State Key Laboratory and Institute of Elemento-Organic Chemistry, Frontiers Science Center for New Organic Matter, Haihe Laboratory of Sustainable Chemical Transformations, College of Chemistry, Nankai University, Tianjin 300071, China
b.
Department of Chemistry, Fudan University, Shanghai 200438, China
Received Date:
22 November 2024 Accepted Date:
28 December 2024 Revised Date:
26 December 2024 Available Online:
15 September 2025
Abstract:
Herein, a metal-free electrochemical demethoxyl-cyanation of methoxyarenes via aromatic nucleophilic substitution (SNAr) using TMSCN as a cheap cyanide source under mild conditions has been presented. This transformation utilizes commercially available reagents, cheap electrodes, and simple equipment. Diverse aryl nitriles were successfully obtained in a direct and efficient way with broad substrate scope, excellent functional group tolerance, and selective C−O bond cleavage. Furthermore, late-stage modification of biorelevant compounds and gram-scale synthesis highlighted the potential application of the strategy. Mechanistic investigations suggest that the arene cation radical was considered as the key intermediate for the transformation, and undergoing the followed SNAr process.
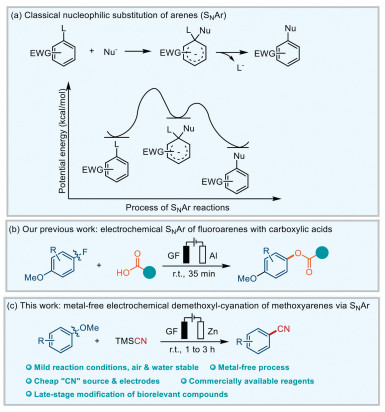
Aromatic nucleophilic substitution (SNAr) is a type of important and basic organic reaction due to its wide range of applications, and its mechanisms have been extensively studied [1-4]. The typical mechanism consists of two steps: firstly, attacking of the nucleophilic species at the ipso carbon of the aromatic ring, forming a Meisenheimer complex or σ-complex, then elimination of the leaving group and regeneration of the aromatic ring (Fig. 1a) [4]. Nowadays, SNAr has become an efficient and convenient approach to converse simple starting materials into valuable fine chemicals. As electron-withdrawing groups reduce the electron cloud density of the aryl rings, which made it conducive for nucleophiles to attack, hence electron-deficient arenes are easier than electron-rich ones to proceed SNAr [5, 6].
Figure 1
Figure 1.
(a) Nucleophilic substitution of arenes (SNAr reaction). (b) Our previous work on electrochemical SNAr of fluoroarenes with carboxylic acids. (c) Metal-free electrochemical demethoxyl-cyanation of methoxyarenes via SNAr.
As traditional SNAr reactions are usually not applicable to electron-rich aromatic compounds, the SNAr reactions for electron-rich aryl derivatives present great challenges [6, 7]. In the past decades, the approaches to activate electron-rich arenes and its derivatives have become a hot research topic, significant advances in the study of electron-rich SNAr reactions have been reported [8-17]. Among them, main SNAr reactions were proceeded using photoredox catalysis and/or transition metal catalysis, such as fluorination [18-20], cyanation [21-24], amination [25, 26], hydroxylation [27-29]. However, synthetic strategies via photocatalysis or transition-metal catalysis usually require the use of special and expensive metal complexes or photocatalysts [22, 30-32]. Inspired by these intriguing works about the approaches of SNAr with electron-rich aromatic compounds, we wondered whether there is a more convenient, efficient, and sustainable strategy.
Here we put forward the way to use electricity as a sustainable and clear energy source [33-41]. Recently, electrochemistry has become a powerful, efficient, and environmental-friendly tool to achieve various types of intractable reactions [42-62]. In our previous work, we reported a new electrochemically driven catalyst-free SNAr reaction of electron-rich methoxyfluoroarenes with carboxylic acids (Fig. 1b) [51]. Based on this work, we would like to continue our work to focus on the substitution of the other functional groups, especially for the challenging methoxy group. We envisioned that arene cation radical intermediate could be the key to solve this issue, which could be formed with electrochemistry.
With our continuous interest in sustainable electrochemistry [51-62], herein, we report an efficient and metal-free electrochemical SNAr demethoxyl-cyanation of methoxyarenes at room temperature (Fig. 1c). The notable features of this protocol are (a) cheap and low toxic cyanide source; (b) metal-free process, mild reaction condition, water and air stable; (c) cheap abundant electrodes and commercially available reagents; (d) late-stage modification of biorelevant compounds. It is noteworthy that this efficient strategy could be applied to directly modify natural products, which further demonstrates the potential practicality and biocompatibility of this reaction.
In our initial studies, 1, 2, 3-trimethoxybenzene (1a) was selected as the model substrate, while TMSCN (1b) was chosen as the CN source to optimize the standard conditions. After systematic investigations, 2, 6-dimethoxybenzonitrile (1) was obtained in an 86% isolated yield under a constant current (20 mA) in a mixed solvent of MeCN/H2O (11:1, v/v) for 3 h. The reaction employed TBAOAc as the supporting electrolyte, K2CO3 as the base, graphite felt (GF) as the anode and Zn plate as the cathode, in an undivided cell (Table 1, entry 1). First, attempts to increase or decrease the current resulted in lower yields (entry 2). Then we tested different cathode electrode materials (entries 3-5), including GF(-), Al(-) and Mg(-), which all gave lower yields (43%-75%) compared to Zn(-). We also examined different bases such as CH3COONa, Cs2CO3, and KOH, but none of them provided a higher yield than that of K2CO3 (entries 6-8). Next, we screened different kinds of solvents, including acetone/H2O (11:1), CH3OH and dry CH3CN (entries 9-11). The results showed that these solvents could decrease the yield of the product, while CH3CN/H2O (11:1, v/v) was found to be the optimal solvent for this conversion. Meanwhile, we investigated various electrolytes (entries 12-14), and the results indicated that TBAClO4 or TBABF4 could obtain the product in somewhat lower yields, while the reaction was suppressed in the presence of NH4PF6. Furthermore, we tried to use 4-cyanopyridine as the CN source instead of TMSCN, but only obtained a yield of 46% for this conversion. Finally, control experiments were conducted in the absence of the base (K2CO3), TBAOAc, or electricity, which gave the yields of 17%, 61%, and no product, respectively (entries 16-18). These results indicating that the base and supporting electrolyte played important roles in this reaction, while the electricity is essential.
a Reaction conditions: undivided cell, graphite felt anode (12 mm ×18 mm × 3 mm), Zn plate cathode (12 mm × 18 mm × 0.20 mm), constant current = 20 mA, 1a (0.3 mmol), 1b (0.6 mmol), K2CO3 (1.0 equiv.), TBAOAc (0.5 equiv.) in CH3CN/H2O (11:1, 6.0 mL), at room temperature for 3 h. b Yields were determined by NMR spectroscopy using 1, 1, 2, 2-tetrachloroethane as the internal standard. c Isolated yield.
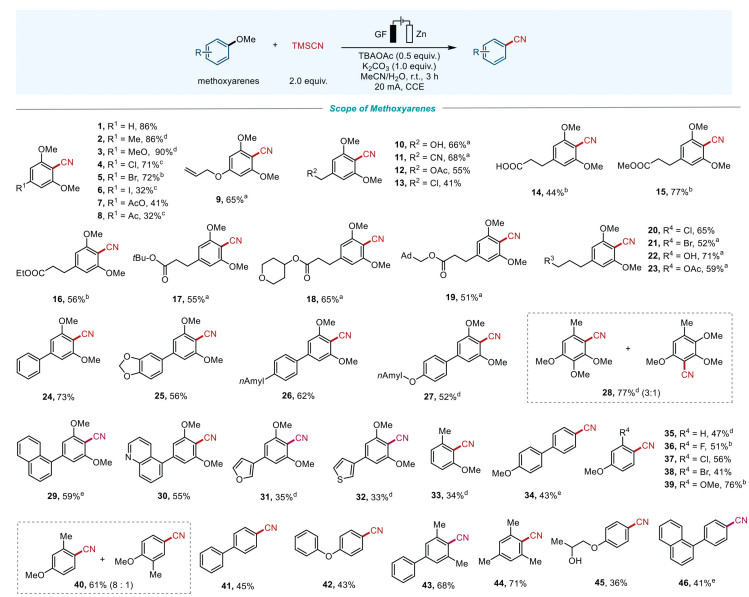
With the optimal reaction conditions in hand, we next investigated the scope and compatibility of various methoxyarenes. Initially, we examined the generality of this strategy with respect to different 5-position functionalized 1, 2, 3-trimethoxy arenes. As shown in Fig. 2, methoxyarenes with electron-donating groups at the 5-position could be converted into 2, 6-dimethoxy aryl nitriles with remarkably high isolated yields (2-3, 9, 65%-90%). When the group at the 5-position was Cl or Br, the isolated yields were also favorable (4-5, 71%-72%), whereas iodine decreased the yield to a low level (6, 32%). In contrast, when 5-position functionalized with electron-withdrawing groups, the isolated yields of relative aryl nitriles would be fairly low (8, 32%) or even no product, such as -COOH, -CN. These results showed that the electron-donating or electron-withdrawing groups exhibited significant influence on the efficiency for this conversion. On the other hand, when electron-withdrawing group was attached to a methylene or a carbon chain instead of directly connected to the aryl ring, the yields of relative products increased significantly. For example, when the 5-position functionalized with a cyanomethyl group, the yield of the corresponding product would be nice (11, 68%). Similarly, when the electron-withdrawing group COOH, -COOR (R = Me, Et, tBu, 4-pyranyl, or adamantane methyl) connected to dicarboalkyl chain, the yields of the corresponding products would be moderate to good (14-19, 44%-77%). For substrates with longer carbon chains, the yields of relative products were also well (20-23, 52%-71%). Moreover, when 5-position was substituted with another aryl ring, the yields of the resulting aryl nitriles would also be favorable (24-27, 52%-73%). Additionally, this synthetic method was also suitable for heterocyclic and polycyclic aromatic derivatives, affording moderate yields of corresponding products (29-32, 33%-59%).
Figure 2
Figure 2.
Substrate scope. Reaction conditions: methoxyarenes (0.3 mmol), TMSCN (0.6 mmol), TBAOAc (0.15 mmol), K2CO3 (0.3 mmol), under 20 mA constant current in an undivided cell at room temperature for 3 h with GF as anode and Zn as cathode, MeCN: H2O = 11:1 (v/v), 6 mL. a The reaction was conducted for 1 h. b With 10 mol% Tempo, K2CO3 (3.0 equiv.), 40 ℃, Pt plate as cathode. c With 10 mol% Tempo, K2CO3 (3.0 equiv.), Pt plate as cathode. d 12 mA constant current. e K2CO3 (3.0 equiv.).
Next, we expanded the substrates that functionalized with fewer methoxy groups. We found that the yields of corresponding products would be somewhat less than that of trimethoxy arenes. For instance, when 2, 3-dimethoxytoluene was used as the reactant, the yield was quite low (33, 34%). Upon adjusting the positions of the substituted groups, we found that the substrates with 1, 4-(para)-substituents on the aromatic ring could produce better yields compared to the substrates with 1, 2-(ortho)-substituents. For instance, 4, 4′-dimethoxybiphenyl could be converted into the corresponding product (34) with a moderate yield (43%). Notably, hydrogen atoms on 1, 4-dimethoxybenzenes could also be substituted with other functional groups, and the yields of corresponding products would not be obviously influenced. For instance, when the 2-position on the aryl ring substituted by halogen atoms (F, Cl, or Br), methoxy or methyl groups, the corresponding products could be obtained in moderate to considerable yields (35-40, 41%-76%). We next explored mono-methoxy-substituted aromatic compounds. The results revealed that the 4-methoxybiphenyl could be converted to 4-cyanobiphenyl (41) with a yield of 45%, which was lower than that of 2, 6-dimethoxy-1-cyanobiphenyl (24, 73%) and 2, 6-dimethyl-1-cyanobiphenyl (43, 68%). Notably, 2, 4, 6-trimethylanisole could be efficiently converted to 2, 4, 6-trimethylbenzonitrile with a remarkable yield (44, 71%). Additionally, 1-(4-methoxyphenoxy)-2-propanol could be transformed into the corresponding product (45) with a yield of 36%, while 1-(4-methoxyphenyl)naphthalene could be obtained with a yield of 41% of the product (46). Overall, all these transformations demonstrated excellent functional group tolerance.
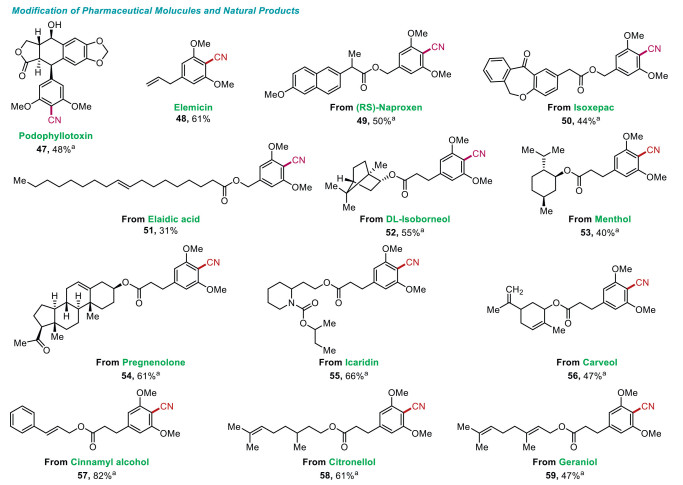
As one of the most important applications, aryl nitriles are most commonly used in pharmaceuticals [63, 64], agrochemicals [65], etc. Late-stage modification has become one of the most widely explored strategies to diversify natural products and drug molecules, which involves installing different functional groups on target molecules to enhance their biological activity [66-69]. As shown in Fig. 3, this electrochemical protocol was further expanded to the late-stage modification of various pharmaceutical molecules and natural products, for instance, Podophyllotoxin, a well-known natural product first found in Podophyllum peltatum L [70]. In 1942, scientists found that venereal warts could be effectively treated by this compound [71]. Due to its significant applications in antitumor and antiviral activities, we sought to modify this compound, and achieved the modified product with a moderate yield (47, 48%). In addition, 1-allyl-3, 4, 5-trimethoxybenzene, also named as Elemicin, is a natural product found in A. dracunculus [72]. It exhibits biological activity against S. aureus, B. subtilis, and C. albicans [73]. Here we chose this natural product to undergo late-stage modification, and successfully gained the product with a good yield (48, 61%).
Figure 3
Figure 3.
Late-stage modification. Reaction conditions: methoxyarenes (0.3 mmol), TMSCN (0.6 mmol), TBAOAc (0.15 mmol), K2CO3 (0.3 mmol), under 20 mA constant current in an undivided cell at room temperature for 3 h with GF as anode and Zn as cathode, MeCN: H2O = 11:1 (v/v), 6 mL. a The reaction was conducted for 1 h.
For carboxylic acidic drug molecular derivatives, such as (RS)-Naproxen (49) and Isoxepac derivative (50) [74], both of which were amenable in this electrochemical system and provided the desired products in moderate yields (44%-50%). In contrast, for modified unsaturated fatty acid derivative, such as Elaidic acid, the desired product was obtained in a low yield (51, 31%). Many kinds of cyclic alcoholic compounds are biologically active products, such as d, l-isoborneol andmenthol [75]. We also proceeded their derivatives under electrochemical condition, and successfully transformed into corresponding products with moderate isolated yields (52-53, 40%-55%). For pharmaceutical intermediate, Pregnenolone [76], its derivative gave the target modified product in a good yield (54, 61%). Some alcoholic compounds such as Icaridin and Carveol are commonly used as insect repellents [77], their derivatives could also be modified to desired products with moderate to good yields (55-56, 47%-66%). Furthermore, we tested alcohols that frequently used as fragrance ingredients [78], their derivatives were successfully converted into corresponding products with moderate to excellent yields (57-59, 47%-82%). All the above results demonstrated the biocompatible for this transformation.
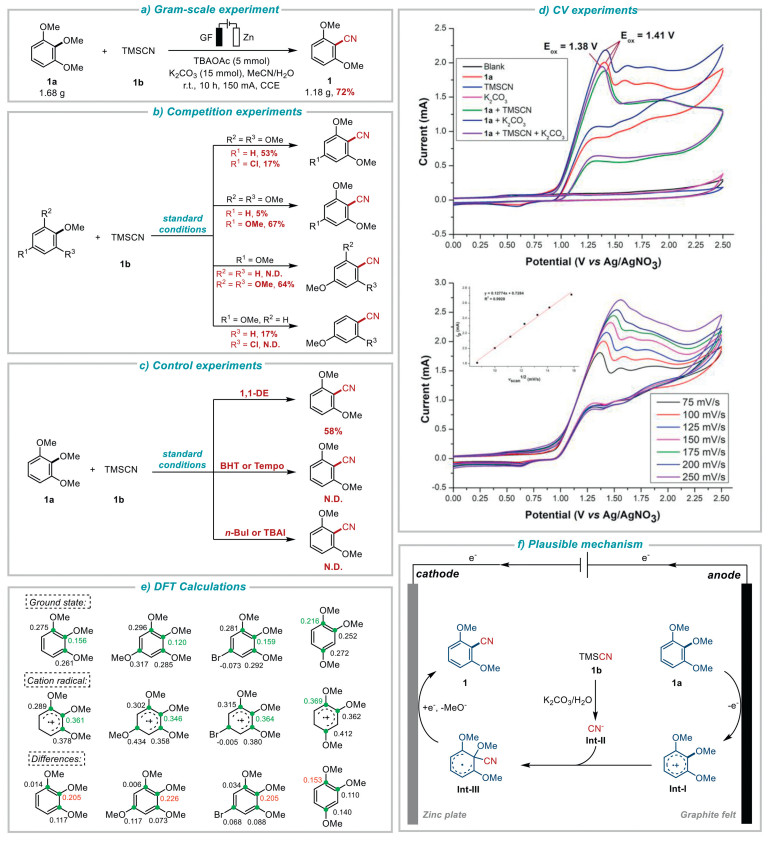
The applicability of this electrochemical cyanation protocol was next demonstrated by gram-scale reaction as shown in Fig. 4a. It can be seen that good yield of the corresponding gram-scale product was obtained (1, 1.18 g, 72%) under a 150 mA constant current (for more details, see Supporting information). This result highlighted the potential broad practicality of our metal-free electrochemical approach. Next, a set of intermolecular competition experiments were conducted to investigate the plausible mechanism (Fig. 4b). First, the competition experiment was undergone between the standard substrate 1, 2, 3-trimethoxybenzene (1a) and the electron-deficient substrate 5-chloro-1, 2, 3-trimethoxybenzene (4a), the yield of 2, 6-dimethoxybenzonitrile (1) was 53%, while 4-chloro-2, 6-dimethoxybenzonitrile (4) could only be obtained in a much lower yield of 17%. A second competition experiment was performed between 1a and the more electron-rich reactant 1, 2, 3, 5-tetramethoxybenzene (3a). This reaction resulted product 1 in only a 5% yield but 3 in a surprisingly high yield (67%). Similarly, when 1, 4-dimethoxybenzene (35a) and the much more electron-rich reactant 3a were conducted, the anisonitrile (35) couldn't be obtained, while 3 could be obtained in 64% yield. When the competition experiment conducted between the substrates that both connected with two methoxy groups, such as 1, 4-dimethoxybenzene (35a) and 2-chloro-1, 4-dimethoxybenzene (37a), the yield of corresponding products 35 is 17%, while 37 could not be detected. All the above results indicated that the presence of electron-withdrawing groups is detrimental to the reaction in this transformation, and the presence of electron-donating groups is highly advantageous for the occurrence of directional nucleophilic substitution reactions under electrochemical conditions, these results also conformed with the electrophilic character of the aromatic radical cation species. Subsequently, control experiments were performed to investigate the plausible mechanism (Fig. 4c). When the radical scavenger 1, 1-DE (1, 1-diphenylethylene) was added under standard condition, the yield of the corresponding product significantly decreased to 58% yield. When excessive TEMPO (2, 2, 6, 6-tetramethylpiperidine-1-oxyl) or BHT (2, 6-di-tert-butyl-4-methylphenol) was added under standard condition, almost no target product 1 was detected, indicating that a radical pathway might be involved in this protocol (for more details, see Supporting information). In addition, when added n-butyl iodide or TBAI to the reaction, no corresponding product was detected. We speculated that the presence of iodine ions may inhibit 1a from being oxidized at the anode.
To further explore the reaction mechanism, we conducted cyclic voltammetry (CV) experiments (Fig. 4d). It can be seen that in the scanning window (0 to +2.5 V), no oxidative potential was observed in the background with a 0.1 mol/L TBAPF6 solution in CH3CN (Fig. 4d, black line). Similarly, when TMSCN or K2CO3 was added, no obvious oxidative peak occurred as well (Fig. 4d, blue line and pink line), indicating that TMSCN or K2CO3 couldn't be easily oxidized. However, the CV of 1, 2, 3-trimethoxybenzene (1a) displayed an obvious oxidation signal at 1.41 V vs. Ag/AgNO3 (Fig. 4d, red line), suggesting that 1a undergone electrochemical oxidation at the anode during the reaction. When TMSCN or K2CO3 was added with 1a respectively (Fig. 4d, green line and dark blue line), the oxidation potentials showed no obvious shifts. However, when 1a, TMSCN and K2CO3 were added together (Fig. 4d, purple line), the oxidative potential downshifted by 0.03 V (from 1.41 V to 1.38 V) suggesting that the co-presence of TMSCN and K2CO3 may promote the oxidation process of 1a. Moreover, we performed CV experiments for 1a with variable scan rates. As shown in Fig. 4d, by increasing scan rates, the oxidation signal would occur in a slightly higher potential, and the current would also be increased. To check out the law between the scan rates and the currents, we performed a linear fit analysis. The results demonstrated a strong linear correlation between vscan1/2 and ip for the oxidation of 1a, with a Pearson's R value of 0.9970 and an R2 value of 0.9928, supported that a diffusion control process might be involved in this reaction.
As supplementary experimental evidence, DFT calculations for selective C–O regiocontrol was conducted (Fig. 4e). The computational data demonstrate that the functionalization to occur at the site which undergoes the largest change in NPA values upon oxidation (for more details, see Supporting information) [79, 80].
Based on all the above mechanistic experiments, herein, we put forward the plausible mechanism of this reaction. As shown in Fig. 4f, firstly, 1a was oxidized at the anode to generate aromatic cation radical Int-Ⅰ. At the same time, the added TMSCN could spontaneously hydrolyze under the condition of water-containing solution and base, producing CN− as Int-Ⅱ. Then the nucleophilic reagent Int-Ⅱ would attack the produced aromatic cation radical species Int-Ⅰ to obtain neutral free aromatic radical intermediate Int-Ⅲ. Subsequently, this aromatic radical species would be reduced at the cathode, and immediately leave its methoxy group to form the final product.
In conclusion, a metal-free electrochemical demethoxyl-cyanation of methoxyarenes via SNAr was reported. This method is efficient, sustainable and easily-equipped, and compatible with diverse electron-rich methoxyarenes. Late-stage modification of biorelevant compounds and gram-scale synthesis demonstrated its potential application for synthetic chemistry. Mechanistic studies suggested that the cyanation of methoxyarenes proceeded through the formation of the aromatic cation radical intermediate. Thus, this electrochemical approach could be potentially applied in organic synthesis.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Financial support from National Key R & D Program of China (No. 2023YFA1507203), National Natural Science Foundation of China (Nos. 22371149, 22188101), the Fundamental Research Funds for the Central Universities (No. 63224098), Frontiers Science Center for New Organic Matter, Nankai University (No. 63181206) and Nankai University are gratefully acknowledged. We thank the Haihe Laboratory of Sustainable Chemical Transformations for financial support.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.110810.
F.A. Carey, R.M. Giuliano, N.T. Allison, et al., Electrophilic and nucleophilic aromatic substitution, in Organic Chemistry ISE, 12th ed., McGraw-Hill Education, New York, 2024, pp. 478-533.
[6]
M. Mąkosza, Chem. Soc. Rev. 39 (2010) 2855-2868. doi: 10.1039/b822559c
K.A. Margrey, J.B. McManus, S. Bonazzi, et al., J. Am. Chem. Soc. 139 (2017) 11288-11299. doi: 10.1021/jacs.7b06715
Figure 1
(a) Nucleophilic substitution of arenes (SNAr reaction). (b) Our previous work on electrochemical SNAr of fluoroarenes with carboxylic acids. (c) Metal-free electrochemical demethoxyl-cyanation of methoxyarenes via SNAr.
Figure 2
Substrate scope. Reaction conditions: methoxyarenes (0.3 mmol), TMSCN (0.6 mmol), TBAOAc (0.15 mmol), K2CO3 (0.3 mmol), under 20 mA constant current in an undivided cell at room temperature for 3 h with GF as anode and Zn as cathode, MeCN: H2O = 11:1 (v/v), 6 mL. a The reaction was conducted for 1 h. b With 10 mol% Tempo, K2CO3 (3.0 equiv.), 40 ℃, Pt plate as cathode. c With 10 mol% Tempo, K2CO3 (3.0 equiv.), Pt plate as cathode. d 12 mA constant current. e K2CO3 (3.0 equiv.).
Figure 3
Late-stage modification. Reaction conditions: methoxyarenes (0.3 mmol), TMSCN (0.6 mmol), TBAOAc (0.15 mmol), K2CO3 (0.3 mmol), under 20 mA constant current in an undivided cell at room temperature for 3 h with GF as anode and Zn as cathode, MeCN: H2O = 11:1 (v/v), 6 mL. a The reaction was conducted for 1 h.
a Reaction conditions: undivided cell, graphite felt anode (12 mm ×18 mm × 3 mm), Zn plate cathode (12 mm × 18 mm × 0.20 mm), constant current = 20 mA, 1a (0.3 mmol), 1b (0.6 mmol), K2CO3 (1.0 equiv.), TBAOAc (0.5 equiv.) in CH3CN/H2O (11:1, 6.0 mL), at room temperature for 3 h. b Yields were determined by NMR spectroscopy using 1, 1, 2, 2-tetrachloroethane as the internal standard. c Isolated yield.

 DownLoad:
DownLoad:


 下载:
下载:





 下载:
下载:
