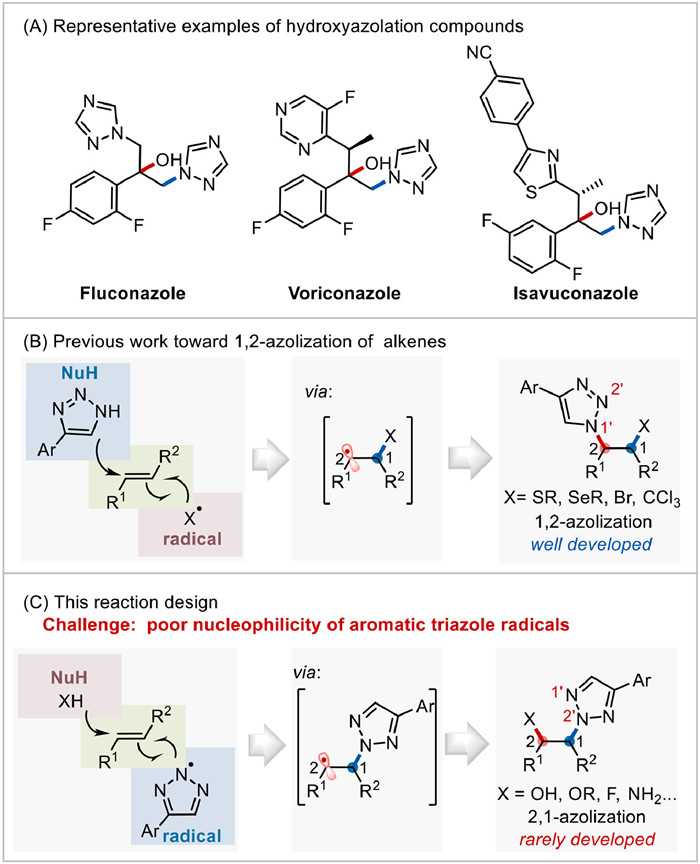
Figure 1.
Background and reaction design. (A) Representative examples highlighting diverse bioactive hydroxyazolation compounds. (B) Previous work toward 1,2-azolization of alkenes. (C) This reaction design.

Nitrogen-containing compounds are structurally prevalent motifs and have been widely found in natural products, functional materials as well as drug molecules [1-5]. Notably, hydroxyazolation compounds, by employing azoles as building blocks, are often used as bioactive molecules in pharmaceuticals, such as clinically important antifungal triazoles, fluconazole, voriconazole, and isavuconazole (Fig. 1A) [6-8]. Consequently, seeking green and efficient strategies for the facile construction of hydroxyazolation compounds by in various C—N bond formations has continuously attracted extensive attention [9, 10].
Radical carboamination of alkenes prove to be one of the most dominant and versatile step-economical and atom-economical approaches for the construction of C—N bonds and complex nitrogen-containing compounds [11]. Among them, strategies represented by metal-catalyzed redox eliminations [12-15] and radical-addition followed by nucleophilic attack process [16-18] have been developed rapidly in recent years. Notably, the latter strategy minimizes the need for functional groups and protecting groups, enabling one-pot construction of multiple chemical bonds [19-21], including various C—N bonds. The efficient strategy is generally promoted by using C/X-centered radicals and N-nucleophiles to conduct 1,2-difunctional amination/azolization of alkenes (Fig. 1B) [22-24]. For example, in 2018, Lei group developed the electrochemical sulfenylation reactions of alkenes with thiols and nucleophiles [25]. Later, Xu group established a photoredox catalytic four-component radical-polar Ritter-type amination of alkenes [26]. Generally, the C/X-centered radicals undergo a radical addition to alkenes and yield relayed carbon- centered radicals (CCRs) which are oxidized as the corresponding cations to be trapped by N-nucleophiles. However, in stark contrast to the extensive research of 1,2-azolization of alkenes, 2,1-difunctional amination/azolization of alkenes by using N-centered radicals (NCRs) and nucleophiles still remains rarely underexplored (Fig. 1C). It is possibly due to the highly active electron properties of NCRs [27] and the relatively poor nucleophilicity of aromatic NCRs to react with arylalkenes [28].
Organic electrochemistry directly uses an anode to remove electrons to achieve the function of oxidants and is increasingly considered as a versatile and environmentally benign method in chemical synthesis [29-33]. One crucial advantage of using electro-oxidation is that the reaction selectivity and efficiency can be conveniently adjusted by manipulating the electric potential or current, enabling various conversions those are otherwise synthetically inaccessible. Under electrochemical conditions, azoles can be formed the corresponding NCRs with high efficiency [34, 35]. Thus, we envision that aromatic N-centered azole radicals could be efficiently generated and activated by modulating the electrochemical conditions to enable the NCR addition to alkenes. The resulting relayed carbon radicals may then be further oxidized as the cations by electro-oxidation to condensate with nucleophiles and enable an unprecedented 2,1-difunctional azolization of alkenes (Fig. 1C).
Inspired by the recent achievements of our group's in diverse transformations of N-sulfonyl-1,2, 3-triazoles [36-43], we herein present an efficient example of electrochemical radical 2,1-azolization of alkenes using azoles as radical precursors (Fig. 2). This electrosynthetic reaction obviates the need for metal catalysts and external chemical oxidants. Furthermore, this reaction features a broad substrate scope, accommodating various alkenes, azoles, and nucleophiles, and delivering the desired products in yields of up to 91%. Significantly, this electrochemical strategy can also be applied to the one-pot construction of C–N and C–X (X = F/N/O) bonds which provides a rapid way to get access to the difunctionalization of alkenes.
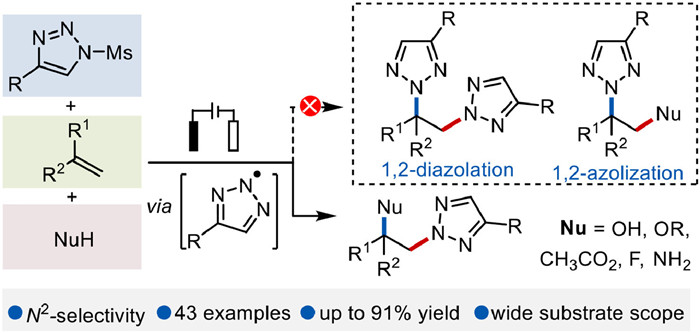
Water is often used as an efficient nucleophilic reagent in electrochemical reactions [44, 45]. Therefore, the electrochemical reaction of 1-(mesyl)-4-phenyl-1H-1,2, 3-triazole 1, 1,1-diphenylethylene 2 and water was conducted by the use of a carbon plate as anode and a platinum-plate as cathode, and nBu4NOAc as an electrolyte, at 5 mA constant current in MeCN at 80 ℃ under nitrogen atmosphere for 5 h (Table 1, entry 1). To our delight, the hydroxyazolation product 4 was afforded in 47% isolated yield with high N2-selectivity, giving NH-1,2, 3-triazole as the main byproduct. Then, the current for this reaction was screened, and the results demonstrated that 8 mA was optimal to deliver the best yield of 89% (entries 2 and 3). Neither N1-/N3-selective product nor 1,2-diazolation/1,2-azolization product was observed (Fig. 2). Control experiments demonstrated that the reaction became less efficient at 50 ℃ (entry 4). Replacing the anode with a platinum plate (entry 5) or conducting the reaction in the absence of electric current (entry 6) completely inhibited the yield of compound 4. No improvement in reaction efficiency was observed when using nBuN4BF4 as electrolytes (entry 7). Subsequently, it was found that conducting the reaction in air led to a decreased yield (74%, entry 8). Furthermore, utilizing acetone as the solvent was found to lower the yield of product 4 (entry 9). Various protecting groups, such as methylsulfonyl, p-toluenesulfonyl, and isopropylsulfonyl, also showed a good tolerance in this electrochemical reaction and gave good to excellent yields (19%−89%, Table S1 in Supporting information).
 DownLoad:
CSV
DownLoad:
CSV
 |
||
| Entry | Deviation from standard conditions | Yield b (%) |
| 1 | 5 mA | 47 c |
| 2 | None | 89 (3.2 F/mol, 62.5%) d |
| 3 | 10 mA | 75 |
| 4 | Reaction at 50 ℃ | 61 |
| 5 | Pt plate (1 cm × 1 cm) as anode | NR |
| 6 | No electricity | NR |
| 7 | nBuN4BF4 instead of nBu4NOAc | 38 |
| 8 | In air | 74 |
| 9 | Acetone as the solvent | 36 |
| a Reaction conditions: undivided cell, graphite rod anode, Pt cathode, 1 (0.5 mmol), 2 (1.0 mmol), MeCN (9.5 mL), H2O (0.5 mL), N2, electrolytes (1.0 mmol), 8 mA, 4–6 h (based on 1). b Yields of isolated products are provided. c Isolated yield. NR = no reaction. d Charge consumption and Faradaic efficiency. |
||
Utilizing optimized reaction conditions, we then examined the generality of our approach in a range of hydroxyazolation of alkenes (Fig. 3). Substituted N-mesyl-1,2,3-triazoles with a diverse array of functional groups, such as methyl, ethyl, methoxyl, halo and ester group at the para-position of the aromatic ring, proceeded smoothly to give the corresponding products 5−13 in good yields (76%–91%). The meta- and ortho-substituted aromatic rings of triazoles also worked well to provide the desired products 14−17 in moderate to high yields (69%–91%). Importantly, triazoles with multi-substituted phenyl ring or bulky group were proven to be viable substrates, resulting in the corresponding products 18−21 in yields of up to 79%. In general, both the electron-donating and electron-withdrawing groups on the benzene ring of N-mesyl-1,2,3-triazoles were well tolerated to this reaction. No N1- or N3-selective product was detected. The structure of 15 was confirmed by X-ray crystallographic analysis as 2,1-azolization products with N2-selectivity (insert in Fig. 3), and the structure of other products were tentatively assigned by analogy. The high N2-selectivity may own to that the relatively more stable N2–1,2,3-triazole radicals (compared to N1–1,2,3-triazole radicals) [39] serve as the key intermediates. It is worth noting that we also successfully achieved the hydroxyazolation of pyrazole, indazole and tetrazole to afford the corresponding products 22−26 in good yields, implying that the present methodology is also applicable to other N-azoles. These results also suggested that the methylsulfonyl may not be an essential functional group in the substrate and NH-1,2,3-trizole may be the key intermediate.
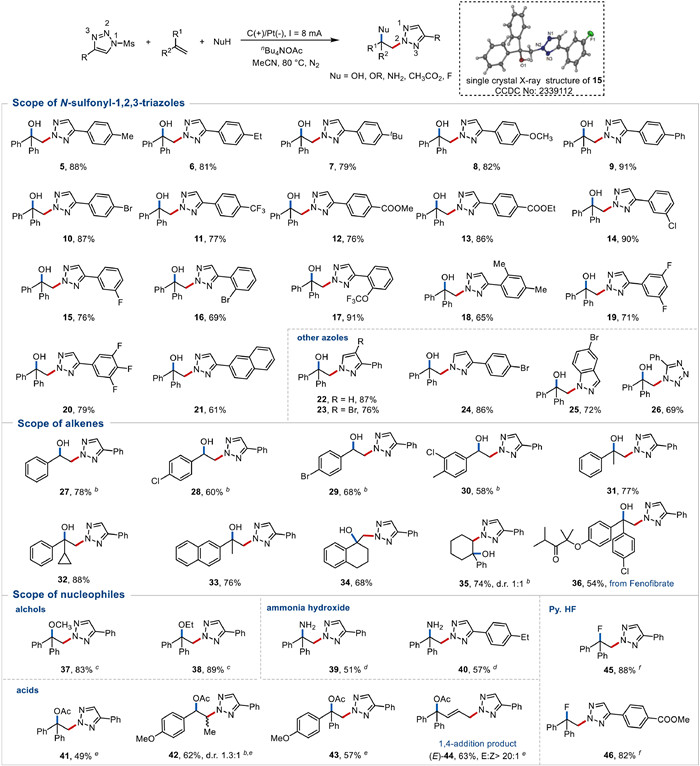
To further investigate the scope of this transformation, we then explored the variation of substituents on the alkenes. The use of monosubstituted styrenylalkenes led to the formation of valuable hydroxyazolation products (27−30). Alkyl-substituted alkenes were also identified as suitable substrates, undergoing the electrochemical alkene difunctionalization reaction to afford products 31−33 in moderate to good yields with high regioselectivity. Furthermore, cycloalkenes also shown as suitable substrates to give corresponding adducts 34–35 in acceptable yields. No diastereoselectivity was observed for 35. Pleasingly, segment of drug molecule (such as fenofibrate) was amenable to this methodology (36), which further highlights the potential value of this protocol. However, the yields of products were significantly suppressed when employing some strongly electron-rich alkenes, such as 2-vinylpyridine, 2-vinylthiophene and 4-methoxystyrene (Scheme S1 in Supporting information). It is possible attributed to that their low oxidation potential interferes with the generation of N-centered radical.
Having successfully revealed this electrochemical oxidative hydroxyazolation of alkenes, we thereafter examined this approach to other readily available nucleophiles. The results indicate an impressive tolerance toward alcohols in MeCN under N2, giving formation of 37 and 38 in 83% and 89% yields, respectively. Of note, this transformation proceeded smoothly with ammonia, which is relatively unexplored in electrochemical difunctionalization reactions, yielding diamination compounds 39 and 40 in 51% and 57% yield, respectively. Due to the large amount of H2O present in ammonia, we also obtained the competitive product using H2O as nucleophilic reagent (30%). Although the redox potential of ammonia (2.06 V) closed to that of 1H-1,2,3-triazole (2.18 V, Fig. S1 in Supporting information), it was triazole but not ammonia to serve as the radical precursor and add to alkenes to yield target products 39 and 40 (2,1-addition products) and no any 1,2-addition products were observed. It may be attributed to that amine radical is highly active and readily quenched in the reaction environment (grabbing H from water to yield NH3 again), giving a much shorter life time than triazole radical. Interestingly, acetic acid even worked equally well in the triazolation difunctionalization reaction, in which AcO- serves as nucleophile to efficiently produce compounds 41−44 in 49%−63% yields. Product 42 gave a poor diastereoselectivity, but 1,4-addition product 44 showed an excellent E/Z selectivity (> 20:1). Further explorations demonstrate that the electrochemical alkene difunctionalization reaction was compatible with pyridine hydrofluoride (Py. HF) as heteroatom nucleophile, performing products 45 and 46 (82% and 88% respectively). The broad substrate scope enables the efficient fluoroamination, diamination and oxoamination of alkenes.
To demonstrate the operational simplicity and scalability of the current electrosynthesis, the process was applied on a 2.0 mmol scale using water and ammonia as nucleophiles under standard conditions. Gratifyingly, the expected product 4 (Fig. 4A) and 39 (Fig. 4B) were isolated in 55% and 39% yield (4 was obtained as by-product in 45% yield), respectively. The hydroxyl group of 4 can be subsequently eliminated to release alkenylated product 47 or substituted by nitrile group to give compound 48 in a nearly quantitative yield. Representative transformations of amine 39 were also performed to afford amides 49 and 50 in good yields. The structure of compound 49 was investigated by the HMBC spectrum (Fig. S2 in Supporting information), further demonstrating the chemo-selectivity of products 39–40. Azole compounds are frequently used as antifungal drugs, such as imazalil, and hydroxyazolation structures are important precursors for their synthesis [46, 47]. These reported approaches to the key intermediates, hydroxyazolation compounds, require multi-step synthesis with a relatively long reaction times (at least 16 h) and give relatively low overall yields (a less total yield of 48%). Here, we presented a green one-pot method to synthesize the key intermediate 51–52 in a yield of up to 68% within 4 h and in a high atom-economical and operationally simplification fashion (Fig. 4C).
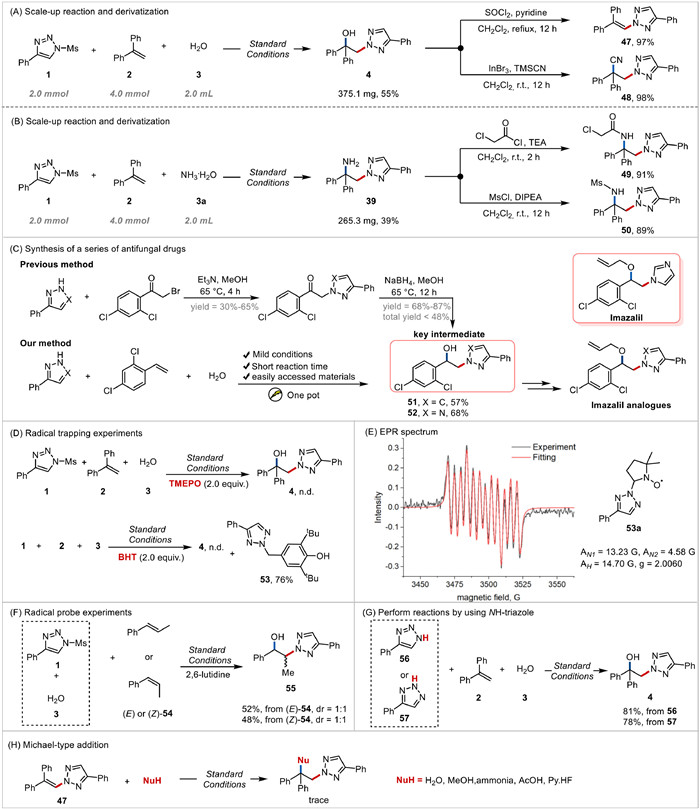
To verify the possible formation mechanism of the desired product, we firstly performed the radical-trapping experiments under standard reaction conditions, with the use of 2,4-di-tert-butyl-4-methylphenol (BHT) and 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO) as the radical scavengers (Fig. 4D). The reaction was significantly quenched and no desired product was observed. The BHT-trapping adduct 53 of triazole radical was obtained in 76% yield. Electron paramagnetic resonance (EPR) experiments were also performed to validate the generation the triazole radicals. As shown in Fig. 4E, the typical signals of a N-centered radical with coupling constants of AN1 = 13.23 G, AN2 = 4.58 G and AH = 14.70 G (g = 2.0060) were observed when 5,5 dimethyl-1-pyrroline N-oxide (DMPO) and N-mesyl-triazole 1 were electrolyzed for 10 min in CH3CN. These observations imply that N-centered triazole radicals are the key intermediates and involved in this electrochemical process. It is different from our previously observation [38] that triazole radical cation is the key intermediate when using 1 to couple with tetrahydropyran under an electrochemical oxidative. We then subjected (Z)- and (E)-β-methylstyrene 54 to the standard reaction conditions (Fig. 4F), respectively. It was observed that both (Z)- and (E)-54 gave the corresponding product 55 in similar yields with the same dr value (1:1), which indicates that the key carbocation intermediate was efficiently generated and trapped by water to give the product. Then, the reaction was tested using 1H-1,2,3-triazole 56 or 2H-1,2,3-triazole 57 instead of N-mesyl-triazole 1 (Fig. 4G). Both 56 and 57 delivered the desired N2-selective product 4 in a good yield (81% or 78% yield), respectively. These outcomes strongly suggest that NH-1,2,3-triazole generated from the hydrolysis of N-mesyl-triazole 1 is the key intermediate in the reaction, and 2H-1,2,3-triazole but not 1H-1,2,3-triazole is the major form under the standard conditions which may be responsible for the high N2-regioselectivity. Subsequently, a series of experiments were performed using 2-(2,2-diphenylvinyl)-4-phenyl-2H-1,2,3-triazole 47 in combination with the nucleophiles, respectively (Fig. 4H). Notably, no products were obtained in all cases in the presence of any nucleophiles, which exclude that the nucleophile lost an electron to generate a radical under standard conditions to undergo radical addition to yield the product.
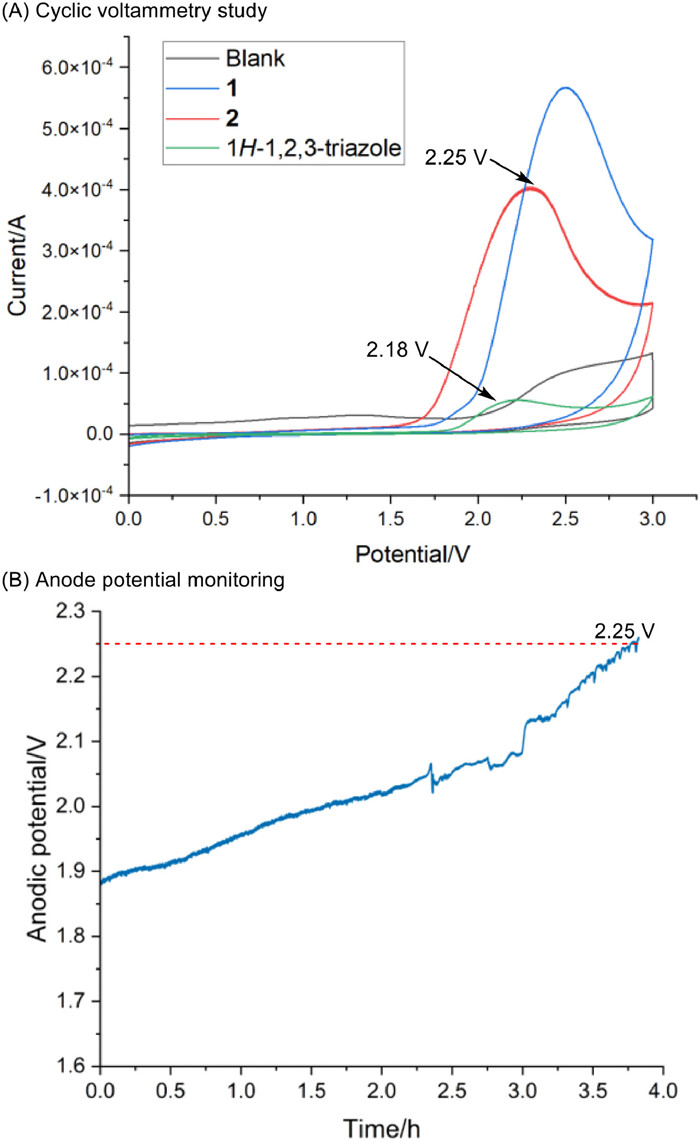
To understand the details of the oxidation process, cyclic voltammetry studies and monitoring of the anode potential were carried out. As shown in Figs. 5A and B, the onset oxidation potentials of N-mesyl-1,2, 3-triazole 1, alkene 2, and 1H-1,2, 3-triazole 49 were determined to be approximately +2.41, +2.25 and +2.18 V (versus SCE), respectively, and the anode potential showed a matched value (about 2.25 V). These results demonstrate that 1H-1,2, 3-triazole was preferentially oxidized under standard conditions. Due to the much higher oxidation potentials of nucleophiles than 1 (Fig. S1 in Supporting information), we can exclude the pathway that a radical generated from nucleophile undergoes an addition to alkene to generate a relayed radical followed by a radical addition to N2 atom in 1 with an aromatic desulfonation process as we previously observed (Fig. S3 in Supporting information) [41, 42]. It is worth to note that, Xu's group reported a highly efficient hydroxy-alkynylation and -alkenylation reactions of arylalkenes via alkene radical cation under electrooxidation [44]. A radical coupling of alkene radical cation and N-centered triazole radical which is followed by a nucleophilic addition may not occur in this reaction due to the higher oxidation potentials of 2 than NH-triazole (Figs. 5A and 6A). The influence of the potential on the reaction was further investigated (Table S2 in Supporting information). When the reaction was performed at a voltage of 2.0–2.2 V, only trace amounts of the desired product was observed after 4–5 h and the current was rapidly decreased to lower than 5 mA within several minutes. By increasing the potential to 2.5 V, the desired product 4 was achieved in 15% yield, and the current was decreased from 10 mA to 5 mA within half an hour. The reason why the current was decreased is not clear, but it obviously slowed the reaction rate and lengthened the reaction time. Further enhancing the potential to 2.6 V, 4 was not detected and unknown compound adherenced on Pt electrode surface were observed. These results show that the reaction is very sensitive to the electrochemical conditions.
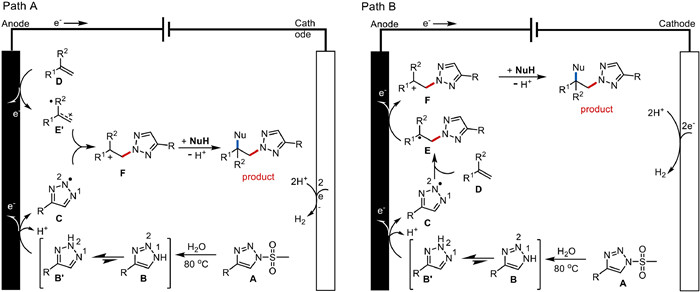
According to the above results, a plausible mechanism for the electrochemical 2,1-difunctional azolization reaction is illustrated in Fig. 6B. First, N-mesyl-triazole A generates 1H-1,2,3-triazole B or 2H-1,2,3-triazole B' by hydrolysis, and the major intermediate B' undergoes deprotonation and anodic oxidation to generate the N-centered triazole radical C [34, 35]. Then, a radical addition of the triazole radical C to alkene D, producing a relayed C-centered radical intermediate E. Further anodic oxidized oxidation of E results in the formation of a carbocation intermediate F. Finally, carbocation F reacts with the nucleophile (NuH) to afford the final desired product after deprotonation. Concomitant cathodic reduction of protons leads to hydrogen evolution. The major intermediate B' is responsible for the high regioselectivity via a steric hindrance effect to provide the desired product.
In conclusion, we have disclosed a green and efficient electrochemical oxidative 2,1-azolization of alkenes with various azoles. The unprecedented reaction is promoted by an aromatic azole radical addition to alkenes followed by further oxidization and nucleophilic addition in high yields and with high regioselectivity. The reaction demonstrates good substrate generality, tolerating various types of nucleophilic sources to one-pot construct C–N and C–X (X = F/N/O) bonds to furnish fluoroamination, diamination and oxoamination of alkenes under electrochemical oxidation conditions. This synthetic strategy can be performed on a gram scale, presenting a highly valuable prospect for the synthesis of azole-substituted compounds by a green electrochemical approach.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yaqi Deng: Writing – review & editing, Writing – original draft, Investigation, Data curation, Conceptualization. Jian Xue: Data curation. Xiang Wu: Conceptualization. Shunying Liu: Writing – review & editing, Writing – original draft, Conceptualization.
We thank the National Science Foundation of China (No. 22071058) and the Fundamental Research Funds for the Central Universities for financial support. The product is available free of charge from the Cambridge Crystallographic Data Centre under CCDC No. 2339112.
Supplementary material associated with this article can be found, in the online version, at doi:
N.T. Chandrika, S.K. Shrestha, H.X. Ngo, et al., J. Med. Chem. 61 (2018) 158–173. doi: 10.1021/acs.jmedchem.7b01138
E. Vitaku, D.T. Smith, J.T. Njardarson., J. Med. Chem. 57 (2014) 10257–10274. doi: 10.1021/jm501100b
R. Elias, P. Basu. J. Med. Chem. 65 (2022) 2361–2373. doi: 10.1021/acs.jmedchem.1c01807
P.H. Zhu, T. Zhou, H. Chen, et al., J. Med. Chem. 66 (2023) 7497–7515. doi: 10.1021/acs.jmedchem.3c00266
R. Hili, A.K. Yudin., Nat. Chem. Biol. 2 (2006) 284–287. doi: 10.1038/nchembio0606-284
P. Björn, W. Viktor, F. Kari, et al., J. Am. Med. Assoc. 319 (2018) 2333–2335. doi: 10.1001/jama.2018.6237
K. James, K.S. Hicks, E.Q. Rod, et al., J. Clin. Oncol. 36 (2018) 6594-6594. doi: 10.1200/JCO.2018.36.15_suppl.6594
H. Arash, S. Rupam, S. Qusai, et al., Clin. Infect. Dis. 76 (2023) 2196–2199. doi: 10.1093/cid/ciad146
J. Bariwal, E.V. Eycken., Chem. Soc. Rev. 42 (2013) 9283–9303. doi: 10.1039/c3cs60228a
G. Evano, N. Blanchard, M. Toumi., Chem. Rev. 108 (2008) 3054–3131. doi: 10.1021/cr8002505
H. Jiang, A. Studer., Chem. Soc. Rev. 49 (2020) 1790–1811. doi: 10.1039/c9cs00692c
N.Gockel S, L.Buchanan T, K.L. Hull, J. Am. Chem. Soc. 140 (2018) 58–61. doi: 10.1021/jacs.7b10529
Y. Zhang, H.D. Liu, L.N. Tang, et al., J. Am. Chem. Soc. 140, (2018) 10695–10699. doi: 10.1021/jacs.8b07023
Z.L. Li, G.C. Fang, Q.S. Gu, et al., Chem. Soc. Rev. 49 (2020) 32–48. doi: 10.1039/c9cs00681h
C.Y. Cai, X.M. Shu, H.C. Xu., Nat. Commun. 10 (2019) 4953–4959. doi: 10.1038/s41467-019-13024-5
J. Li, W.H. Huang, J.Z. Chen, et al., Angew. Chem. Int. Ed. 57 (2018) 5695–5698. doi: 10.1002/anie.201801106
F.Yang D, Z.P. Guan, Y.N. Peng, et al., Nat. Commun. 14 (2023) 1476–1482. doi: 10.1038/s41467-023-37032-8
N. Chen, H.C. Xu., Green Synth. Catal. 2 (2021) 165–178.
R. Francke, R.D. Little., Chem. Soc. Rev. 43 (2014) 2492–2521. doi: 10.1039/c3cs60464k
F. Nian, G.S. Sauer, A. Saha, et al., Science 357 (2017) 575–579. doi: 10.1126/science.aan6206
Z.P. Guan, D.F. Yang, Z. Liu, et al., Chin. J. Catal. 52 (2023) 144–153. doi: 10.1016/S1872-2067(23)64510-3
J. Huang, Y.Y. Liang, X.H. Yang, et al., Org. Chem. Front. 8 (2021) 7009–7014. doi: 10.1039/d1qo01263k
X.L. Wang, H.J. Li, M. Zhu, et al., RSC Adv. 7 (2017) 15709–15714. doi: 10.1039/C6RA27202A
K. Sun, B.X. Luan, Z.H. Liu, et al., Org. Biomol. Chem. 17 (2019) 4208–4211. doi: 10.1039/c9ob00317g
Y. Yuan, Y.X. Chen, S. Tang, et al., Sci. Adv. 4 (2018) eaat5312. doi: 10.1126/sciadv.aat5312
S.Q. Guo, H.Q. Yang, A.L. Wang, et al., Green Chem. 23 (2021) 9571–9576. doi: 10.1039/d1gc03048e
T. Xiong, Q. Zhang., Chem. Soc. Rev. 45 (2016) 3069–3087. doi: 10.1039/C5CS00852B
A.Y. Rulev., Russ. Chem. Rev. 80 (2011) 197–218. doi: 10.1070/RC2011v080n03ABEH004162
L. Song, N. Fu, B.G. Ernst, et al., Nat. Chem. 12 (2020) 747–754. doi: 10.1038/s41557-020-0469-5
Y.Y. Jiang, K. Xu, C.C. Zeng, Chem. Rev. 118 (2018) 4485-454.
C.Y. Cai, Y.T. Zheng, J, F. Li, et al., J. Am. Chem. Soc. 144 (2022) 11980–11985;. doi: 10.1021/jacs.2c05126
M. Yan, Y. Kawamata, P.S. Baran., Chem. Rev. 117 (2017) 13230–13319. doi: 10.1021/acs.chemrev.7b00397
M. Liu, T. Feng, Y.W. Wang, et al., Nat Commun. 14 (2023) 6467–6476. doi: 10.1038/s41467-023-42106-8
C.C. Sun, K. Xu, C.C. Zeng, A.C.S. Sustain. Chem. Eng. 7 (2019) 2255–2261. doi: 10.1021/acssuschemeng.8b04934
Z.H. Wan, D. Wang, Z.X. Yang, et al., Green Chem. 22 (2020) 3742–3748. doi: 10.1039/d0gc00687d
S.Y. Liu, W.F. Yao, Y. Liu, et al., Sci. Adv. 3 (2017) e1602467. doi: 10.1126/sciadv.1602467
Z. Li, Q.H. Wei, L.L. Song, et al., Org. Lett. 21 (2019) 6413–6417. doi: 10.1021/acs.orglett.9b02269
C. Guan, J.B. Yin, J. Ji, et al., Org. Lett. 25 (2023) 5383–5388. doi: 10.1021/acs.orglett.3c01896
Y.Q. Deng, Z.J. Hu, J. Xue, et al., Org. Lett. 26 (2024) 933–938. doi: 10.1021/acs.orglett.3c04291
J. Ji, J.H. Liu, C. Guan, et al., Chin. J. Org. Chem. 43 (2023) 1168–1176. doi: 10.6023/cjoc202209021
J. Ji, C. Guan, Q.H. Wei, et al., Org. Lett. 24 (2022) 1–5. doi: 10.1021/acs.orglett.1c02935
J. Ji, X.W. Chen, Z.J. Hu, et al., Org. Chem. Front. 11 (2024) 3066–3071;. doi: 10.1039/d4qo00246f
J. Ji, Z.J. Hu, C. Guan, et al., ACS Sustain. Chem. Eng. 11 (2023) 16240–16248. doi: 10.1021/acssuschemeng.3c04487
P. Xiong, Hao. Long, J.S. Song, et al., J. Am. Chem. Soc. 140 (2018)16387–16391. doi: 10.1021/jacs.8b08592
Y. Wang, L.L. Deng, H.B. Mei, et al., Green Chem. 20 (2018) 3444–3450. doi: 10.1039/C8GC01337C
Z.Z. Yan, Y.X. Huang, D.Z. Zhao, et al., J. Med. Chem. 66 (2023) 13247–13265. doi: 10.1021/acs.jmedchem.3c01254
H. Xu, Y.H. Mou, M.B. Guo, et al., Eur. J. Med. Chem. 243 (2022) 114707. doi: 10.1016/j.ejmech.2022.114707

Figure 1 Background and reaction design. (A) Representative examples highlighting diverse bioactive hydroxyazolation compounds. (B) Previous work toward 1,2-azolization of alkenes. (C) This reaction design.

Figure 3 Scope of azoles, alkenes and nucleophiles. a Reaction conditions from Table 1, entry 1, alkene (1.0 mmol), triazole (0.5 mmol), H2O (0.5 mL), nBuN4OAc (1.0 mmol), MeCN (9.5 mL), carbon rod as anode, platinum as cathode, undivided cell, constant current = 8 mA, 80 ℃, N2, 4–6 h. Isolated yields were reported. b 2,6-Lutidine (1.0 mmol) was added. c Used alcohol (0.5 mL) instead of H2O. d Used ammonium hydroxide (1.0 mL) instead of H2O, 4 Å MS. e Used HOAc (0.5 mL) instead of H2O. f Used pyridine hydrofluoride (Py. HF) (0.5 mL) instead of H2O. See the Supporting information for detailed reaction condition.

Figure 4 Scale-up reaction and derivatization and mechanistic studies. (A, B) Scale-up reaction and derivatization. (C) Synthesis of a series of antifungal drug-like molecules. (D) Radical trapping experiments. (E) EPR spectrum. (F) Radical probe experiments. (G) Perform reactions by using NH-triazole. (H) Michael-type addition.
Table 1. Optimization of the reaction conditions.a
|
||
| Entry | Deviation from standard conditions | Yield b (%) |
| 1 | 5 mA | 47 c |
| 2 | None | 89 (3.2 F/mol, 62.5%) d |
| 3 | 10 mA | 75 |
| 4 | Reaction at 50 ℃ | 61 |
| 5 | Pt plate (1 cm × 1 cm) as anode | NR |
| 6 | No electricity | NR |
| 7 | nBuN4BF4 instead of nBu4NOAc | 38 |
| 8 | In air | 74 |
| 9 | Acetone as the solvent | 36 |
| a Reaction conditions: undivided cell, graphite rod anode, Pt cathode, 1 (0.5 mmol), 2 (1.0 mmol), MeCN (9.5 mL), H2O (0.5 mL), N2, electrolytes (1.0 mmol), 8 mA, 4–6 h (based on 1). b Yields of isolated products are provided. c Isolated yield. NR = no reaction. d Charge consumption and Faradaic efficiency. |
||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们



 下载:
下载: