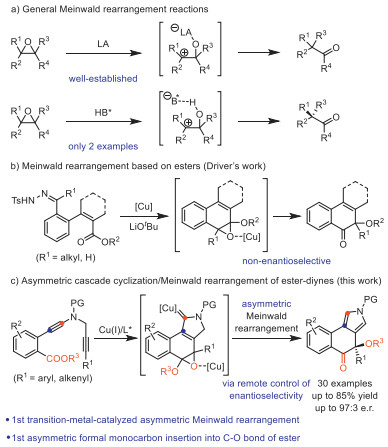
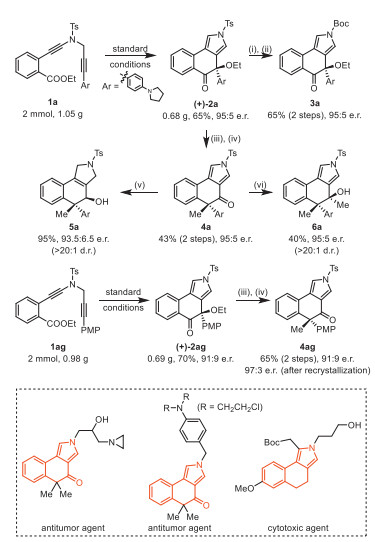
Scheme 1.
Meinwald rearrangement reactions.
Copper-catalyzed asymmetric cascade diyne cyclization/Meinwald rearrangement
Ji-Jia Zhou , Li-Gao Liu , Zhen-Tao Zhang , Hao-Xuan Dong , Xin Lu , Zhou Xu , Xin-Qi Zhu , Bo Zhou , Long-Wu Ye

The Meinwald rearrangement (MR) refers to the process of conversion of epoxides to carbonyl compounds via the 1, 2-migration of one of the substituents of the epoxide ring, and has been widely used in the synthesis of biologically active molecules [1-14]. Although significant achievements have been made in the past decade [15-26], these protocols mostly rely on the use of epoxides directly as precursors, which may limit the practical application of this approach because the stability of epoxides is generally poor, and strong oxidants are typically needed during the preparation process of epoxides. Moreover, examples of catalytic asymmetric MRs, which are able to transform racemic epoxides into enantioenriched α-substituted ketones, are still quite scarce and limited to chiral Brønsted acid (BA) catalysis (Scheme 1a) [27-32]. For example, Zhu and co-workers in 2019 demonstrated an elegant protocol for chiral phosphoramide-catalyzed enantioselective MR of tetrasubstituted epoxides, leading to the formation of chiral 2, 2-diarylcyclohexanones [27]. Simultaneously, Sun and co-workers achieved the asymmetric MR of tetrasubstituted epoxides by chiral phosphoric acid (CPA) catalysis for the synthesis of both cyclic and acyclic ketones [28]. In 2015, Driver's group disclosed the in situ generation of tetrasubstituted epoxides by the reaction of copper carbenes with the ester group, followed by copper-catalyzed MR, affording tricyclic 2H-naphthalenones containing an oxa-quaternary carbon center (Scheme 1b) [33]. Thus, this method provides a new strategy for MR by the direct conversion of esters into ketones [34-42] via monocarbon insertion into C–O bond of ester. Despite these achievements, such an ester-based MR and the related protocols [43-46] are extremely rare. Therefore, the exploration of new strategies for MR, especially in a practical and asymmetric approach, is highly desirable.
Due to their unique carbene-like reactivity, the chemistry of vinyl cations has received particular attention over the past decade [47, 48]. However, the exploration of enantioselective transformations of vinyl cations remains elusive but highly desirable [49]. Over the past several years, our laboratory has developed an effective approach to the formation of highly electrophilic vinyl cations intermediates via copper-catalyzed diyne cyclization [50-61]. On the basis of this general strategy, various useful asymmetric transformations have been established via a remote control of enantioselectivity, including intramolecular aromatic C(sp2)–H functionalization [50], vinylic C(sp2)–H functionalization [52], unactivated C(sp3)–H functionalization [58], cyclopropanation [50], one-carbon ring expansion [60], and [1, 2]-Stevens-type rearrangement [53, 55, 61], and intermolecular annulations with styrenes [51] and ketones [54], and atroposelective cyclization [56, 57, 59]. Despite these advances, no Meinwald-type rearrangement involving vinyl cations has been reported to date. We surmised that the vinyl cations, generated from copper-catalyzed cyclization of COOEt-substituted N-propargyl ynamides [62-79], might be trapped by the intramolecular ester group to afford epoxide intermediates. Subsequent asymmetric Meinwald rearrangement catalyzed by chiral copper catalyst would deliver the corresponding chiral tricyclic pyrroles (Scheme 1c). However, realizing this multiple cascade reaction is highly challenging. First, the ester group, a weak nucleophile, may not be nucleophilic enough to trap the vinyl cations. Second, chiral copper-catalyzed enantioselective MR has not been documented. Herein, we disclose a copper-catalyzed cascade cyclization/MR via in situ-generated epoxides from 1, 5-diynes. Furthermore, such a catalytic asymmetric version with high enantioselectivities has also been achieved via remote control of enantioselectivity, enabling practical access to functionalized chiral tricyclic pyrroles containing a chiral oxa-quaternary carbon center. To our best knowledge, this protocol not only represents the first transition-metal-catalyzed asymmetric MR, but also constitutes the first asymmetric formal monocarbon insertion into C–O bond of ester.
We started the investigations by using COOEt-substituted N-propargyl ynamide 1a [80-82] as the model substrate to demonstrate our designed cascade cyclization/MR reaction, and selected results are listed in Table 1. To our delight, the desired 2, 4-dihydro-5H-benzo[e]isoindol-5-one 2a bearing an oxa-quaternary carbon stereocenter could be obtained in 30% yield in the presence of Cu(MeCN)4PF6 (10 mol%) as the catalyst (Table 1, entry 1), albeit together with a large number of hydrolysis side product 2a (40%). Then, we investigated the effect of NaBArF4 and ligand, and found that the addition of NaBArF4 increased the reactivity (Table 1, entry 2), while the addition of both NaBArF4 and ligand could significantly inhibit the formation of unwanted 2a (Table 1, entry 3). Next, other typical copper(I) catalysts such as Cu(MeCN)4BF4, Cu(MeCN)4OTf, CuOTf·(C6H6)0.5 and CuOTf were evaluated, but the use of these catalysts led to a slightly decreased yield (Table 1, entries 4–7). Of note, CuCl and CuI could not catalyze this cascade cyclization effectively (Table 1, entries 8 and 9). Further studies revealed that the use of 3 Å molecular sieve (MS) as the additive completely prohibited the formation of side product 2a, and the desired tricyclic product 2a was obtained in 81% yield (Table 1, entry 10). However, replacing 3 Å MS with 4 Å or 5 Å MS resulted in a slight decrease in yield (Table 1, entries 11 and 12). Subsequently, various typical solvents including DCM, toluene and Et2O were screened, but failed to give better results (Table 1, entries 13–15). Without catalyst, the reaction failed to give even a trace of 2a, and > 99% of 1a remained unreacted.
 DownLoad:
CSV
DownLoad:
CSV
 |
|||
| Entry | [Cu] | Reaction conditions | Yield (%)b |
| 1c | Cu(MeCN)4PF6 | DCE, 25 ℃, 1 h | 30 |
| 2d | Cu(MeCN)4PF6 | DCE, 25 ℃, 0.5 h | 40 |
| 3 | Cu(MeCN)4PF6 | DCE, 25 ℃, 8 h | 68 |
| 4 | Cu(MeCN)4BF4 | DCE, 25 ℃, 8 h | 67 |
| 5 | Cu(MeCN)4OTf | DCE, 25 ℃, 1 h | 61 |
| 6 | CuOTf·(C6H6)0.5 | DCE, 25 ℃, 1 h | 67 |
| 7 | CuOTf | DCE, 25 ℃, 1 h | 56 |
| 8 | CuCl | DCE, 25 ℃, 24 h | < 10 |
| 9 | CuI | DCE, 25 ℃, 24 h | < 10 |
| 10e | Cu(MeCN)4PF6 | DCE, 25 ℃, 8 h | 81 |
| 11f | Cu(MeCN)4PF6 | DCE, 25 ℃, 8 h | 61 |
| 12g | Cu(MeCN)4PF6 | DCE, 25 ℃, 8 h | 60 |
| 13e | Cu(MeCN)4PF6 | DCM, 25 ℃, 8 h | 74 |
| 14e | Cu(MeCN)4PF6 | Toluene, 25 ℃, 8 h | 73 |
| 15e | Cu(MeCN)4PF6 | Et2O, 25 ℃, 8 h | 74 |
| a Reaction conditions: 1a (0.05 mmol), Cu(MeCN)4PF6 (0.005 mmol), (±)-BINAP (0.006 mmol), NaBArF4 (0.006 mmol), solvent (1 mL), in vials. b Yields are measured by 1H NMR using 1, 3, 5-trimethoxybenzene as the internal standard. c Without (±)-BINAP and NaBArF4. d Without (±)-BINAP. e Using 3 Å MS (30 mg) as additive. f Using 4 Å MS (30 mg) as additive. g Using 5 Å MS (30 mg) as additive. |
|||
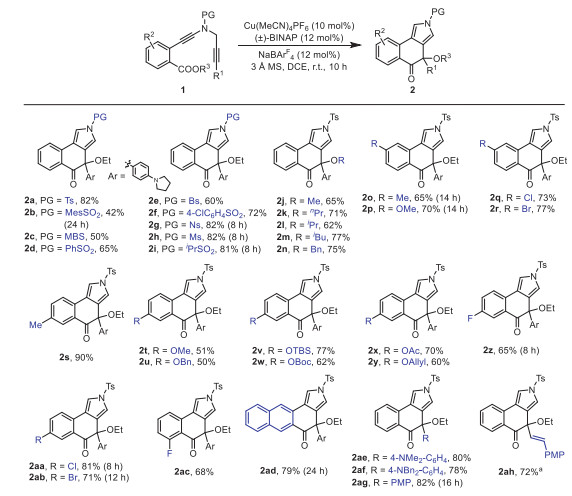
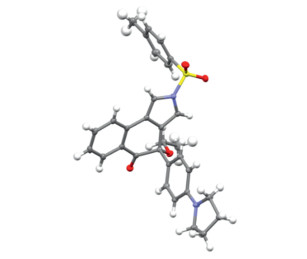
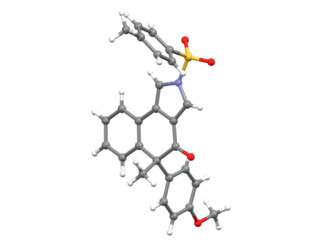
After identification of the optimal reaction conditions (Table 1, entry 10), we next sought to explore the scope of this cascade cyclization/MR reaction. As depicted in Scheme 2, we initially investigated the scope of different groups of the N-propargyl ynamides 1. Apart from the Ts protecting group, other aryl sulfonyl groups such as MesSO2, MBS (4-methoxybenzenesulfonyl), PhSO2, Bs (4-bromobenzenesulfonyl), 4-ClC6H4SO2 and Ns (4-nitrobenzenesulfonyl), could be well tolerated in this reaction, providing the corresponding tricyclic products 2b–2g bearing an oxa-quaternary carbon stereocenter in 42%–82% yields. In addition, diynes with the alkyl sulfonyl groups were also suitable substrates for this cascade cyclization, affording the desired products 2h–2i in 81%–82% yields. Subsequently, we studied the effect of different ester groups on the reaction, and found that various substituted ester groups, including methyl, propyl, isopropyl, isobutyl and benzyl groups, reacted smoothly to deliver the expected tricyclic pyrroles 2j–2n in 62%−77% yields. Then, various aryl-substituted ynamides were screened, and it was found that the reaction occurred efficiently with a wide range of substituents including both the electron-donating and -withdrawing groups on the phenyl linker, furnishing the corresponding products 2o–2ac with yields ranging from 50% to 90%. Of note, the naphthyl-substituted diyne could also be readily converted into the desired product 2ad in 79% yield. Further studies revealed that other electron-rich substituents of diynes such as 4-NMe2C6H4, 4-NBn2C6H4 and PMP, could also undergo smooth cyclization/rearrangement to obtain the target products 2ae–2ag in 78%−82% yields. Interestingly, we were pleased to find that alkenyl-substituted diyne was compatible with the reaction conditions and the target product 2ah was afforded in 72% yield. The molecular structure of 2a was confirmed by X-ray diffraction analysis (Fig. 1). Attempts to expand this cascade cyclization/Meinwald rearrangement to an intermolecular reaction with ethyl benzoate have been unsuccessful as yet (see Supporting information for details).
Moreover, other mother ring structures were also applicable to this cascade cyclization. For example, the reaction was successfully extended to the indole and aliphatic alkyl-linked ester-diynes 1ai–1aj, leading to the expected products 2ai and 2aj in 40% and 65% yields, respectively (Eqs. 1 and 2). It is notable that similar indole-fused tetracyclic molecules could display their bioactivity as antitumor agents [80, 81]. In addition, the seven membered ring-fused pyrrole product 2ak containing oxa-quaternary carbon stereocenter could also be formed in 67% yield under the optimal reaction conditions (Eq. 3).
|
|
(1) |
|
|
(2) |
|
|
(3) |
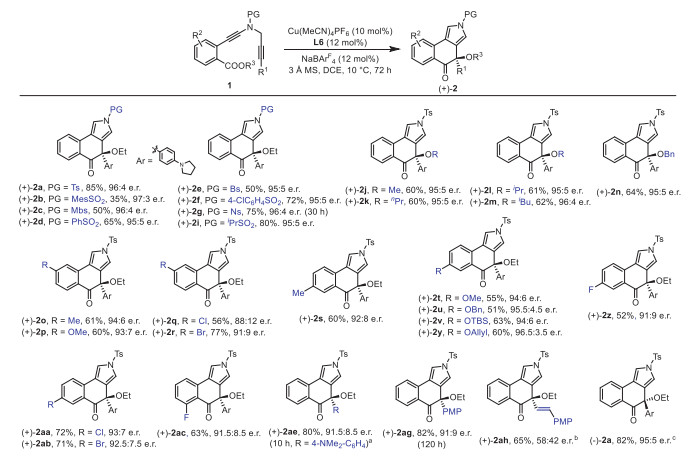
After verifying the feasibility and universality of this cascade cyclization/MR reaction of ester-substituted N-propargyl ynamides, we attempted to further explore its asymmetric catalytic version for the enantioselective construction of oxa-quaternary carbon stereocenters (see Supporting information for details). As summarized in Table 2, we were pleased to find that replacing the (±)-BINAP with a commercially available (R)-BINAP on the basis of racemic reaction conditions could lead to the desired product (+)−2a in 68% yield with an e.r. of 79.5:20.5 (entry 1, see Supporting information for details). Encouraged by this preliminary result, we next investigated assorted types of chiral biphosphine ligands. Attempts to modify the phenyl group on phosphine to 4-tolyl led to a slight decrease in its enantioselectivity (entry 2). Then, other typical chiral biphosphine ligands L3–L7 were screened (entries 3–7), and the results showed that the chiral ligand L6 with a SEGPHOS skeleton could afford the desired (+)−2a in 75% yield with an e.r. of 94.5:5.5 (entry 6). In addition, it was found that the use of GARPHOS ligands L8–L10 failed to further improve the enantioselectivities (entries 8–10). Of note, the use of chiral biphosphine ligands bearing sterically hindered Ar groups led to significantly decreased yields and enantioselectivities (entries 7 and 10). Gratifyingly, decreasing the reaction temperature to 10 ℃ allowed for the formation of (+)−2a in 78% yield with an e.r. of 95.5:4.5 (entry 11).
DownLoad:
CSV
 |
||||
| Entry | L | Conditions | Yield (%)b | e.r.c |
| 1 | L1 | DCE, 25 ℃, 10 h | 68 | 79.5:20.5 |
| 2 | L2 | DCE, 25 ℃, 10 h | 74 | 78.5:21.5 |
| 3 | L3 | DCE, 25 ℃, 6 h | 68 | 57.5:42.5 |
| 4 | L4 | DCE, 25 ℃, 8 h | 70 | 64.5:35.5 |
| 5 | L5 | DCE, 25 ℃, 10 h | 70 | 90:10 |
| 6 | L6 | DCE, 25 ℃, 14 h | 75 | 94.5:5.5 |
| 7 | L7 | DCE, 25 ℃, 72 h | < 10 | 53:47 |
| 8 | L8 | DCE, 25 ℃, 22 h | 70 | 94.5:5.5 |
| 9 | L9 | DCE, 25 ℃, 15 h | 30 | 79.5:20.5 |
| 10 | L10 | DCE, 25 ℃, 72 h | < 10 | 51.5:48.5 |
| 11 | L6 | DCE, 10 ℃, 72 h | 78 | 95.5:4.5 |
| a Reactions run in vials; 1a (0.05 mmol), Cu(MeCN)4PF6 (0.005 mmol), L (0.006 mmol), NaBArF4 (0.006 mmol), solvent (1 mL). b Yields are measured by 1H NMR using 1, 3, 5-trimethoxybenzene as the internal standard. c Determined by HPLC analysis. |
||||
Having established the optimal conditions for the asymmetric version of this tandem cyclization/MR reaction (Table 2, entry 11), we next examined the substrate scope for synthesis of chiral tricyclic pyrroles. As shown in Scheme 3, the asymmetric cascade cyclization occurred smoothly with ynamides bearing a variety of aromatic sulfonyl protecting groups, allowing the formation of enantioenriched tricyclic pyrroles (+)−2a–(+)−2g in 35%–85% yields with excellent enantioselectivities. Additionally, the alkyl sulfonyl protecting group was also proven to be well tolerated in this reaction, and the target product (+)−2i was formed in 80% yield with an e.r. of 95:5. Subsequently, ynamide substrates with different ester groups were investigated, and the expected chiral products (+)−2j–(+)−2n were obtained in 60%–64% yields with the e.r. of 95:5–96:4. We then studied different substituents on the mother ring, and found that all these substrates with electron-donating and -withdrawing substituents could undergo smooth asymmertric process, delivering the desired chiral products (+)−2o–(+)−2ac with 51%–77% yields and e.r. of 88:12–96.5:3.5. Diynes containing other electron-rich substituents such as 4-NMe2C6H4 and PMP were also suitable substrates, and were readily converted into the corresponding products (+)−2ae (80%, 91.5:8.5 e.r.) and (+)−2ag (82%, 91:9 e.r.), respectively. Of note, our attempts to extend the reaction to the alkenyl-substituted diyne only led to the formation of the desired (+)−2ah in low enantioselectivity (see Supporting information for details). Finally, it was found that the reaction also proceeded efficiently to produce the desired (-)−2a in 82% yield and e.r. of 95:5 with the opposite enantioselectivity by employing ent‑L6 as chiral ligand. Thus, this method also represents an unprecedented example of formal asymmetric monocarbon insertion into C–O bond of ester [34-42].
In order to explore the synthetic utility of this method, we then synthesized the chiral tricyclic pyrrole (+)−2a on a preparative-scale and performed a series of transformations, as depicted on the Scheme 4. Pleasingly, the preparative-scale reaction of N-propargyl ynamide 1a also achieved excellent enantioselectivity, albeit with a slight decrease in yield. Subsequently, the Ts group of (+)−2a was readily removed by the treatment with tBuOK, followed by protection with the Boc group, affording the corresponding product 3a (65%, 2 steps, 95:5 e.r.). In addition, (+)−2a could react with MeLi to obtain the corresponding chiral tertiary alcohol, which underwent further pinacol rearrangement under acidic conditions (see Supporting information for details), leading to the pyrrole-conjugated ketone 4a (43%, 2 steps, 95:5 e.r.) bearing an all-carbon quaternary stereocenter. This tricyclic ketone 4a could be further converted into the desired reduction product 5a (95%, > 20:1 dr., 93.5:6.5 e.r.) and new tertiary alcohol 6a (40%, > 20:1 dr., 95:5 e.r.) by treatment with NaBH3CN and MeLi, respectively. Preparative-scale reaction of diyne 1ag was also feasible to produce the expected (+)−2ag (70%, 91:9 e.r.), which underwent similar methylation and pinacol rearrangement, delivering the corresponding product 4ag (65%, 2 steps, 91:9 e.r.). After recrystallization of 4ag, we determined its absolute configuration by X-ray diffraction (Fig. 2), which also determined the absolute configuration of chiral tricyclic pyrroles (+)−2 (see Supporting information for details). Importantly, the enantioselectivity was well maintained and excellent diastereoselectivities were achieved in all these transformations. It is notable that these tricyclic pyrrole skeletons can be found in a variety of bioactive molecules with antitumor and cytotoxic activity [83-91] and may be of medicinal interest.
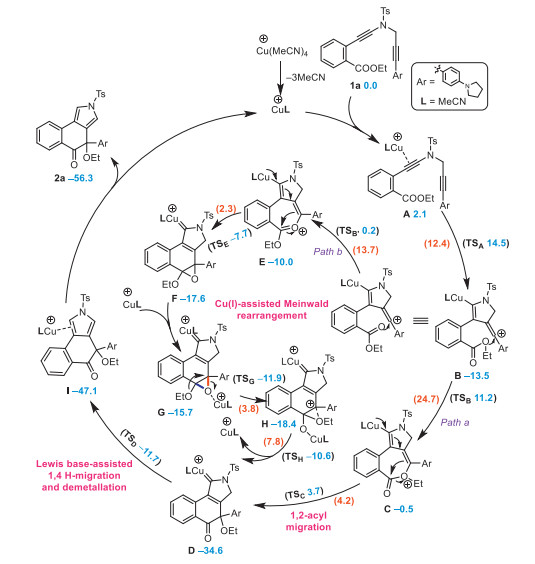
Based on the above experimental results, density functional theory (DFT) computations have been performed to gain more insights into the mechanism for the synthesis of product 2a from 1a and elucidate the origin of enantioselectivity. As is exhibited in Scheme 5, at the beginning of the reaction the coordination of CuL (L = MeCN) copper species to diyne 1a leads to a [Cu]-tethered intermediate A, which subsequently triggers an intramolecular cyclization to afford the vinyl cation intermediate B via a transition state TS. After the formation of the vinyl cation intermediate B, path a (a nucleophilic addition and a concerted C–O bond cleavage and C–C coupling process) and path b (two consecutive nucleophilic addition and a [Cu]-assisted MR process) have been considered to afford the copper-carbene intermediate D, and path b is thermodynamically more favorable with a largest barrier height of 13.7 kcal/mol compared to 24.7 kcal/mol of path a. Finally, similar to our previously reported studies [50-61], a Lewis base (H2O, for example)-assisted formal [1, 4]-H migration and demetallation process (barrier height: 22.9 kcal/mol) affords the product 2a. It is worth mentioning that different from Driver's protocol [33], involving a [1, 2]-alkyl shift in the MR, the [1, 2]-OEt shift is involved in the present MR as the C–O bond in red is more inclined to be cleaved rather than the C–O bond in blue due to the presence of the electron-rich aryl group (pushing the electron back to facilitate the C–O bond cleavage). Above all, the reaction can proceed smoothly under the given conditions, via the Meinwald rearrangement-involved mechanism depicted as path b, with the [1, 4]-H migration step being the rate-determining step.
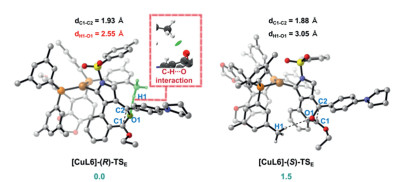
Furthermore, the origin of enantioselective synthesis of (+)−2a is also computationally explored by using the chiral L6 ligated to the CuI center in the enantio‑determining irreversible epoxidation step in path b (Fig. 3). The free energy of the transition state [CuL6]-(R)-TS is predicted to be 1.5 kcal/mol lower than the [CuL6]-(S)-TS, which well supports the experimental ee value of 91%. Further transition-state structural analysis indicates the C–H···O hydrogen bonding interaction between the substrate and the branched 3, 5-dimethylphenyl group of the bulky chiral ligand L6 is critical to the stereoselectivity, which is then verified and visualized by independent gradient model (IGM) analysis [92-94]. In addition, the chiral copper-carbene on the N-heterocycle should exert a remote stereocontrol effect [50-61] on subsequent copper-assisted MR.
In summary, we have developed a copper-catalyzed enantioselective cascade cyclization/MR of 1, 5-diynes via vinyl cations. Importantly, this protocol not only represents the first example of transition-metal-catalyzed enantioselective Meinwald rearrangement, but also constitutes the first asymmetric formal monocarbon insertion into C–O bond of ester and the first trapping of vinyl cations by the weakly nucleophilic ester group. This method enables the divergent, practical and atom-economic synthesis of a range of tricyclic pyrroles bearing a chiral oxa-quaternary carbon stereocenter in moderate to good yields with excellent enantioselectivities (up to 97:3 e.r.) under mild conditions. Furthermore, theoretical calculations provide further evidence for this vinyl cation-involved cascade cyclization and elucidate the origin of enantioselectivity. We envision that the above findings will open up new perspectives in the field of asymmetric catalysis based on MR and vinyl cations.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Ji-Jia Zhou: Validation, Investigation. Li-Gao Liu: Validation, Investigation. Zhen-Tao Zhang: Validation, Investigation. Hao-Xuan Dong: Investigation. Xin Lu: Writing – review & editing, Supervision. Zhou Xu: Writing – original draft, Validation, Supervision. Xin-Qi Zhu: Writing – original draft, Investigation. Bo Zhou: Writing – original draft, Investigation. Long-Wu Ye: Writing – review & editing, Supervision.
We are grateful for financial support from the National Natural Science Foundation of China (Nos. 22125108, 22101238, 22331004 and 22121001), Yunnan Fundamental Research Project (No. 202401CF070024), Natural Science Foundation of Jiangsu Province (No. BK20211059), the Project of Science and Technology of Xuzhou Government (No. KC22080), and NFFTBS (No. J1310024). Dedicated to Professor Yong Tang on the occasion of his 60th birthday.
Supplementary material associated with this article can be found, in the online version, at doi:
S. Meninno, A. Lattanzi, A.C.S. Org. Inorg. Au 2 (2022) 289–305. doi: 10.1021/acsorginorgau.2c00009
J.P.S. Choo, Z. Li, Org. Process Res. Dev. 26 (2022) 1960–1970. doi: 10.1021/acs.oprd.1c00473
X.M. Zhang, B.S. Li, S.H. Wang, et al., Chem. Sci. 12 (2021) 9262–9274. doi: 10.1039/d1sc02386a
V.L. Mamedova, G.Z. Khikmatova, Chem. Heterocycl. Comp. 53 (2017) 976–978. doi: 10.1007/s10593-017-2158-x
C.Y. Huang, A.G. Doyle, Chem. Rev. 114 (2014) 8153–8198. doi: 10.1021/cr500036t
B. Wang, Y.Q. Tu, Acc. Chem. Res. 44 (2011) 1207–1222. doi: 10.1021/ar200082p
Z.L. Song, C.A. Fan, Y.Q. Tu, Chem. Rev. 111 (2011) 7523–7556. doi: 10.1021/cr200055g
T.J. Snape, Chem. Soc. Rev. 36 (2007) 1823–1842. doi: 10.1039/b709634h
J.A. Marshall, Chem. Rev. 89 (1989) 1503–1511. doi: 10.1021/cr00097a006
H.G. Cheng, S. Jia, Q. Zhou, Acc. Chem. Res. 56 (2023) 573–591. doi: 10.1021/acs.accounts.2c00781
D. Li, W. Zang, M.J. Bird, C.J.T. Hyland, M. Shi, Chem. Rev. 121 (2021) 8685–8755. doi: 10.1021/acs.chemrev.0c00624
B. Delayre, Q. Wang, J. Zhu, ACS Cent. Sci. 7 (2021) 559–569. doi: 10.1021/acscentsci.1c00075
V.I. Kalinin, A.S. Silchenko, S.A. Avilov, V.A. Stonik, Steroids 147 (2019) 42–51. doi: 10.1016/j.steroids.2018.11.010
X.M. Zhang, Y.Q. Tu, F.M. Zhang, Z.H. Chen, S.H. Wang, Chem. Soc. Rev. 46 (2017) 2272–2305. doi: 10.1039/C6CS00935B
W. Zhao, D. Zhang, Y. Wang, M. Yang, J. Am. Chem. Soc. 145 (2023) 27160–27166. doi: 10.1021/jacs.3c12249
X.T. An, X.M. Ge, X.Y. Liu, et al., J. Am. Chem. Soc. 145 (2023) 9233–9241. doi: 10.1021/jacs.3c01572
W.W.L. See, Z. Li, ACS Catal. 13 (2023) 13215–13224. doi: 10.1021/acscatal.3c03456
J.P.S. Choo, F.L. Sirota, W.W.L. See, B. Eisenhaber, Z. Li, ACS Catal. 13 (2023) 11268–11276. doi: 10.1021/acscatal.3c02777
C. Muller, F. Horký, M. Vayer, et al., Chem. Sci. 14 (2023) 2983–2989. doi: 10.1039/d2sc06692k
X. Long, J. Li, F. Gao, H. Wu, J. Deng, J. Am. Chem. Soc. 144 (2022) 16292–16297. doi: 10.1021/jacs.2c07198
M. Alekseychuk, S. Adrian, R.C. Heinze, P. Heretsch, J. Am. Chem. Soc. 144 (2022) 11574–11579. doi: 10.1021/jacs.2c05358
R. Xin, W.W.L. See, H. Yun, X. Li, Z. Li, Angew. Chem. Int. Ed. 61 (2022) e202204889. doi: 10.1002/anie.202204889
A. Meza, M.E. Campbell, A. Zmich, et al., ACS Catal. 12 (2022) 10700–10710. doi: 10.1021/acscatal.2c02369
G. Wang, H. Huang, W. Guo, C. Qian, J. Sun, Angew. Chem. Int. Ed. 59 (2020) 11245–11249. doi: 10.1002/anie.201916727
T. Song, Z. Ma, P. Ren, et al., ACS Catal. 10 (2020) 4617–4629. doi: 10.1021/acscatal.9b05197
Y. Chen, J. Hu, L.D. Guo, et al., Angew. Chem. Int. Ed. 58 (2019) 7390–7394. doi: 10.1002/anie.201902908
H. Wu, Q. Wang, J. Zhu, J. Am. Chem. Soc. 141 (2019) 11372–11377. doi: 10.1021/jacs.9b04551
D. Ma, C.B. Miao, J. Sun, J. Am. Chem. Soc. 141 (2019) 13783–13787. doi: 10.1021/jacs.9b07514
Y.M. Shen, B. Wang, Y. Shi, Angew. Chem. Int. Ed. 45 (2006) 1429–1432. doi: 10.1002/anie.200501520
J. Xu, Y. Song, J. Yang, et al., Angew. Chem. Int. Ed. 62 (2023) e202217887. doi: 10.1002/anie.202217887
J. Zhang, W.L. Yang, H. Zheng, Y. Wang, W.P. Deng, Angew. Chem. Int. Ed. 61 (2022) e202117079. doi: 10.1002/anie.202117079
J. Xu, Y. Song, J. He, et al., Angew. Chem. Int. Ed. 60 (2021) 14521–14527. doi: 10.1002/anie.202102054
N. Su, J.A. Theorell, D.J. Wink, T.G. Driver, Angew. Chem. Int. Ed. 54 (2015) 12942–12946. doi: 10.1002/anie.201505993
R. Takise, K. Muto, J. Yamaguchi, Chem. Soc. Rev. 46 (2017) 5864–5888. doi: 10.1039/C7CS00182G
P.S. Fier, R.A. Roberts, R.T. Larson, Org. Lett. 25 (2023) 3131–3135. doi: 10.1021/acs.orglett.3c00992
X. Xi, Y. Luo, W. Li, et al., Angew. Chem. Int. Ed. 61 (2021) e202114731.
Y.L. Zheng, P.P. Xie, O. Daneshfar, et al., Angew. Chem. Int. Ed. 60 (2021) 13476–13483. doi: 10.1002/anie.202103327
B. Lee, P.J. Chirik, J. Am. Chem. Soc. 142 (2020) 2429–2437. doi: 10.1021/jacs.9b11944
Y.L. Zheng, S.G. Newman, Angew. Chem. Int. Ed. 58 (2019) 18159–18164. doi: 10.1002/anie.201911372
J. Masson-Makdissi, J.K. Vandavasi, S.G. Newman, Org. Lett. 20 (2018) 4094–4098. doi: 10.1021/acs.orglett.8b01646
A. Chatupheeraphat, H.H. Liao, W. Srimontree, et al., J. Am. Chem. Soc. 140 (2018) 3724–3735. doi: 10.1021/jacs.7b12865
L. Hie, N.F.F. Nathel, X. Hong, et al., Angew. Chem. Int. Ed. 55 (2016) 2810–2814. doi: 10.1002/anie.201511486
Y. Wu, C. Hu, T. Wang, L. Eberle, A.S.K. Hashmi, Adv. Synth. Catal. 364 (2022) 1233–1238. doi: 10.1002/adsc.202101469
X. Tian, L. Song, K. Farshadfar, et al., Angew. Chem. Int. Ed. 59 (2020) 471–478. doi: 10.1002/anie.201912334
R.L. Sahani, R.S. Liu, Angew. Chem. Int. Ed. 56 (2017) 12736–12740. doi: 10.1002/anie.201707423
A.S.K. Hashmi, M. Rudolph, J.P. Weyrauch, et al., Angew. Chem. Int. Ed. 44 (2005) 2798–2801. doi: 10.1002/anie.200462672
X.J. Liu, Y. Xu, C. Tang, P.C. Qian, L.W. Ye, Sci. China Chem. 65 (2022) 20–30. doi: 10.1007/s11426-021-1117-2
M. Niggemann, S. Gao, Angew. Chem. Int. Ed. 57 (2018) 16942–16944. doi: 10.1002/anie.201810701
S.K. Nistanaki, C.G. Williams, B. Wigman, et al., Science 378 (2022) 1085–1091. doi: 10.1126/science.ade5320
F.L. Hong, Z.S. Wang, D.D. Wei, et al., J. Am. Chem. Soc. 141 (2019) 16961–16970. doi: 10.1021/jacs.9b09303
F.L. Hong, Y.B. Chen, S.H. Ye, et al., J. Am. Chem. Soc. 142 (2020) 7618–7626. doi: 10.1021/jacs.0c01918
X.Q. Zhu, P. Hong, Y.X. Zheng, et al., Chem. Sci. 12 (2021) 9466–9474. doi: 10.1039/d1sc02773e
F.L. Hong, C.Y. Shi, P. Hong, et al., Angew. Chem. Int. Ed. 61 (2022) e202115554. doi: 10.1002/anie.202115554
L.J. Qi, C.T. Li, Z.Q. Huang, et al., Angew. Chem. Int. Ed. 61 (2022) e202210637. doi: 10.1002/anie.202210637
J.J. Zhou, Y.N. Meng, L.G. Liu, et al., Chem. Sci. 14 (2023) 3493–3500. doi: 10.1039/d2sc06152j
Y.B. Chen, L.G. Liu, C.M. Chen, et al., Angew. Chem. Int. Ed. 62 (2023) e202303670. doi: 10.1002/anie.202303670
C.T. Li, L.J. Qi, L.G. Liu, et al., Nat. Commun. 14 (2023) 7058. doi: 10.1038/s41467-023-42805-2
Y.B. Chen, L.G. Liu, Z.Q. Wang, et al., Nat. Commun. 15 (2024) 2232. doi: 10.1038/s41467-024-46288-7
H.H. Chen, Y.B. Chen, J.Z. Gao, et al., Angew. Chem. Int. Ed. 63 (2024) e202411709. doi: 10.1002/anie.202411709
F.S. Li, X.Y. Zou, T.Q. Hu, et al., Sci. Adv. 10 (2024) eadq7767. doi: 10.1126/sciadv.adq7767
C.Y. Shi, Q. Wang, L.G. Liu, et al., Org. Chem. Front. 11 (2024) https://doi.org/10.1039/d4qo01623h. doi: 10.1039/d4qo01623h
L. Hu, J. Zhao, Acc. Chem. Res. 57 (2024) 855–869. doi: 10.1021/acs.accounts.3c00743
Y.C. Hu, Y. Zhao, B. Wan, Q.A. Chen, Chem. Soc. Rev. 50 (2021) 2582–2625. doi: 10.1039/d0cs00283f
C.C. Lynch, A. Sripada, C. Wolf, Chem. Soc. Rev. 49 (2020) 8543–8583. doi: 10.1039/d0cs00769b
Y.B. Chen, P.C. Qian, L.W. Ye, Chem. Soc. Rev. 49 (2020) 8897–8909. doi: 10.1039/d0cs00474j
F.L. Hong, L.W. Ye, Acc. Chem. Res. 53 (2020) 2003–2019. doi: 10.1021/acs.accounts.0c00417
J. Luo, G.S. Chen, S.J. Chen, et al., ACS Catal. 10 (2020) 13978–13992. doi: 10.1021/acscatal.0c04180
B. Zhou, T.D. Tan, X.Q. Zhu, M. Shang, L.W. Ye, ACS Catal. 9 (2019) 6393–6406. doi: 10.1021/acscatal.9b01851
G. Evano, C. Theunissen, M. Lecomte, Aldrichimica Acta 48 (2015) 59–70.
X.N. Wang, H.S. Yeom, L.C. Fang, et al., Acc. Chem. Res. 47 (2014) 560–578. doi: 10.1021/ar400193g
E.H. Huang, L.G. Liu, Y.W. Yin, et al., Sci. China Chem. 67 (2024) 2982–2988. doi: 10.1007/s11426-024-1990-y
X. Liu, L.G. Liu, C.M. Chen, et al., Angew. Chem. Int. Ed. 62 (2023) e202216923. doi: 10.1002/anie.202216923
Y. Xu, G.L. Qian, D.Q. Cui, et al., ACS Catal. 13 (2023) 8803–8812. doi: 10.1021/acscatal.3c01680
H.H. Li, Y.N. Meng, C.M. Chen, et al., Sci. China Chem. 66 (2023) 1467–1473. doi: 10.1007/s11426-022-1536-9
Z.X. Zhang, X. Wang, J.T. Jiang, et al., Chin. Chem. Lett. 34 (2023) 107647. doi: 10.1016/j.cclet.2022.06.070
G.Y. Zhu, J.J. Zhou, L.G. Liu, et al., Angew. Chem. Int. Ed. 61 (2022) e202204603. doi: 10.1002/anie.202204603
Z.S. Wang, L.J. Zhu, C.T. Li, et al., Angew. Chem. Int. Ed. 61 (2022) e202201436. doi: 10.1002/anie.202201436
Y.Q. Zhang, Y.B. Chen, J.R. Liu, et al., Nat. Chem. 13 (2021) 1093–1100. doi: 10.1038/s41557-021-00778-z
P.F. Chen, B. Zhou, P. Wu, B. Wang, L.W. Ye, Angew. Chem. Int. Ed. 60 (2021) 27164–27170. doi: 10.1002/anie.202113464
A. Ahrens, J. Schwarz, D.M. Lustosa, et al., Chem. Eur. J. 26 (2020) 5280–5287. doi: 10.1002/chem.202000338
S. Tavakkolifard, K. Sekine, L. Reichert, et al., Chem. Eur. J. 25 (2019) 12180–12186. doi: 10.1002/chem.201902381
T. Wurm, J. Bucher, S.B. Duckworth, et al., Angew. Chem. Int. Ed. 56 (2017) 3364–3368. doi: 10.1002/anie.201700057
Z.J. Ma, J. Wang, W. Ding, Y. Zhang, T. Gao, Patent, CN 113768925 A, 2021.
R.J. Carmosin, J.R. Carson, P.M. Pitis, Patent, WO 9965911, 1999.
M. Odagi, K. Hosoya, Y. Yamamoto, K. Nagasawa, Synlett 28 (2017) 1305–1309. doi: 10.1055/s-0036-1588151
H. Spreitzer, C. Puschmann, Molbank 2012 (2012) M781.
H. Spreitzer, C. Puschmann, Molbank 2012 (2012) M772.
M. Boes, H. Stadler, J. Wichmann, Patent, WO 9830546 A1, 1998.
M. Bös, H. Stadler, J. Wichmann, et al., Helv. Chim. Acta 81 (1998) 525–538. doi: 10.1002/hlca.19980810306
K. Kim, F. Basha, A. Hancock, J.F. Debernardis, Pharm. J. Sci-Us. 82 (1993) 521–525. doi: 10.1002/jps.2600820518
A.A. Hancock, M.D. Meyer, J.F. Debernardis, J. Receptor Res. 11 (1991) 177–196. doi: 10.3109/10799899109066398
T. Lu, F. Chen, J. Comput. Chem. 33 (2012) 580–592. doi: 10.1002/jcc.22885
C. Lefebvre, G. Rubez, H. Khartabil, et al., ChemPhysChem 19 (2017) 17928–17936.
T. Lu, Q.X. Chen, J. Comput. Chem. 43 (2022) 539–555. doi: 10.1002/jcc.26812

Scheme 2 Reaction scope for the formation of tricyclic pyrroles 2. Reactions run in vials; 1 (0.2 mmol), Cu(MeCN)4PF6 (0.02 mmol), (±)-BINAP (0.024 mmol), NaBArF4 (0.024 mmol), DCE (4 mL), 3 Å MS (120 mg), r.t., 8–24 h; yields are those for the isolated products. a Replacing Cu(MeCN)4PF6 with CuTc and 3 Å MS with H2O (2 equiv.). Mes = mesityl, MBS = 4-methoxybenzenesulfonyl, Bs = 4-bromobenzenesulfonyl, Ns = 2-nitrobenzenesulfonyl, Ms = methanesulfonyl, PMP = 4-methoxyphenyl.

Scheme 3 Reaction scope for the formation of tricyclic pyrroles (+)−2. Reactions run in vials; 1 (0.2 mmol), Cu(MeCN)4PF6 (0.02 mmol), L6 (0.024 mmol), NaBArF4 (0.024 mmol), DCE (4 mL), 3 Å MS (120 mg), 10 ℃, 8–24 h; yields are those for the isolated products; e.r. is determined by HPLC analysis. a At 0 ℃. b CuTc (0.02 mmol), L (0.024 mmol) [79], NaBArF4 (0.024 mmol), DCE (4 mL), H2O (0.4 mmol), 40 ℃, 15 h. c Ent-L6 instead of L6.

Scheme 4 Scale-up reaction and product elaboration. Reagents and conditions: (i) tBuOK (1.2 equiv.), THF, r.t., 2 h; (ii) DMAP (20 mol%), (Boc)2O (1.3 equiv.), Et3N (1.3 equiv.), DCM, r.t., 8 h; (iii) MeLi (1 equiv.), THF, 0 ℃, 1 h; (iv) HCOOH (10 equiv.), H2O: 1, 4-dioxane = 1:1, 60 ℃, 2 h; (v) NaBH3CN (5 equiv.), TFA, r.t., 2 h; (vi) MeLi (0.8 equiv.), THF, 0 ℃, 1 h.

Figure 3 The geometries and relative free energies (ΔΔG, in kcal/mol) of the transition state [CuL6]-(R)-TSE and [CuL6]-(S)-TSE were computed at the SMD (solvent = dichloroethane)-M06-D3/6–311++G(d, p)-SDD//B3LYP-D3/6–31G(d)-LANL2DZ level of theory. Color code: red = O; white = H; gray = C; yellow = S; blue = N; orange = P; brown = Cu.
Table 1. Optimization of reaction conditions for racemic copper-catalyzed cyclization of ynamide 1a.a
|
|||
| Entry | [Cu] | Reaction conditions | Yield (%)b |
| 1c | Cu(MeCN)4PF6 | DCE, 25 ℃, 1 h | 30 |
| 2d | Cu(MeCN)4PF6 | DCE, 25 ℃, 0.5 h | 40 |
| 3 | Cu(MeCN)4PF6 | DCE, 25 ℃, 8 h | 68 |
| 4 | Cu(MeCN)4BF4 | DCE, 25 ℃, 8 h | 67 |
| 5 | Cu(MeCN)4OTf | DCE, 25 ℃, 1 h | 61 |
| 6 | CuOTf·(C6H6)0.5 | DCE, 25 ℃, 1 h | 67 |
| 7 | CuOTf | DCE, 25 ℃, 1 h | 56 |
| 8 | CuCl | DCE, 25 ℃, 24 h | < 10 |
| 9 | CuI | DCE, 25 ℃, 24 h | < 10 |
| 10e | Cu(MeCN)4PF6 | DCE, 25 ℃, 8 h | 81 |
| 11f | Cu(MeCN)4PF6 | DCE, 25 ℃, 8 h | 61 |
| 12g | Cu(MeCN)4PF6 | DCE, 25 ℃, 8 h | 60 |
| 13e | Cu(MeCN)4PF6 | DCM, 25 ℃, 8 h | 74 |
| 14e | Cu(MeCN)4PF6 | Toluene, 25 ℃, 8 h | 73 |
| 15e | Cu(MeCN)4PF6 | Et2O, 25 ℃, 8 h | 74 |
| a Reaction conditions: 1a (0.05 mmol), Cu(MeCN)4PF6 (0.005 mmol), (±)-BINAP (0.006 mmol), NaBArF4 (0.006 mmol), solvent (1 mL), in vials. b Yields are measured by 1H NMR using 1, 3, 5-trimethoxybenzene as the internal standard. c Without (±)-BINAP and NaBArF4. d Without (±)-BINAP. e Using 3 Å MS (30 mg) as additive. f Using 4 Å MS (30 mg) as additive. g Using 5 Å MS (30 mg) as additive. |
|||
 下载: 导出CSV
下载: 导出CSV
Table 2. Optimization of reaction conditions for asymmetric copper-catalyzed cyclization of ynamide 1a.a
|
||||
| Entry | L | Conditions | Yield (%)b | e.r.c |
| 1 | L1 | DCE, 25 ℃, 10 h | 68 | 79.5:20.5 |
| 2 | L2 | DCE, 25 ℃, 10 h | 74 | 78.5:21.5 |
| 3 | L3 | DCE, 25 ℃, 6 h | 68 | 57.5:42.5 |
| 4 | L4 | DCE, 25 ℃, 8 h | 70 | 64.5:35.5 |
| 5 | L5 | DCE, 25 ℃, 10 h | 70 | 90:10 |
| 6 | L6 | DCE, 25 ℃, 14 h | 75 | 94.5:5.5 |
| 7 | L7 | DCE, 25 ℃, 72 h | < 10 | 53:47 |
| 8 | L8 | DCE, 25 ℃, 22 h | 70 | 94.5:5.5 |
| 9 | L9 | DCE, 25 ℃, 15 h | 30 | 79.5:20.5 |
| 10 | L10 | DCE, 25 ℃, 72 h | < 10 | 51.5:48.5 |
| 11 | L6 | DCE, 10 ℃, 72 h | 78 | 95.5:4.5 |
| a Reactions run in vials; 1a (0.05 mmol), Cu(MeCN)4PF6 (0.005 mmol), L (0.006 mmol), NaBArF4 (0.006 mmol), solvent (1 mL). b Yields are measured by 1H NMR using 1, 3, 5-trimethoxybenzene as the internal standard. c Determined by HPLC analysis. |
||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




 下载:
下载: