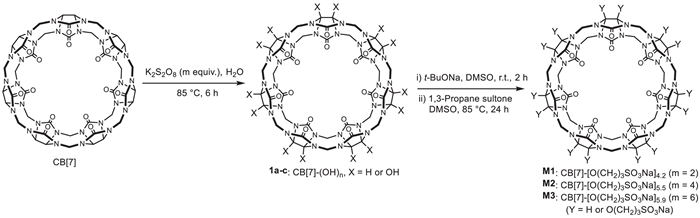
Scheme 1.
The synthesis of M1-M3, with n representing the number of the side chains.
Sulfonatoproxylated cucurbit[7]urils as highly water-soluble and biocompatible excipients for solubilizing poorly soluble drugs and improving the bioavailability of indomethacin
Pei-Pei Liu , Jia-Bin Xing , Yue-Yang Liu , Ke Feng , Hui Wang , Dan-Wei Zhang , Wei Zhou , Gang Zhao , Jiaheng Zhang , Zhan-Ting Li

Over the past few decades, host-guest chemistry has made significant strides in both the development of new host-guest systems and their functional applications [1-7]. In practical applications, natural cyclodextrins and their derivatives as hosts have achieved unprecedented success [8-10]. Prominent examples include sulfonatobutylether-β-cyclodextrin Captisol as a solubilizing excipient in drug formulations and carboxyethylthio-ether-γ-cyclodextrin Sugammadex as an antagonist for rapid reversal of the blockade of neuromuscular blocking agents like rocuronium and vecuronium [11,12]. However, the binding affinity and selectivity of cyclodextrins for organic guests are relatively low [13]. Therefore, supramolecular chemists have a long-standing interest in developing synthetic receptors, such as calixarenes [14,15], cucurbiturils [16,17] pillararenes [18-20], and other various macrocycles and molecular cages [21-23], that offer enhanced binding affinity and selectivity for different kinds of guests. Advances along this line have open many opportunities for the design of new functional supramolecular architectures for, such as, separation, sensing, bioimaging, and drug delivery [6,24].
Since the early 20th century, small-molecule drugs have played a central role in drug development, and their use has continued to grow in recent years [25]. However, many clinically used drugs and new active pharmaceutical ingredients with specific target affinities are poorly soluble [26], which has been a challenge in drug formulation. The use of excipients to encapsulate and solubilize these drugs has proven to be an effective approach, as demonstrated by Captisol [11]. However, the search for new excipients has been active in host-guest chemistry. In this context, cucurbit[7]uril (CB[7]) has been successfully used to solubilize clinically relevant drugs, improve their bioavailability and/or reduce their toxicity [27-32]. Although CB[7] demonstrates good biosafety [35,36], its modest water solubility (20–30 mmol/L) limits its wider utility [37,38]. Previously, we sulfonatopropoxylated cucurbit[8]uril to prepare water-soluble derivatives for antagonizing neuromuscular blockers [33]. The key modification step involved multihydroxylation of the backbone through radical oxidation [34]. Isaacs et al. prepared a CB[7] derivative bearing two sulfonatobutyl groups, but found that these hydrophilic groups did not improve the water solubility of the resulting derivatives [35]. We envisioned that multiple sulfonatopropoxy groups of randomly-appended to the outer surface of the macrocyclic backbone might create steric hindrance to reduce intermolecular aggregation and consequently improve the solubility of the resulting derivatives. We thus synthesized sulfonatopropoxy-attached CB[7]s M1-M3 (Scheme 1), starting with multihydroxylation of CB[7] followed by sulfonate-propylation of the multihydroxylated intermediates with 1,3-propane sultone. Approximate 4–6 hydrophilic sulfonatepropoxy groups were introduced to the "equator" carbons of CB[7] and its solubility increased by up to 26-fold. These resulting derivatives exhibit excellent biocompatibility, significantly solubilize eighteen poorly soluble drugs, and improve the therapeutic efficacy of indomethacin while weakening its side effect of causing gastriculcer.
The amount of potassium persulfate was used to control the average number of hydroxyl groups introduced to the backbone. With the use of 2, 4, and 6 equiv. of the oxidizing agent, averagely 4.2, 5.5, and 5.9 sulfonatopropoxy groups were introduced to M1, M2, and M3, respectively, as determined by analyzing the relative content of N and S elements in the samples. The average number of the side chains was further estimated by comparing the integrative intensities of the CH2 units of CB[7] and the CH2CHCH2 units of the side chains in the 1H NMR spectra, with results in general agreement (Table S1 in Supporting information). Since all fourteen NCHN units of the backbone can be oxidized, both the intermediates and final products are expected to be a mixture of macrocycles bearing different numbers of side chains at different positions. This was reflected in the 13C NMR spectra, as seen with M1, where the CB[7] signals around 156, 96, 71, and 52 ppm split into multiple peaks (Figs. S1-S3 in Supporting information). High performance liquid chromatographic (HPLC) analysis of M1-M3 displayed one broad major peak and one or more minor peaks, even though the major broad peak was expected to be formed by different derivatives (Fig. S4 in Supporting information). The solubility of M1-M3 in water was determined to be 829, 738, and 607 mmol/L, which corresponded to 1520, 1513 and 1280 mg/mL, respectively. Compared to the solubility of CB[7] (20–30 mmol/L), the molar solubilities of the three derivatives increased by 26.6-, 23.6-, and 19.2-fold, respectively. In phosphate buffer solution (10 mmol/L, pH7.4), the solubilities of M1 and M2 was determined to be as high as 804 and 715 mmol/L, respectively.
Although bearing the largest number of side chains, M3 exhibited the lowest solubility. We thus focused on the potential of M1 and M2 as excipients for solubilizing poorly soluble drugs. Prior to this, we assessed their biocompatibility. The maximum tolerated dose for ICR mice (3 female and 3 male) via intravenous injection was determined to be at least 1000 mg/kg, which is 4 times higher than that of CB[7] (250 mg/kg) under same conditions [36]. When converted to molar quantities, the maximum tolerated doses of M1 and M2 were 3.5- and 3.3-fold that of CB[7]. Mice treated with the 1000 mg/kg dose of M1 or M2 was monitored for 14 days, with body weight changes recorded (Fig. S5 in Supporting information). Mice treated with M1 suffered about < 10% decrease of the body weight on day 1. The body weight then recovered and increased steadily. No such weight loss was observed in the M2-treated group, and the body weight continuously increased throughout the observation period.
Encouraged by the high water-solubility and biocompatibility of M1 and M2, we then investigated their ability to solubilize eighteen poorly soluble drugs (Scheme 2). For comparison, their concentration was kept at 5 mmol/L, and the concentration of the solubilized drugs was determined using HPLC according to reported methods [37]. The results are provided in Table 1. It can be found that M1 increased the solubility of 16 of the 18 drugs, while M2 improved the solubility of all the drugs. For nine drugs, M1 exhibited better solubilizing efficiency, while M2 was superior for eight others. For estriol, both M1 and M2 achieved comparable results. The S/S0 ratios of both excipients showed the highest solubilization efficiency for β-estradiol.
 DownLoad:
CSV
DownLoad:
CSV
| Drugs | S0 (mmol/L) | S (mmol/L) | |||
| M1 | S/S0 | M2 | S/S0 | ||
| Corticosterone | 0.44 | 1.58 | 3.59 | 1.60 | 3.64 |
| Fluticasone propionate | −a | −a | 0.011 | ||
| Testosterone | 0.033 | 1.55 | 47.0 | 1.38 | 41.8 |
| Megestrol acetate | 0.0018 | 0.12 | 66.7 | 0.11 | 61.1 |
| β-Estradiol | 0.0084 | 1.27 | 151 | 0.69 | 82.1 |
| Estriol | 0.064 | 0.92 | 14.3 | 0.92 | 14.3 |
| Dapagliflozin | 2.87 | 4.53 | 1.58 | 5.00 | 1.74 |
| Melphalan | 1.33 | 2.08 | 1.56 | 2.71 | 2.04 |
| Canagliflozin | −a | 0.17 | 0.43 | ||
| Indomethacin | 0.084 | 5.00 | 59.5 | 3.60 | 42.9 |
| Carbamazepine | 0.27 | 2.13 | 7.89 | 1.34 | 4.92 |
| Emodin | −a | 0.90 | 0.59 | ||
| Donepezil | 0.073 | 3.67 | 50.3 | 3.15 | 43.2 |
| Aripiprazole | 0.011 | 0.41 | 37.3 | 0.55 | 50.0 |
| (+)-Camptothecin | 0.0071 | 0.76 | 107 | 0.36 | 50.7 |
| Remdesivir | −a | 0.67 | 0.23 | ||
| Posaconazole | −a | 0.002 | 0.003 | ||
| Tamoxifen | −a | −a | 0.006 | ||
| a No detectable concentration was obtained. | |||||
For dapagliflozin, M1 and M2 could improve its solubility from 2.87 mmol/L to 4.53 or 5.00 mmol/L. Nuclear Overhauser effect spectroscopic (NOESY) 1H NMR spectrum of the 1:1 solution of dapagliflozin and M1 in D2O revealed intermolecular nuclear Overhauser effect (NOE) between the CH3 protons of dapagliflozin and the outward protons of the CH2 groups of M1 (Fig. S6 in Supporting information), supporting that ethoxy-bearing benzene ring was encapsulated by the macrocycle of M1 which shortened the distance of these two groups of protons. Transmission electron microscopic (TEM) image of the example prepared from the evaporation of the 1:1 solution of M1 and dapagliflozin (1 mmol/L) in water did not reveal the formation of any observable nanoparticles. Dynamic light scattering spectrum of their 1:1 solution (0.5 mmol/L) in water did not exhibit the formation of nanoparticles with hydrodynamic diameter of ≥5 nm. Diluting their 1:1 solution in D2O from 4.5 mmol/L to 0.5 mmol/L did not cause observable shifting of the signals in the 1H NMR spectra. All these results indicated that the CB[7] derivative and the guest molecule did not assemble into well-defined nanoparticles due to the introduction of the hydrophilic side chains.
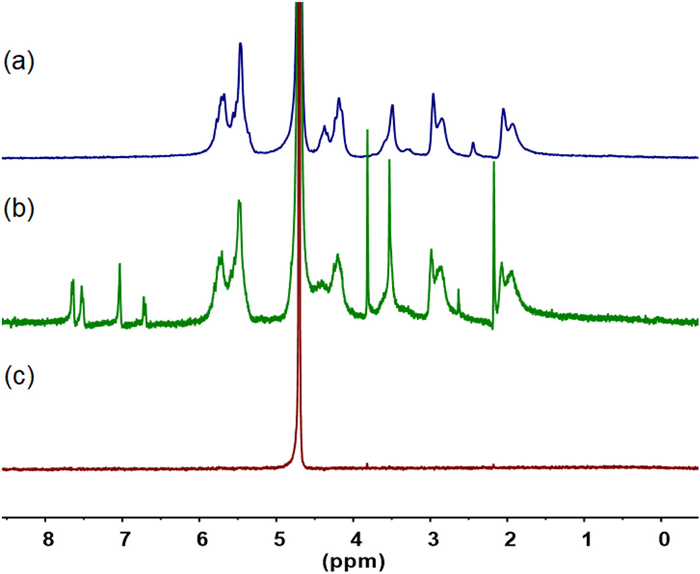
For indomethacin (IMC), M1 and M2 increased the solubility from 0.084 mmol/L to 5.00 and 3.60 mmol/L, respectively, which indicated that M1 achieved a complete availability. 1H NMR experiments in D2O showed that, at a concentration of 5 mmol/L, M1 could increase the concentration of IMC to 4.7 mmol/L (Fig. 1) [38]. Considering the accuracy of the 1H NMR spectroscopy, this result further supports the high availability of M1 for solubilizing IMC. For drugs with relatively high initial solubility, the S/S0 ratios were generally lower, but the solubilizing effect remained apparent. Given the high solubility of IMC achieved by M1 and M2, we further evaluated the stability of the two 1:1 complexes formed between them using the phase solubility diagram method (Fig. S7 in Supporting information) [39]. The binding constants (Ka) for the M1·IMC and M2·IMC complexes were determined to be 1.7 × 106 and 3.2 × 105 L/mol, respectively [40].
Considering the high binding affinity and efficient solubilization of M1 for IMC, we further investigated if M1 could comprehensively improve IMC's anti-inflammatory activity in Sprague-Dawley rats with adjuvant-induced arthritis [41]. Nine rats (male, 200 ± 20 g) were randomly divided into three groups and fasted but given free access to water for 24 h prior to gastric administration of normal saline, IMC (15 mg/kg), and IMC (15 mg/kg)/M1 (85 mg/kg), respectively. The administration volume was approximately 1 mL, adjusted according to each rat's body weight. The right hind paw volume was measured using a toe volume measuring instrument and recorded as the baseline (0 h). 30 min after administration, edema was induced by injecting 0.1 mL of complete Freund's adjuvant into the right hind paw pad. At 1, 2, 4, 6, and 8 h after injection, swelling of the rat paw was measured. The swelling rate and swelling inhibition were calculated according to the reported method [42,43]. After the final measurement at 8 h, rats were sacrificed by 5% isoflurane, and right hind paw and stomach were taken for histological analysis.
The swelling rate of the saline group increased significantly over time and reached 68% after 8 h (Fig. 2a), indicating successful model establishment. The IMC-treated group displayed a much lower swelling rate than the saline group, consistent with its inherent anti-inflammatory properties. Notably, the IMC/M1 group exhibited the lowest swelling rates, with reductions of 14%, 32%, 19%, 29%, and 20% compared to the IMC group at 1, 2, 4, 6, and 8 h, respectively. The inhibition rates of the IMC/M1 group were 39% and 52% at 1 and 2 h, compared to 21% and 27% for the IMC group, revealed that M1 improved inhibition efficiency, particularly during the first 2 h (Fig. 2b). Given that IMC was used as a suspension due to its low solubility, the enhanced swelling inhibition in the IMC/M1 group can be attributed to the solubilization of M1 for IMC, which might speed up the absorption of IMC.
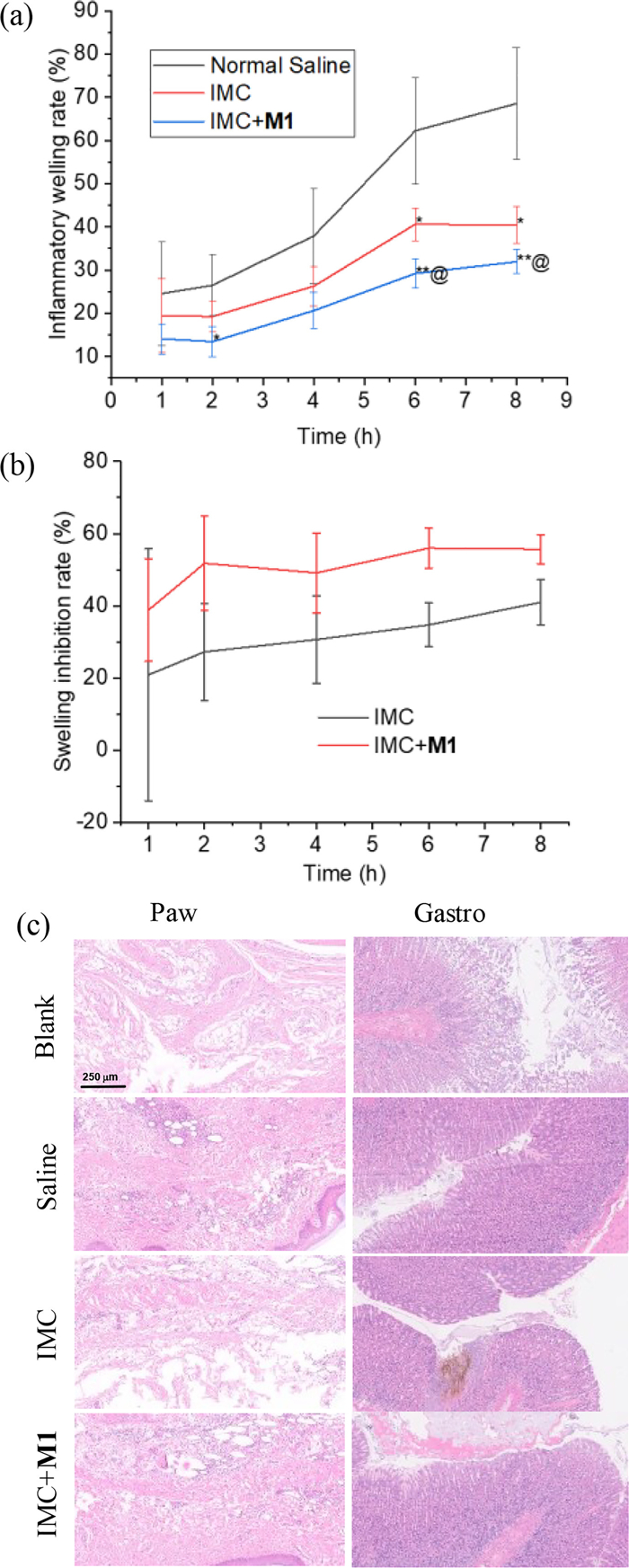
H&E staining revealed severe inflammatory infiltration and edema in the toe sections of the saline and M1 groups (Fig. 2c). The inflammatory infiltration in the IMC group was significantly reduced, but edema remained severe. In contrast, the IMC/M1 group displayed much milder tissue damage, aligning with the paw swelling results. Examination of the stomach sections showed no signs of necrosis or exudation in the saline group, while the IMC group exhibited obvious hemorrhagic mucosal erosion and gastric ulcers, consistent with the known gastric irritation side effect of IMC. In the IMC/M1 group, there was almost no evidence of mucosal erosion or hemorrhage, suggesting that the inclusion of M1 for IMC reduced its irritation to the gastric mucosa. This effect may also decrease the adhesion of dispersed IMC particles to the gastric wall, promoting their passage into the intestinal tract. Thus, in addition to enhancing its anti-inflammatory efficacy, M1 was able to mitigate its gastrointestinal side effects. The serum concentrations of two inflammatory cytokines, i.e., TNF-α and IL-1β, were assessed using ELISA. However, no significant difference was observed between the normal and model groups (Fig. S8 in Supporting information), which might be attributed to the fact that 8 h was not long enough for the adjuvant-induced plantar inflammation to trigger the whole body's immunity response.
Finally, we evaluated the hemolytic and cytotoxic effects of M1 (Fig. S9 in Supporting information) [44]. It was found that even at concentrations of up to 2.5 mg/mL, the hemolysis rates of rat and human red blood cells remained below 5%. The cytotoxicity of M1 on rat cardiomyocytes (H9C2) and mouse fibroblasts (L929) was also assessed. The results showed that both cell types could maintain survival rates above 80% in the presence of 2.0 mg/mL of M1 (Fig. S10 in Supporting information).
In conclusion, we have developed a simple strategy to significantly improve the water solubility of CB[7] by introducing multiple sulfonatopropoxy groups to its "equator" carbons. The resulting derivatives exhibit increased maximum tolerated doses and function as biocompatible excipients capable of significantly enhancing the solubility of eighteen poorly soluble drugs. In vivo experiments using an adjuvant-induced arthritis rat model showed that one derivative not only increased the anti-inflammatory activity of indomethacin but also alleviated its side effect of gastric irritation. This kind of multi-sulfonatopropoxylated CB[7]-derived excipients might be a potential safe drug delivery system to improve the bioactivity and bioavailability of poorly soluble drugs.
Animal experiments were performed in agreement with the guidelines of the Animal Care and Use Committee of Fudan University (No. 2024-CHEM-026). Scientific research with human whole blood, which was provided by the Shanghai Blood Center, was approved by the Shanghai Municipal Commission of Health and Family Planning (No. HXB-2024–15).
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Pei-Pei Liu: Writing – original draft, Investigation. Jia-Bin Xing: Investigation. Yue-Yang Liu: Investigation, Formal analysis. Ke Feng: Investigation. Hui Wang: Supervision. Dan-Wei Zhang: Supervision. Wei Zhou: Writing – review & editing, Supervision, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. Gang Zhao: Supervision, Resources, Data curation. Jiaheng Zhang: Supervision, Resources, Formal analysis, Data curation. Zhan-Ting Li: Writing – review & editing, Supervision, Resources, Project administration, Methodology, Funding acquisition, Formal analysis, Data curation, Conceptualization.
We thank the National Natural Science Foundation of China (Nos. 21921003 and 22201293) and the National Key R & D Program of China (No. 2023YFC3503400) for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
D.J. Cram, Angew. Chem. Int. Ed. 27 (1988) 1009–1020. doi: 10.1002/anie.198810093
X. Ma, Y. Zhao, Chem. Rev. 115 (2015) 7794–7839. doi: 10.1021/cr500392w
Y.L. Ma, S. Yan, X.J. Xu, H. Cao, R. Wang, Chin. Chem. Lett. 35 (2024) 108645. doi: 10.1016/j.cclet.2023.108645
S. Liu, Y. Lin, D. Yan, Sci. China Chem. 66 (2023) 3532–3538. doi: 10.1007/s11426-023-1656-y
G. Li, R.W. Lewis, R. Eelkema, CCS Chem. 6 (2024) 27–40. doi: 10.31635/ccschem.023.202303177
Y.Y. Liu, X.Y. Yu, Y.C. Pan, et al., Sci. China Chem. 67 (2024) 1397–1441. doi: 10.1007/s11426-024-1971-4
S.B. Yu, W. Zhou, J. Tian, et al., Sci. Sin. Chim. 53 (2023) 2345–2356. doi: 10.1360/SSC-2023-0134
M. Inam, M.F. Sareh Sadat, W. Chen, Chem. Res. Chin. Univ. 39 (2023) 857–861. doi: 10.1007/s40242-023-3204-0
Y. Sun, L. Jiang, Y. Chen, Y. Liu, Chin. Chem. Lett. 35 (2024) 108644. doi: 10.1016/j.cclet.2023.108644
G. Crini, S. Fourmentin, É. Fenyvesi, et al., Environ. Chem. Lett. 16 (2018) 1361–1375. doi: 10.1007/s10311-018-0763-2
V.J. Stella, R.A. Rajewski, Int. J. Pharm. 583 (2020) 119396. doi: 10.1016/j.ijpharm.2020.119396
G.M. Keating, Drugs 76 (2016) 1041–1052. doi: 10.1007/s40265-016-0604-1
M.V. Rekharsky, Y. Inoue, Chem. Rev. 98 (1998) 1875–1918. doi: 10.1021/cr970015o
D.S. Guo, Y. Liu, Chem. Soc. Rev. 41 (2012) 5907–5921. doi: 10.1039/c2cs35075k
Y.C. Pan, X.Y. Hu, D.S. Guo, Angew. Chem. Int. Ed. 60 (2021) 2768–2794. doi: 10.1002/anie.201916380
P. Balaram, ACS Omega 9 (2024) 4162–4165. doi: 10.1021/acsomega.3c07196
S.J. Barrow, S. Kasera, M.J. Rowland, J. del Barrio, O.A. Scherman, Chem. Rev. 115 (2015) 12320–12406. doi: 10.1021/acs.chemrev.5b00341
T. Ogoshi, T.A. Yamagishi, Y. Nakamoto, Chem. Rev. 116 (2016) 7937–8002. doi: 10.1021/acs.chemrev.5b00765
Z.D. Tang, X.M. Sun, T.T. Huang, et al., Chin. Chem. Lett. 34 (2023) 107698. doi: 10.1016/j.cclet.2022.07.041
W. Yang, W. Zhang, J. Chen, J. Zhou, Chin. Chem. Lett. 35 (2024) 108740. doi: 10.1016/j.cclet.2023.108740
H. Zhu, L. Chen, B. Sun, et al., Nat. Rev. Chem. 7 (2023) 768–782. doi: 10.1038/s41570-023-00531-9
X.Y. Lou, S. Zhang, Y. Wang, Y.W. Yang, Chem. Soc. Rev. 52 (2023) 6644– 6663. doi: 10.1039/D3CS00506B
G. Montà-González, E. Ortiz-Gómez, R. López-Lima, et al., Molecules 29 (2024) 1621. doi: 10.3390/molecules29071621
Y. Zhang, G. Zhang, X. Xiao, Q. Li, Z. Tao, Coord. Chem. Rev. 514 (2024) 215889. doi: 10.1016/j.ccr.2024.215889
M.W.Y. Southey, M. Brunavs, Front. Drug Discov. 3 (2023) 1314077. doi: 10.3389/fddsv.2023.1314077
L. Di, P.V. Fish, T. Mano, Drug Discov. Today 17 (2012) 486–495. doi: 10.1016/j.drudis.2011.11.007
K.I. Kuok, S. Li, I.W. Wyman, R. Wang, Ann. N. Y. Acad. Sci. 1398 (2017) 108–119. doi: 10.1111/nyas.13376
J.J. Chu, M.G. Apps, N.J. Wheate, Supramol. Chem. 26 (2014) 648–656. doi: 10.1080/10610278.2014.926361
X. Yang, Z. Wang, Y. Niu, et al., MedChemComm 7 (2016) 1392–1397. doi: 10.1039/C6MD00239K
S. Li, J.Y.W. Chan, Y. Li, et al., Org. Biomol. Chem. 14 (2016) 7563–7569. doi: 10.1039/C6OB01060A
Y. Wang, X. Yang, J. Luo, et al., Int. J. Pharm. 660 (2024) 124351. doi: 10.1016/j.ijpharm.2024.124351
C.H.T. Kwong, J. Mu, S. Li, et al., Chin. Chem. Lett. 32 (2021) 3019–3022. doi: 10.1016/j.cclet.2021.04.008
H.K. Liu, F. Lin, S.B. Yu, et al., J. Med. Chem. 65 (2022) 16893–16901. doi: 10.1021/acs.jmedchem.2c01677
S.Y. Jon, N. Selvapalam, D.H. Oh, et al., J. Am. Chem. Soc. 125 (2003) 10186–10187. doi: 10.1021/ja036536c
E.L. Robinson, P.Y. Zavalij, L. Isaacs, Supramol. Chem. 27 (2015) 288–297. doi: 10.1080/10610278.2014.940952
V.D. Uzunova, C. Cullinane, K. Brix, W.M. Nau, A.I. Day, Org. Biomol. Chem. 8 (2010) 2037–2042. doi: 10.1039/b925555a
P.S. Lee, J.Y. Han, T.W. Song, et al., Int. J. Pharm. 316 (2006) 29–36. doi: 10.1016/j.ijpharm.2006.02.035
B. Zhang, P.Y. Zavalij, L. Isaacs, Org. Biomol. Chem. 12 (2014) 2413–2422. doi: 10.1039/C3OB42603C
D. Ma, G. Hettiarachchi, D. Nguyen, et al., Nat. Chem. 4 (2012) 503–510. doi: 10.1038/nchem.1326
B. Zhang, L. Isaacs, J. Med. Chem. 57 (2014) 9554–9563. doi: 10.1021/jm501276u
K. Gou, X. Guo, Y. Wang, et al., Mater. Des. 214 (2022) 110359. doi: 10.1016/j.matdes.2021.110359
J. Li, Y. Guo, H. Li, L. Shang, S. Li, Artif. Cells Nanomed. Biotechnol. 46 (2017) 1085–1094.
J. Zhou, F. Zhu, J. Li, Y. Wang, Mater. Sci. Eng. C 90 (2018) 314–324. doi: 10.1016/j.msec.2018.04.071
I. Sæbø, M. Bjørås, H. Franzyk, E. Helgesen, J. Booth, Int. J. Mol. Sci. 24 (2023) 2914. doi: 10.3390/ijms24032914

Figure 1 1H NMR spectrum (500 MHz) of (a) M1 (5.0 mmol/L), (b) M1 (5.0 mmol/L) + indomethacin (4.7 mmol/L), and (c) indomethacin (0.084 mmol/L) in D2O.

Figure 2 (a) Swelling rate versus time, (b) swelling inhibition rate versus time, and (c) H & E staining results of the stomach and toe of rats in each group. Compared with the swelling rate of the saline group, *P < 0.05; **P < 0.01; compared with the swelling rate of IMC, @P < 0.05.
Table 1. Solubility of drugs in the absence (S0, mmol/L) and presence (S, mmol/L) of M1 and M2 both at 5 mmol/L.
| Drugs | S0 (mmol/L) | S (mmol/L) | |||
| M1 | S/S0 | M2 | S/S0 | ||
| Corticosterone | 0.44 | 1.58 | 3.59 | 1.60 | 3.64 |
| Fluticasone propionate | −a | −a | 0.011 | ||
| Testosterone | 0.033 | 1.55 | 47.0 | 1.38 | 41.8 |
| Megestrol acetate | 0.0018 | 0.12 | 66.7 | 0.11 | 61.1 |
| β-Estradiol | 0.0084 | 1.27 | 151 | 0.69 | 82.1 |
| Estriol | 0.064 | 0.92 | 14.3 | 0.92 | 14.3 |
| Dapagliflozin | 2.87 | 4.53 | 1.58 | 5.00 | 1.74 |
| Melphalan | 1.33 | 2.08 | 1.56 | 2.71 | 2.04 |
| Canagliflozin | −a | 0.17 | 0.43 | ||
| Indomethacin | 0.084 | 5.00 | 59.5 | 3.60 | 42.9 |
| Carbamazepine | 0.27 | 2.13 | 7.89 | 1.34 | 4.92 |
| Emodin | −a | 0.90 | 0.59 | ||
| Donepezil | 0.073 | 3.67 | 50.3 | 3.15 | 43.2 |
| Aripiprazole | 0.011 | 0.41 | 37.3 | 0.55 | 50.0 |
| (+)-Camptothecin | 0.0071 | 0.76 | 107 | 0.36 | 50.7 |
| Remdesivir | −a | 0.67 | 0.23 | ||
| Posaconazole | −a | 0.002 | 0.003 | ||
| Tamoxifen | −a | −a | 0.006 | ||
| a No detectable concentration was obtained. | |||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: