Fig. S6b in Supporting information).
MSPE-HPLC-UV method based on [PBA]3/4−MCD-MON-0.04 has a wide linear range of 0.2–500 µg/L, low limit of detection (LOD, S/N=3.0) of 0.05–0.5 µg/L, low limit of quantification (LOQ, S/N=10) of 0.2–1.5 µg/L, large enrichment factor of 92.5–96.1, and good intra-day, inter-day, and batch-to-batch precisions of 0.4%−7.6% for SUHs in cucumber and river water samples (Tables S3 and S4 in Supporting information). The adsorption of SUHs on [PBA]3/4−MCD-MON-0.04 followed the Langmuir model (R2 > 0.991) with monolayer and homogeneous adsorption processes (Fig. S7 in Supporting information) [40]. Their adsorption capacities for THSM, MSM, and HSM, were of 145.0, 169.5, and 140.8 mg/g, respectively (Table S5 in Supporting information). The proposed method also consumed lowest adsorbent amounts and required lesser extraction time compared with most reported methods (Table S6 in Supporting information). The LOD of the proposed method was almost similar to the methods that used TpBD-(NH2)2@Fe3O4, AL-MoS2@Fe3O4, PVE@anode, PMED@cathode, and Fe3O4@PDA-DES and MIPs as the adsorbents, and was considerably lower than those of Nd2Fe14B/UIO-66 (Zr)-NH2−MSBSE-HPLC-UV and MGO@PDA-MIPs-MSPE-HPLC-UV, respectively. These findings confirmed that the proposed method had the advantages of low LOD, adsorbent dosage, and extraction time. In addition to SUHs, [PBA]3/4−MCD-MON-0.04 also exhibited high recoveries (82.7%−99.4%) for benzimidazole fungicides, sulfonamides, and phenylurea herbicides (Table S7 in Supporting information), indicating that it is a good adsorbent for N-containing compounds.
Subsequently, the SUH extraction mechanisms were evaluated. Fig. 3e showed that [PBA]0/7−MCD-MON-0.04 without EPBA doping yielded high SUH recoveries (>70%), suggesting the existence of π-π, host-guest, and hydrogen bond interactions. The recoveries gradually increased with increasing doping ratios, indicating the positive role of PBA molecules and the synergistic extraction mechanisms between [PBA]3/4−MCD-MON-0.04 and SUHs. Combining these results with the pH-influencing factors, we believe that π-π, hydrogen bond, host-guest interactions as well as B-N coordination are involved in SUH extraction.
FT-IR and XPS were performed to prove these extraction mechanisms (Fig. 5 and Fig. S8 in Supporting information). The XPS spectra of [PBA]3/4−MCD-MON-0.04 before and after THSM extraction were compared. New N 1s peak appeared after the extraction, revealing the successful uptake of THSM on [PBA]3/4−MCD-MON-0.04 (Fig. 5a and Fig. S8). The C 1s peaks for C=C/C—C, C≡C, and C—O—C shifted from 284.27, 285.42, and 288.49 eV to 283.99, 284.98, and 288.05 eV (Figs. 5b and c), respectively, after MSPE, demonstrating the π-π and host-guest interaction between THSM and [PBA]3/4−MCD-MON-0.04 [31]. Moreover, the O 1s peaks for C—OH/B-OH and C—O—C at 531.12 and 529.79 eV were separately shifted to 531.06 and 529.67 eV after MPSE, suggesting the presence of hydrogen bonding interaction and B-N coordination (Figs. 5d and e). The B 1s peak shifted from 189.72 to 188.60 eV after THSM extraction, indicating B-N coordination between [PBA]3/4−MCD-MON-0.04 and THSM (Figs. 5f and g).
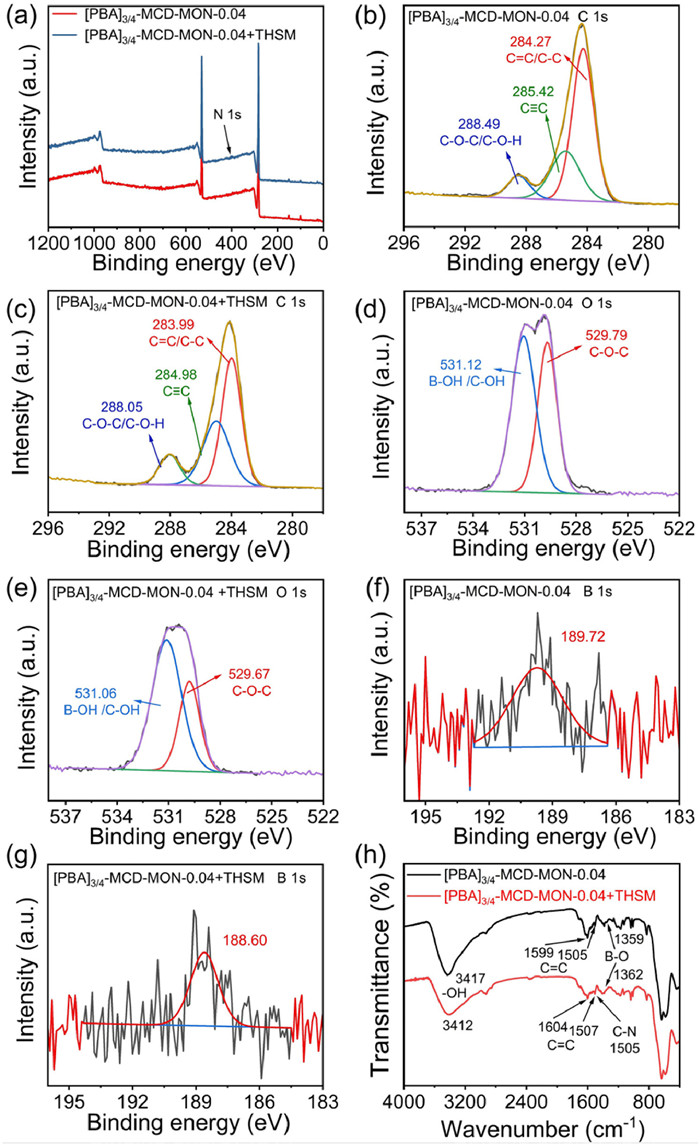
These findings were also supported by FT-IR data (Fig. 5h). The FT-IR spectrum of [PBA]3/4−MCD-MON-0.04 showed a C—N peak, confirming successful THSM extraction. The peaks corresponding to -OH stretching at 3417 cm−1 and C=C stretching at 1505 and 1599 cm−1 for [PBA]3/4−MCD-MON-0.04 individually shifted to 3412 and 1507 and 1604 cm−1 after extraction, indicating possible hydrogen bonding and π-π interaction between [PBA]3/4−MCD-MON-0.04 and THSM. The B-O stretching peak for [PBA]3/4−MCD-MON-0.04 shifted from 1359 to 1362 cm−1 after extraction, confirming B-N coordination between [PBA]3/4−MCD-MON-0.04 and THSM. CDs are a class of supramolecular compounds that can provide multiple host-guest recognition sites for SUH extraction. These results revealed the presence of hydrogen bonding, host-guest, and π-π interactions as well as B-N coordination during MSPE (Fig. S9 in Supporting information).
The feasibility and practicability of [PBA]3/4−MCD-MON-0.04-based MSPE-HPLC-UV method were evaluated by analyzing its performance in SUH extraction from food and environmental water samples (Fig. S10 in Supporting information). The matrix effect values of real samples were between 91.3% and 102.5%, revealing their insignificant matrix interference for SUHs using the proposed method [41]. The recoveries of SUHs in river water, lake water, cucumber, tomato, and carrot at spiking levels of 5, 10, and 20 µg/L were 85.4%−111% (Table S8 in Supporting information). This confirmed the good applicability of the proposed method and the potential of [PBA]3/4−MCD-MON-0.04 for enriching trace SUHs from complex food and environment samples.
In summary, a novel one-pot in situ doping strategy was proposed herein to synthesize [PBA]3/4−MCD-MON-0.04 for efficient MSPE and monitoring trace amounts of SUHs in complex samples. The extraction efficiency was facilely regulated by varying the doping ratios and shell thickness. The prepared adsorbent exhibited numerous π-π, host-guest, B-N coordination, and hydrogen bonding interaction sites, which facilitated the sensitive and selective detection of SUHs in diverse real samples. Based on the specific B-N coordination sites within structures, [PBA]3/4−MCD-MON-0.04 served as an advanced candidate for enriching N-containing compounds. This study provides a simple and feasible strategy for synthesizing novel multifunctional CD-MON-based magnetic adsorbents and reveals the significant roles of B-N coordination for the selective enrichment of trace N-containing compounds from complex samples. The following limitations will be addressed in future studies: (1) Complex preparation of Fe3O4 should be simplified; (2) the universality of one-pot in situ doping synthesis method should be evaluated in detail; and (3) UV detectors should will be replaced with MS detector to considerably improve the sensitivity.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Chun-Ying Xu: Writing – original draft, Investigation, Data curation. Xiao-Lin Luan: Data curation. Yuan-Yuan Cui: Supervision, Funding acquisition. Cheng-Xiong Yang: Writing – review & editing, Supervision, Funding acquisition, Conceptualization.
This work was supported by the National Natural Science Foundation of China (Nos. 22174071 and 22206114), the Natural Science Foundation of Shandong Province (Nos. ZR2022YQ08 and ZR2022QB085), the Innovation Team of Shandong Higher School Youth Innovation Technology Program (No. 2023KJ344), and the Academic Promotion Program (No. 2019LJ003) and Joint Innovation Team for Clinical & Basic Research (No. 202401) of Shandong First Medical University.
Supplementary material associated with this article can be found, in the online version, at doi:
Y.R. Yamada, M. Murase, Y. Goto, et al., J. Agric. Food Chem. 71 (2023) 5006–5015. doi: 10.1021/acs.jafc.2c09077
C.S. Li, N. Zhang, J.X. Chen, et al., Environ. Pollut. 255 (2019) 113150. doi: 10.1016/j.envpol.2019.113150
Y. de Lafontaine, C. Beauvais, A.J. Cessn, et al., Sci. Total Environ. 479-480 (2014) 1–10. doi: 10.1016/j.scitotenv.2014.01.094
M. Singh, A. Srivastava, Y.K. Sharma, S. Singh, S.P. Singh, Microchim. Acta 187 (2020) 490. doi: 10.1007/s00604-020-04464-8
J.P. Ma, S. Li, G.G. Wu, et al., J. Colloid Interf. Sci. 553 (2019) 834–844. doi: 10.1016/j.jcis.2019.06.082
X.L. Guo, T.Z. Ren, J.C. Ji, Y. Yang, X. Di, Food Chem. 396 (2022) 133652. doi: 10.1016/j.foodchem.2022.133652
M.J. Lerma-Garcıá, E.F. Simó-Alfonso, M. Zougagh, Á. Ríos, Talanta 105 (2013) 372–378. doi: 10.1016/j.talanta.2012.10.056
Y. Peng, Y. Xie, J. Luo, et al., Anal. Chim. Acta 674 (2010) 190–200. doi: 10.1016/j.aca.2010.06.022
S.H. Zhang, X.F. Yin, Q. Yang, C. Wang, Z. Wang, Anal. Bioanal. Chem. 401 (2011) 1071–1081. doi: 10.1007/s00216-011-5138-5
A. Gure, F.J. Lara, A.M. García-Campaña, N. Megersa, M. del Olmo-Iruela, Food Chem. 170 (2015) 348–353. doi: 10.1016/j.foodchem.2014.08.065
K.J. Tang, X.H. Gu, Q.S. Luo, et al., Food Chem. 150 (2014) 106–112. doi: 10.1016/j.foodchem.2013.10.152
J.Y. Wu, S.Y. Luo, X.J. Huang, Chem. Eng. J. 455 (2023) 140786. doi: 10.1016/j.cej.2022.140786
J.H. Yang, C.X. Cui, L.B. Qu, et al., Microchem. J. 141 (2018) 369–376. doi: 10.1016/j.microc.2018.05.049
J.P. Ma, L.H. Jiang, G.G. Wu, et al., J. Chromatogr. A 1466 (2016) 12–20. doi: 10.1016/j.chroma.2016.08.065
H. Qi, L.Y. Jiang, Q. Jia, Chin. Chem. Lett. 32 (2021) 2629–2636. doi: 10.1016/j.cclet.2021.01.037
C. Tian, Z.K. Wu, M. He, B.B. Chen, B. Hu, J. Sep. Sci. 45 (2022) 1746–1756. doi: 10.1002/jssc.202200055
Z.Y. He, D.H. Liu, R.H. Li, Z.Q. Zhou, P. Wang, Anal. Chim. Acta 747 (2012) 29–35. doi: 10.1016/j.aca.2012.08.015
S.S. Miao, M.S. Wu, H.G. Zuo, et al., J. Agric. Food Chem. 63 (2015) 3634–3645. doi: 10.1021/jf506239b
Y.F. Zhou, M.X. Zhao, Z. Meng, et al., Microchim. Acta 186 (2019) 486. doi: 10.1007/s00604-019-3536-0
M. Bouri, M. Gurau, R. Salghi, et al., Anal. Bioanal. Chem. 404 (2012) 1529–1538. doi: 10.1007/s00216-012-6221-2
Y. Qi, M.F. Wan, A.M. Abd El-Aty, et al., Microchim. Acta 187 (2020) 279. doi: 10.1007/s00604-020-04243-5
D.D. Wang, Y. Zhao, M.N. Ou Yang, H.M. Guo, Z.H. Yang, J. Chromatogr. A 1601 (2019) 53–59. doi: 10.1016/j.chroma.2019.05.011
Y.Y. Cui, X.Q. He, C.X. Yang, X.P. Yan, Trends Anal. Chem. 139 (2021) 116268. doi: 10.1016/j.trac.2021.116268
S.H. Li, L.L. Bian, C.Y. Yang, A.V. Schepdael, X. Wang, J. Hazard. Mater. 438 (2022) 129505. doi: 10.1016/j.jhazmat.2022.129505
L. Liu, H.C. Tu, F. Liu, et al., Chem. Eng. J. 442 (2022) 136171. doi: 10.1016/j.cej.2022.136171
X.H. Li, Y.Y. Cui, X. Wu, A. Abdukayum, C.X. Yang, Food Chem. 429 (2023) 136808. doi: 10.1016/j.foodchem.2023.136808
X.Q. He, Y.Y. Cui, C.X. Yang, ACS Appl. Mater. Interfaces 13 (2021) 39905–39914. doi: 10.1021/acsami.1c11148
X.H. Li, Y.Y. Cui, S.L. Ji, A. Abdukayum, C.X. Yang, Food Chem. 443 (2024) 138559. doi: 10.1016/j.foodchem.2024.138559
C.Y. Li, J.M. Liu, Z.H. Wang, et al., J. Hazard. Mater. 384 (2020) 121348. doi: 10.1016/j.jhazmat.2019.121348
Y.Y. Cui, Y.P. Bi, C.X. Yang, Chem. Eng. J. 435 (2022) 134829. doi: 10.1016/j.cej.2022.134829
C.Y. Xu, C.Q. Zhen, Y.J. He, Y.Y. Cui, C.X. Yang, J. Chromatogr. A 1728 (2024) 464991. doi: 10.1016/j.chroma.2024.464991
L. Yu, J. Ding, Y.L. Wang, P. Liu, Y.Q. Feng, Anal. Chem. 88 (2016) 1286–1293. doi: 10.1021/acs.analchem.5b03720
Z.Y. Li, B. Luo, L.Z. Yu, F. Lan, Y. Wu, J. Mater. Chem. B 9 (2021) 453. doi: 10.1039/d0tb01901a
J.S. Chen, X.W. Min, P. Li, et al., Anal. Chim. Acta 879 (2015) 41–47. doi: 10.1016/j.aca.2015.03.058
S.S. Chi, C. Peng, Y.H. Lan, et al., J. Chromatogr. A 1590 (2019) 10–18. doi: 10.1016/j.chroma.2018.12.067
Y. Zhang, M. Mei, X.J. Huang, D.X. Yuan, Anal. Chim. Acta 899 (2015) 75–84. doi: 10.1159/000374054
M. Pei, X.Y. Zhu, X.J. Huang, J. Chromatogr. A 1531 (2018) 13–21. doi: 10.1016/j.chroma.2017.11.030
Z.X. Cai, X. Zhou, Y.S. Yang, et al., Chem. Eng. J. 466 (2023) 143315. doi: 10.1016/j.cej.2023.143315
S.C. Jiang, Z. Li, X.M. Yang, et al., Food Chem. 404 (2023) 134652. doi: 10.1016/j.foodchem.2022.134652
M.M. Xu, J.M. Wang, L.H. Zhang, et al., J. Hazard. Mater. 429 (2022) 128288. doi: 10.1016/j.jhazmat.2022.128288
Z.X. Cai, Z. Li, Q.Q. Wang, et al., J. Hazard. Mater. 462 (2024) 132746. doi: 10.1016/j.jhazmat.2023.132746

Figure 2 SEM images of Fe3O4 (a) and [PBA]X−MCD-MON-0.04 with different X ([EPBA]/[TEPB]): (b) 0/7, (c) 1/6, (d) 2/5, (e) 3/4, and (f) 4/3; and [PBA]3/4−MCD-MON-c with different c: (g) 0.01, (h) 0.02, and (i) 0.08. TEM images of (j) [PBA]3/4−MCD-MON-0.04.

Figure 3 Characterization of [PBA]X−MCD-MON-c: (a) FT-IR spectra, (b) magnetic hysteresis loops, (c) XRD patterns, and (d) N2 adsorption-desorption isotherms; recoveries of SUHs on [PBA]X−MCD-MON-c with different X (e) and c (f).

Figure 4 MSPE parameters optimization: (a) elution solvent, (b) eluent volume, (c) adsorbent dosage, (d) extraction time, (e) pH and (f) ionic strength. Error bars show the standard deviations for three replicate extractions.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

 下载:
下载: