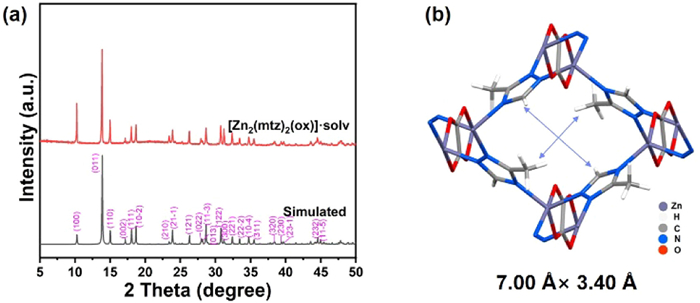
Figure 1.
(a) PXRD patterns of [Zn2(mtz)2(ox)]·solv. (b) Three-dimensional pillared-layer frameworks of [Zn2(mtz)2(ox)]·solv (for clarity, solvents were removed).
Elucidation of the CO2 adsorption mechanism of [Zn2(mtz)2(ox)] using neutron powder diffraction
Lingxiang Bao , Jing-Hong Li , Rui-Biao Lin , Jianyuan Wu , Zhenhong Tan , Wu Xie , Wenhai Ji , Dong Zhang , Anucha Koedtruad , Jingjing Ma , Wang Hay Kan , Feng Pan , Toru Ishigaki , Takashi Kamiyama , Ping Miao

Metal-organic frameworks (MOFs) have gained great interest due to their potential applications in catalysis [1-3], gas adsorption and separation [4-6], and drug delivery [7-9]. MOFs are usually assembled through solvothermal reactions with various metal ions and organic linkers, leading to structural diversity and functional adjustability [10-13]. In general, the properties and pore size of these porous materials can be adjusted for specific applications by using different metal ions or organic ligands. For instance, when using different metal ions, MAF-123-Cd showed highest usable storage capacity of C2H2 compared with MAF-123-Zn and MAF-123-Mn [14]. Alternatively, by substitution of organic ligands with different lengths, the pore sizes as well as performances of MOFs can also be adjusted, for example, the effective window size of [Cu(bpy-n)2(SiF6)] (bpy-1 = 4,4′-bipyridine; bpy-2 = 1,2-bis (4 pyridyl) ethane) is ca. 8 and 10.6 Å, respectively. At room temperature and ambient pressure, [Cu(bpy-1)2(SiF6)] exhibits a higher CO2 capacity (23.1 wt%) and CO2/CH4 selectivity (10.5) than [Cu(bpy-2)2(SiF6)] [15].
Recently, MOF [Zn2(tz)2(ox)] (CALF-20) has attracted great attention due to its excellent ability to capture carbon dioxide, leading to the emergence of many similar adsorbents, such as Zn-ox-mtz and NCU-20 [16-20]. It is interesting that NCU-20 and CALF-20 exhibit significantly different properties despite of similar crystal structures, coordination modes and unit formulas. The different properties might originate from delicate changes in the unit cell and atomic positions accompanied by the variation of structures and pore sizes. Therefore, exploring the structure-properties relationship of these MOFs is of great significance for developing new MOF adsorbents. However, most of these MOF samples cannot retain in single crystal when the solvent is removed, so that it is difficult to characterize the crystal structure by single crystal X-ray diffraction (SXRD). Moreover, the detection of hydrogen atom position is always a problem for X-rays.
Neutron bears the merit of high sensitivity to light atom and good penetration, and has been proved to be an essential tool for determining both the structural details of MOF host and the locations of adsorbed gas molecules [21-23]. This work reports the synthesis and structure characterization of [Zn2(mtz)2(ox)], which exhibits good CO2 adsorption capacity. Neutron powder diffraction (NPD) experiments were performed on the solvated, the activated and CO2 loaded samples. The strong incoherent neutron scattering to hydrogen sometimes causes high background in NPD data, but in our NPD experiment, the incoherent neutron scattering is rather weak due to a small atomic fraction of hydrogen in [Zn2(mtz)2(ox)]. With the reasonable-background NPD data, we have precisely determined the structural transformation as well as the interactions between CO2 and the host framework, which was corroborated by the Grand Canonical Monte Carlo simulations. As a result, we finally unveiled the CO2 adsorption mechanism and structure-properties relationship of [Zn2(mtz)2(ox)].
[Zn2(mtz)2(ox)] was prepared according to the reported method with slight modifications [17]. ZnCO3 (250 mg), oxalic acid (187 mg), 3-methyl-1H-1,2,4-triazole (830 mg), H2O (12 mL) and n-BuOH (4 mL) was added in a 50 mL Teflon autoclave. The mixture was vigorously stirred for 30 min at room temperature and heated in an oven at 453 K for 3 days. The obtained white precipitate was separated by centrifuge, washed several times with methanol, and dried under vacuum.
Sorption isotherms for CO2 (99.99%, from Mulai air) was measured at 298 K on the ASAP 2020 instrument. The sorbent [Zn2(mtz)2(ox)] was activated at 373 K for 8 h under dynamic vacuum before sorption measurements.
X-ray powder diffraction (PXRD) of [Zn2(mtz)2(ox)]·solv were measured at room temperature using a Rigaku Smartlab powder diffractometer. Neutron powder diffraction (NPD) data of the solvated ([Zn2(mtz)2(ox)]·solv), the activated ([Zn2(mtz)2(ox)]), and the CO2 loaded samples ([Zn2(mtz)2(ox)]·0.28CO2) were collected using the time-of-flight neutron powder diffractometer, iMATERIA, at the Materials and Life Science Experimental Facility of the Japan Proton Accelerator Research Complex (J-PARC) and the time-of-flight high-resolution neutron diffractometer (TREND) at China Spallation Neutron Source (CSNS). Activation of the samples was carried out at 373 K for 8 h in vacuum prior to the neutron experiments. CO2 was loaded using a gas handling system at 1 bar and 298 K for 0.5 h. The data collecting was performed after all the gas had been adsorbed, allowing sufficient time to achieve thermal equilibrium. All the NPD data were collected at room temperature.
Rietveld refinements on the NPD patterns of the MOFs were performed using the TOPAS software package [24], in which the initial framework model was geometrically constrained. The CO2 guests were treated as a rigid body. The mass center, orientation, and occupancies of the guest molecule were first refined, followed by the refinements comprised all free structural variables from both the framework and guest molecules.
GCMC simulations were performed to generate CO2 positions in the MOFs using the Sorption module in Material Studio software package. The structure of [Zn2(mtz)2(ox)]·0.28CO2 obtained from the NPD experiment were used as the starting configuration. The solvent and gas molecules were removed from the framework prior to the simulations, denoted as [Zn2(mtz)2(ox)]·0.28CO2 (removed), which is analogous to the activation in the experiment. Both MOF framework and CO2 molecules were regarded as rigid bodies. The adsorption site simulation was performed at 298 K and 1.0 bar by the fixed loading task and Metropolis method. The atomic partial charges of the host MOF framework and guest CO2 molecules were obtained by the QEq method. The framework-CO2 interaction and the gas-gas interaction were taken from the universal force field (UFF). The cut-off radius used for the Lennard-Jones interactions is 15.5 Å and the electrostatic interactions were calculated based on the Ewald summation method. Each simulation consists of an equilibration period of 5.0 × 106 iterations followed by a production run of further 1.0 × 107 iterations.
Both PXRD and NPD confirms that the synthesized sample ([Zn2(mtz)2(ox)]·solv) is of high purity and the crystal structure is a isomer of Zn-ox-mtz (Figs. 1a and 2a) [17].
[Zn2(mtz)2(ox)]·solv crystallizes into a three-dimensional framework structure with the monoclinic space group P21/c (Fig. 1b). The Zn(Ⅱ) ion is coordinated by two O-donors from oxalate group [Zn-O = 2.162(6) and 1.965(3) Å] and three N-donors from bridging triazole units [Zn-N = 2.061(9), 2.038(6) and 2.077(9) Å]. The coordination induces a one-dimensional channel made of zig–zag segment running through the framework, which contains uncoordinated water (1.0 per Zn). So the chemical formula for the as-synthesized material is [Zn2(mtz)2(ox)]·2H2O. Although the space group and unit formula of [Zn2(mtz)2(ox)]·solv resemble those of Zn-ox-mtz·solv [17], the structure of [Zn2(mtz)2(ox)]·solv differs slightly. For instance, due to the rotation of triazole ring accompanied by the shift of atomic positions in [Zn2(mtz)2(ox)]·solv, the pore aperture for [Zn2(mtz)2(ox)]·solv is 7.00 × 3.40 Å2, which is significantly larger than 5.30 × 3.50 Å2 found in Zn-ox-mtz·solv [17]. There are also small differences in the lattice parameters between [Zn2(mtz)2(ox)]·solv and Zn-ox-mtz·solv (Table 1) [17]. In [Zn2(mtz)2(ox)]·solv, the a, b and β expand while the c contracts compared with Zn-ox-mtz·solv. As a result, the cell volume of [Zn2(mtz)2(ox)]·solv is larger that of Zn-ox-mtz·solv.
 DownLoad:
CSV
DownLoad:
CSV
| Isomer | Space group | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | V (Å3) |
| Zn-ox-mtz·solv [17] | P21/c | 8.6166(2) | 7.8948(1) | 10.5066(2) | 90 | 96.97(2) | 90 | 709.45(2) |
| [Zn2(mtz)2(ox)]·solv | P21/c | 8.6881(1) | 8.0947(1) | 10.4423(2) | 90 | 98.51(1) | 90 | 726.31(1) |
| [Zn2(mtz)2(ox)] | P21/c | 8.7207(1) | 8.2027(1) | 10.3689(1) | 90 | 99.58(1) | 90 | 731.38(1) |
| [Zn2(mtz)2(ox)]·0.28CO2 | P21/c | 8.7581(1) | 8.3420(1) | 10.2752(1) | 90 | 100.83(1) | 90 | 737.34(1) |
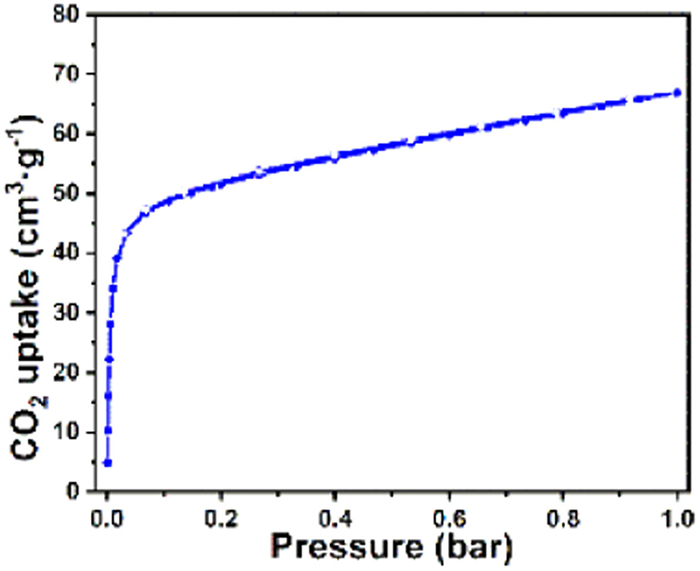
The CO2 adsorption capacity of desolvated [Zn2(mtz)2(ox)] was obtained through CO2 sorption experiments at 298 K in the pressure range of 0.0–1.0 bar It can be seen from Fig. 3 that [Zn2(mtz)2(ox)] exhibits Type-Ⅰ behavior with reversible uptake. The maximum CO2 uptake of [Zn2(mtz)2(ox)] is 66.9 cm3/g (2.99 mmol/g) at 298 K and 1.0 bar, which is slightly lower to Zn-ox-mtz (68.8 cm3/g or ~3.07 mmol/g).
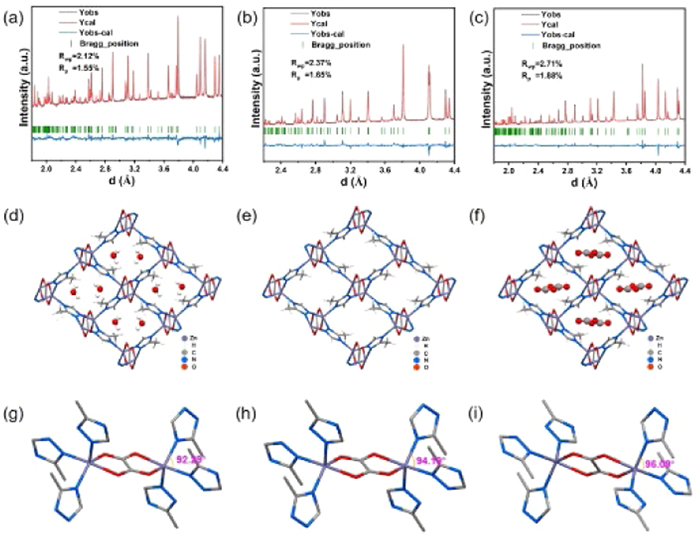
To determine the structure of the framework and the interaction between the adsorbed CO2 molecules and the framework, the solvated ([Zn2(mtz)2(ox)]·solv), the activated ([Zn2(mtz)2(ox)]), and the CO2 loaded samples ([Zn2(mtz)2(ox)]·0.28CO2) were studied using NPD (Figs. 2a-f). Compared to [Zn2(mtz)2(ox)]·solv, the cell volume of [Zn2(mtz)2(ox)] expands by 0.69% while that of [Zn2(mtz)2(ox)]·0.28CO2 expands by 1.5% (Table 1). The volume expansions are due to the rotation of one of the triazole rings in the frameworks that elongates lattice constant a and b. The ∠NZnN angle also increases during the rotation of the ring, which is 92.29°, 94.19° and 96.09° in [Zn2(mtz)2(ox)]·solv, [Zn2(mtz)2(ox)] and [Zn2(mtz)2(ox)]·0.28CO2, respectively (Figs. 2g-i). Therefore, the MOFs’ structures show the dynamic response of the framework to external stimuli such as heat or guest molecules. Moreover, since all CALF-20 analogues feature the triazole rings, such ring rotation might occur in CALF-20 analogues as well.
NPD studies on CO2 loaded [Zn2(mtz)2(ox)] also determined the location of the adsorbed gas molecule, which resides toward the center of the pore (Site Ⅰ), as shown in Figs. 4a and b. The CO2 molecule not only interacts with the triazole forming three C–H···O hydrogen bonds (H–O lengths: 2.578, 2.702 and 3.040 Å) but also with three methyl groups to form five C–H···O hydrogen bonds (H–O lengths: 2.593, 2.709, 3.404, 3.048, and 3.096 Å). The occupancies for these CO2 molecules at Site Ⅰ refined to values of 0.14, yielding the overall formula [Zn2(mtz)2(ox)]·0.28CO2 for the CO2 loaded [Zn2(mtz)2(ox)] samples. The value is lower than the experimental uptake from the CO2 isotherm at 298 K, which gives the overall formula of [Zn2(mtz)2(ox)] 1.14CO2 (Fig. 3). The discrepancy can be attributed to the presence of disordered CO2 molecules in the cavity, as indicated in nuclear density maps of CO2 loaded [Zn2(mtz)2(ox)] data obtained from our Maximum Entropy Method (MEM) analysis. Nevertheless, this discrepancy does not prevent us from determining the binding mechanisms. To compare the CO2 adsorption sites determined computationally and experimentally, the experimental CO2 positions are superimposed on three-dimensional adsorption density distribution maps for CO2 generated by GCMC simulations in Fig. 4c. The symmetry of the probability clouds suggests one distinct adsorption sites, which is consistent with the experiment result. In light of the adsorption density distribution, the favorable adsorption site of CO2 is in excellent agreement with the experimentally observed position.
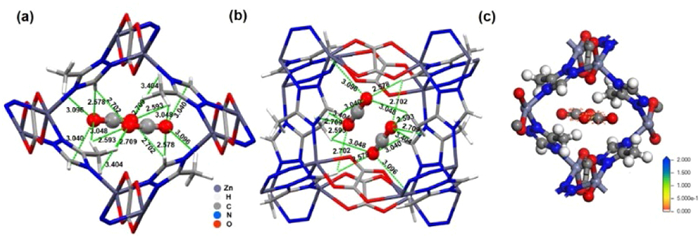
Site Ⅰ determined experimentally gives a profound insight into the effect of the structure transformation on the CO2 adsorption capacity. Since Site Ⅰ locates near the center of the pore, the strength of its interaction with the framework (C–H···O bonds) depends on the volume of the framework. As a result, a more compact framework facilitates stronger host-guest interaction, which triggers higher CO2 adsorption capacity. This is exactly what happens in [Zn2(mtz)2(ox)] and Zn-ox-mtz. The CO2 sorption isotherm of Zn-ox-mtz [17] exhibits higher CO2 uptake than that of isomorphous [Zn2(mtz)2(ox)], whereas the cell volume of Zn-ox-mtz·solv is smaller than that of [Zn2(mtz)2(ox)]·solv. Since the two isomers were obtained by only different solvents, the cell volume as well as the CO2 adsorption capacity of similar MOFs can be tuned by varying the solvents.
In conclusion, NPD studies of [Zn2(mtz)2(ox)]·solv, [Zn2(mtz)2(ox)] and CO2 loaded [Zn2(mtz)2(ox)] revealed, [Zn2(mtz)2(ox)] exhibits delicate softness to external stimuli. The expansion of [Zn2(mtz)2(ox)] during the gas loading was attributed to the rotation of triazole rings. In the molecular level, preferred binding site within the pores of [Zn2(mtz)2(ox)] determined by NPD was in good agreement with the results of GCMC simulations, i.e., CO2 locates toward the center of the pore via C–H···O hydrogen bonding interactions with methyl group or triazole. As a result, [Zn2(mtz)2(ox)] analogues with more compact structure bear stronger hydrogen bonding interaction, inducing better adsorption performance. The primary cell volumes as well as the CO2 adsorption capacity of similar MOFs can be tuned by varying the solvents in the synthesis.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Lingxiang Bao: Writing – original draft, Validation, Investigation, Funding acquisition, Formal analysis, Data curation. Jing-Hong Li: Writing – original draft, Investigation, Formal analysis. Rui-Biao Lin: Writing – review & editing, Methodology, Conceptualization. Jianyuan Wu: Investigation, Formal analysis. Zhenhong Tan: Software. Wu Xie: Software. Wenhai Ji: Software. Dong Zhang: Investigation. Anucha Koedtruad: Visualization. Jingjing Ma: Resources. Wang Hay Kan: Supervision, Conceptualization. Feng Pan: Resources. Toru Ishigaki: Resources, Formal analysis, Data curation. Takashi Kamiyama: Supervision, Data curation. Ping Miao: Writing – review & editing, Validation, Supervision, Funding acquisition, Conceptualization.
The authors acknowledge the support of the National Natural Science Foundation of China (NSFC, Nos. 12005243, 22205239, 12304183, and 22375221) and the Guangdong Basic and Applied Basic Research Foundation (Nos. 2022B1515120014, 2023B0303000003, 2023A1515110785, and 2023B1515120060) The authors appreciate the neutron beamtime at iMATERIA of J-PARC (Proposal No 2024PM3003) and TREND (
Supplementary material associated with this article can be found, in the online version, at doi:
H. Jiang, W. Zhang, X. Kang, et al., J. Am. Chem. Soc. 142 (2020) 9642–9652.
N. Antil, N. Akhtar, R. Newar, et al., ACS Catal. 11 (2021) 10450–10459. doi: 10.1021/acscatal.1c02529
X.B. Liu, C.Y. Zhu, M.Y. Li, et al., Angew. Chem. Int. Ed. 63 (2024) e202412408. doi: 10.1002/anie.202412408
R.B. Lin, L. Li, H. Wu, et al., J. Am. Chem. Soc. 139 (2017) 8022–8028. doi: 10.1021/jacs.7b03850
C. Gu, N. Hosono, J.J. Zheng, et al., Science 363 (2019) 387–391. doi: 10.1126/science.aar6833
S.Q. Yang, B. Xing, L.L. Wang, et al., Chem. Bio. Eng. 1 (2024) 245–251. doi: 10.1021/cbe.3c00073
I.A. Lazaro, C.J.R. Wells, R.S. Forgan, Angew. Chem. Int. Ed. 59 (2020) 5211–5217. doi: 10.1002/anie.201915848
A. Vadivelmurugan, R. Sharmila, W.L. Pan, et al., ACS Omega 8 (2023) 41909–41917. doi: 10.1021/acsomega.3c06991
X.W. Liu, J. Obacz, G. Emanuelli, et al., Chem. Mater. 36 (2024) 3588–3603. doi: 10.1021/acs.chemmater.3c02954
H.C. Zhou, J.R. Long, O.M. Yaghi, Chem. Rev. 112 (2012) 673–674. doi: 10.1021/cr300014x
X. Cui, K.J. Chen, H.B. Xing, et al., Science 353 (2016) 141–144. doi: 10.1126/science.aaf2458
R.B. Lin, S. Xiang, W. Zhou, et al., Chem 6 (2020) 337–363. doi: 10.1016/j.chempr.2019.10.012
K.J. Chen, D.G. Madden, T. Pham, et al., Angew. Chem. Int. Ed. 55 (2016) 10268–10272. doi: 10.1002/anie.201603934
C.T. He, Z.M. Ye, Y.T. Xu, et al., Chem. Sci. 8 (2017) 7560–7565. doi: 10.1039/C7SC03067C
S.D. Burd, S.Q. Ma, J.A. Perman, et al., J. Am. Chem. Soc. 134 (2012) 3663–3666. doi: 10.1021/ja211340t
J.B. Lin, T.T.T. Nguyen, R. Vaidhyanathan, et al., Science 374 (2021) 1464–1469. doi: 10.1126/science.abi7281
S.Q. Yang, R. Krishna, H.W. Chen, et al., J. Am. Chem. Soc. 145 (2023) 13901–13911. doi: 10.1021/jacs.3c03265
X.L. Wang, M. Alzayer, A.J. Shih, et al., J. Am. Chem. Soc. 146 (2024) 3943–3954. doi: 10.1021/jacs.3c11671
Z.N. Deng, L.S. Yang, H.T. Xiong, et al., Small Methods 9 (2025) 2400838. doi: 10.1002/smtd.202400838
Y.T. Li, W.L. Li, L.P. Zhang, et al., Adv. Funct. Mater. 35 (2025) 2411951. doi: 10.1002/adfm.202411951
Y.T. Zhou, A. Koedtruad, Z.H. Tan, et al., Chin. J. Struct. Chem. 42 (2023) 100059.
L. Yu, X. Han, H. Wang, et al., J. Am. Chem. Soc. 143 (2021) 19300–19305. doi: 10.1021/jacs.1c10423
S.S. Liu, X. Han, Y.C. Chai, et al., Angew. Chem. Int. Ed. 60 (2021) 6526–6532. doi: 10.1002/anie.202014680
A.A. Coelho, J. Appl. Cryst. 51 (2018) 210–218. doi: 10.1107/s1600576718000183

Figure 1 (a) PXRD patterns of [Zn2(mtz)2(ox)]·solv. (b) Three-dimensional pillared-layer frameworks of [Zn2(mtz)2(ox)]·solv (for clarity, solvents were removed).

Figure 2 NPD patterns, structure and ∠NZnN angle of MOFs: (a, d, g) [Zn2(mtz)2(ox)]·solv, (b, e, h) [Zn2(mtz)2(ox)] and (c, f, i) [Zn2(mtz)2(ox)]·0.28CO2.

Figure 4 (a) Site Ⅰ in [Zn2(mtz)2(ox)]·0.28CO2 for CO2. (b) Side view of Site Ⅰ in [Zn2(mtz)2(ox)]·0.28CO2 for CO2. (c) Adsorption density distribution calculated by GCMC simulation at 298 K and 1.0 bar and Site Ⅰ in [Zn2(mtz)2(ox)]·0.28CO2 for CO2.
Table 1. Lattice parameters of Zn-ox-mtz·solv [17], [Zn2(mtz)2(ox)]·solv, [Zn2(mtz)2(ox)] and [Zn2(mtz)2(ox)]·0.28CO2 obtained from NPD.
| Isomer | Space group | a (Å) | b (Å) | c (Å) | α (°) | β (°) | γ (°) | V (Å3) |
| Zn-ox-mtz·solv [17] | P21/c | 8.6166(2) | 7.8948(1) | 10.5066(2) | 90 | 96.97(2) | 90 | 709.45(2) |
| [Zn2(mtz)2(ox)]·solv | P21/c | 8.6881(1) | 8.0947(1) | 10.4423(2) | 90 | 98.51(1) | 90 | 726.31(1) |
| [Zn2(mtz)2(ox)] | P21/c | 8.7207(1) | 8.2027(1) | 10.3689(1) | 90 | 99.58(1) | 90 | 731.38(1) |
| [Zn2(mtz)2(ox)]·0.28CO2 | P21/c | 8.7581(1) | 8.3420(1) | 10.2752(1) | 90 | 100.83(1) | 90 | 737.34(1) |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: