State Key Laboratory of Chemical Oncogenomics, School of Chemical Biology and Biotechnology, Peking University Shenzhen Graduate School, Shenzhen 518055, China
b.
Pingshan translational medicine center, Shenzhen Bay Laboratory, Shenzhen 518055, China
c.
Institute of Chemical Biology, Shenzhen Bay Laboratory, Shenzhen 518132, China
d.
Department of Pharmacy, the First Affiliated Hospital of Shenzhen University, Shenzhen Second People's Hospital, Shenzhen 518035, China
e.
College of Health Science and Environmental Engineering, Shenzhen Technology University, Shenzhen 518118, China
f.
Office of Core Facilities, Shenzhen Bay Laboratory, Shenzhen 518055, China
g.
Tianfu Jincheng Laboratory, Chengdu 610212, China
h.
Department of Chemistry, Faculty of Science, Hong Kong Baptist University, Hong Kong SAR, China
lizg@pkusz.edu.cn (Z. Li). 1 These authors contribute equally to the work.
Received Date:
05 October 2024 Accepted Date:
19 December 2024 Revised Date:
05 December 2024 Available Online:
15 September 2025
Abstract:
Developing novel building blocks with predictable side-chain orientations and minimal intramolecular interactions is essential for peptide-based self-assembling materials. Traditional structures like α-helices and β-sheets rely on such interactions for stability, limiting control over exposed interacting moieties. Here, we reported a novel, frame-like peptide scaffold that maintains exceptional stability without intramolecular interactions. This structure exposes its backbone and orients side chains for hierarchical self-assembly into micron-scale cubes. By introducing mutations at specific sites, we controlled packing orientations, offering new options for tunable self-assembly. Our scaffold provides a versatile platform for designing advanced peptide materials, with applications in nanotechnology and biomaterials.
Peptide-based self-assembly plays a critical role in modern nanotechnology [1-4]. The importance of polypeptides as molecules for the rational design of self-assembled materials lies in their well-defined backbone conformations [5-7]. Previous studies have focused on self-assembly based on α-helix and β-sheet, each characteristic of its own spatial alignment of backbone peptide bonds that lead to inter-molecular hydrogen bond (HB) interaction networks with special structural order over a long range [8-15]. However, the majority of the peptide bonds in α-helix and β-sheet form intramolecular HBs to maintain the stability of the structure itself [16], limiting the forces and orientation options for self-assembly.
Numerous efforts have been made to design novel conformations that can exist stably while minimizing intramolecular HBs, and increasing the spatial orientation specificity to provide more potential elements for self-assembly control [17-19]. One effective method involves introducing non-coded amino acids [20], followed by cyclization to yield stable conformations. This method has been used to construct various self-assemblies including sphere [21], fiber [22], and two-dimensional (2D) material [23]. Previously, our group proposed a cyclopentapeptide (CP) scaffold with a thioether-containing bridge exhibiting switchable conformation precisely controlled by a single in-tether chiral center [24, 25]. The absolute configuration of the chiral center (the γ position to the peptide C terminus) dictated the peptide conformation, with an (R)-configuration resulting in an α-helix conformation. Properly substituted R-configured CPs (R-CPs) can form assemblies with various morphologies [24, 26]. In contrast to R-CPs, the S-configured CPs (S-CPs) (Fig. 1a) exhibit a unique circular dichroism (CD) spectrum that is distinct from either the α-helix or β-sheet conformations.
Figure 1
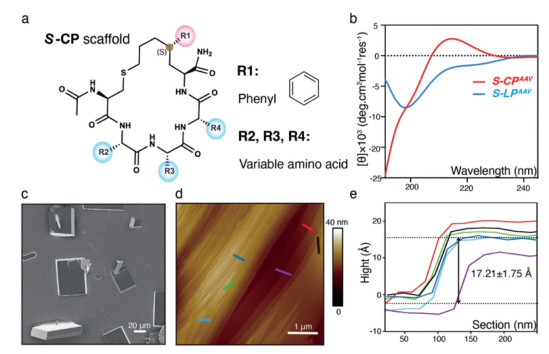
Figure 1.
Self-assembly exploration of S-CP. (a) Chemical structure of S-CP scaffold. (b) CD spectra of S-CPAAV and S-LPAAV in deionized water. (c) SEM image of the S-CPAAV cuboid assemblies (scale bar: 20 µm). (d) AFM image of the S-CPAAV cuboid assemblies (scale bar: 1 µm). (e) The height measurement of layers depicted in panel d by different colors. Six independent measurements were conducted. The mean layer height was calculated to be 1.721 µm with a standard deviation of 0.175 µm.
Here, we showed that the S-CP could self-assemble into highly ordered cubic structures by annealing process. Single-crystal X-ray diffraction (SCXRD) analysis and molecular dynamics (MD) simulations results revealed that the S-CP shows a rigid frame-like structure, which lacks intramolecular interactions and is highly robust upon amino acid substitutions. Further structural analysis revealed that the self-assembly of S-CP adopted a highly order hierarchical packing mechanism. Moreover, we achieved a mode switch between molecular packings through single atomic-level modulation of site-specific residues. This study highlights the potential for developing more geometrically structured basic units and paves the way for bottom-up peptide design and self-assembly.
To assess the self-assembly potential of S-CPs, we synthesized three variants, S-CPVAA, S-CPAVA, S-CPAAV (Figs. S1a-c in Supporting information), as previous research suggested that hydrophobic amino acids facilitate the self-assembly of CPs systems [26]. A linear pentapeptide (S-LPAAV) was also synthesized to examine the impact of cyclization (Fig. S1d in Supporting information). Compared to the linear state, the CD spectra of the all S-CPs were similar and exhibited a unique pattern unmatched by α-helix and β-sheet (Fig. 1b, Figs. S1e and g in Supporting information), indicating that the conformation of S-CP is a highly conserved and special. These appealing properties promoted us to examine if the S-CP could be applied as a building block for self-assemble.
To this end, we dissolved the samples in deionized water and annealed them to encourage the formation of ordered structures. Scanning electronic microscope (SEM) analysis revealed that only S-CPAAV formed ordered cuboid morphologies (Fig. 1c), while the other two S-CPs and S-LPAAV formed amorphous aggregates, suggesting that a special structure or sequence alone is insufficient for ordered assembly (Figs. S1f and h-i). The S-CPAAV crystals measured up to approximately 65 µm × 50 µm × 15 µm (Fig. 1c). Atomic force microscopy (AFM) analysis revealed staircase-like structures on the cuboid surfaces, perpendicular to the c-axis, with each layer about 17 Å thick (Figs. 1d and e). This indicates that the crystals grow through a layer-by-layer stacking mechanism [27, 28]. Together, these findings demonstrate that S-CP can spontaneously assemble into a layered 3D cuboid structure.
Next, the SCXRD analysis was conducted to determine the precise structure of S-CPAAV (Fig. S1j and Table S1 in Supporting informaiton). The analysis revealed a unique frame-like structure, with all peptide side chains positioned on the outer surface. Specifically, the Ac group, aminoacyl group, side chains of the second alanine (Ala2) and fourth valine (Val4) residues are located at the corners, while the side chains of the third alanine (Ala3) and the phenyl group of the S5-Ph5 residue are positioned along the long edges (Fig. 2a). Notably, S-CPAAV lacks intramolecular interactions in its crystalline state. Due to the constraint of the chiral center within the tether, the main chain adopts a distinctive dihedral angle sequence, "β·α·PII·PII·PII" (Fig. 2b), which orients the adjacent peptide bond planes nearly perpendicular to one another (Fig. S2 in Supporting information). This arrangement allows all side chains and backbone peptide bonds to point in different directions, providing sufficient non-covalent bonding sites for self-assembly.
Figure 2
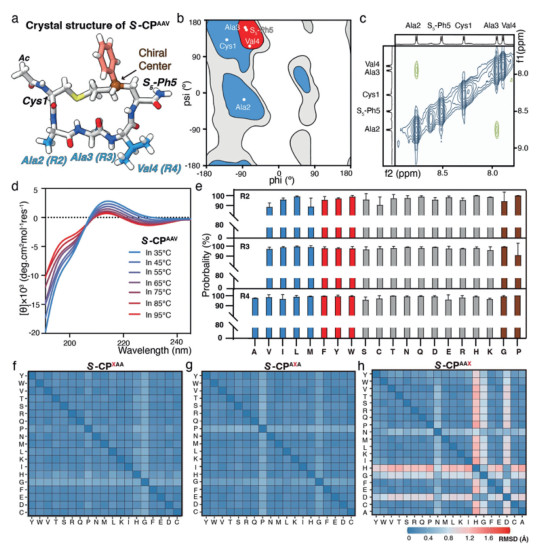
Figure 2.
Structure characteristics of S-CP. (a) Crystal structure of S-CPAAV. Off-white: C, white: H, dark blue: N, red: O. The residues, the Ac group, the phenyl and the chiral center are labeled. (b) Ramachandran plot of the residues in S-CPAAV crystal structure. (c) ROESY map of S-CPAAV. (d) Variable temperature CD of S-CPAAV. (e) The high possibility (at least > 80%) of S-CP scaffold peptides preferring a predominant conformational state. (f-h) Structure robustness of the S-CP scaffold. The substitutions for R2, R3, and R4 are shown from left to right, and the main structures of the corresponding mutants are compared with S-CPAAV.
To determine if the solution structure of S-CPAAV mirrors its crystalline state, we examined the peptide's conformation in solution using Rotating-frame nuclear Overhauser Effect Spectroscopy (ROESY). The results showed that only the amide protons of Ala2 and Ala3 are in spatial proximity (Fig. 2c). This finding was supported by 1H spectroscopy and total correlation spectroscopy (TOCSY) (Fig. S3 in Supporting information). To further confirm the solution conformation, we conducted MD simulations with full atomistic detail using enhanced sampling techniques [29]. The simulations revealed that S-CPAAV maintained a frame structure consistent with experimental data, showing a root mean square distance (RMSD) of 0.515 Å (Fig. S4 in Supporting information). Simulated ramachandran analysis of the peptide bonds also aligned well with the experimental results, except for the last residue, which exhibited both PPII and α conformations due to free rotation in the simulated state (Fig. 2b, and Figs. S5a-e in Supporting information). In contrast, simulations of the linear form peptide S-LPAAV showed a disordered structure (Figs. S5f-i in Supporting information). These findings suggest that such frame-like structure is predominates for S-CPAAV in solution.
To shed more light on the nature of this frame-shape structure, we performed variable temperature CD characterization experiments and MD simulations. The CD spectra of S-CPAAV, but not linear form, remained consistent across a broad temperature range (Fig. 2d, and Fig. S6 in Supporting information), indicating excellent thermal stability and rigidity of such frame scaffold. Given the uniformity of CD spectra observed across all previously studied S-CP derivatives [24, 25], we hypothesized that the structure of S-CPs is robust to various amino acid substitutions. Experimentally scanning these sequence variations is impractical, so we conducted extensive simulations to examine the conformational ensembles of S-CPs with substitutions at positions R2 to R4. The simulations showed that over 80% of the variants maintained a frame-like conformation (Fig. 2e), with most of the RMSDs < 1 Å compared to S-CPAAV (Figs. 2f-h, and Fig. S7a in Supporting information). The exceptions included S-CPAAH, S-CPAAD and S-CPAAG, where mutations were introduced at R4 and caused the local backbone of this part to assume an αL conformer, leading the formation of a twisted ring structure of S-CP (Figs. S7b and c in Supporting information). These findings indicated that the S-configured staple effectively preserves the frame conformation, even with diverse amino acid substitutions, making S-CPs a promising building block for self-assembly applications.
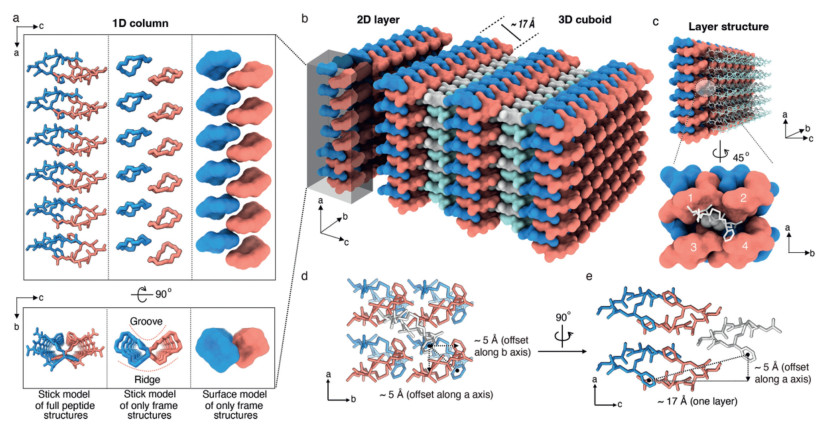
To understand the molecular hierarchy in S-CPAAV assemblies, we dissected the molecular arrangement of the cuboid crystals and found that the molecular packing in the cuboid assemblies was characterized by the stacking of S-CP frame structure. The S-CP molecules stacked on top of each other, forming 1D columns in the direction of one of the long edges of the cuboids (the a-axis) (Fig. 3a). There was also an alternating pattern of S-CP stacking in these columns, with every pair of neighboring rings using their N-terminal corners and phenyl groups in the staple to form a staggered stacking structure with two-fold symmetry (Fig. S8 in Supporting information). In addition, each 1D column structure exhibited a groove and a ridge, with the staple parts located on the groove side and the backbone parts located on the ridge side (Fig. 3a). These columns were associated laterally in the direction of the other long edge of cuboids (the b-axis), with one column using its ridge part to dock onto the groove region of another (Fig. S9 in Supporting information). This lateral packing of the columns gave rise to a two-dimensional (2D) layer that was parallel to the ab plane (Fig. 3b). This 2D layer was estimated to be ~17.0 Å in thickness, according to the lattice constant for the short edge (the c-axis) (Fig. 3b and Fig. S10 in Supporting information), which is consistent with the former AFM analysis. Multiple 2D layers further stacked in the direction of the c-axis with an offset of about 5 Å between neighboring layers in both the a and b direction so that the layers could be interdigitated in a "knobs-into-holes" interaction pattern, where every four adjacent Val sidechains in one layer form a pocket that is filled with a Val sidechain from the opposite layer (Figs. 3c-e).
Figure 3
Figure 3.
Hierarchical assembly rules of S-CPAAV. (a) One-dimension column structure formation. Two adjacent molecules alternate in the a-axis to form a 1D column represented by full or only the frame structure in stick or surface model. The distinct groove and ridge are indicated by the red dash curves. (b) Two-dimension layer and three-dimension cuboid structure formation. The 1D columns form an ab 2D plane along the b-axis by nesting groove and ridge with a thickness of ~17 Å, and multiple 2D layers are further stacked in the c-axis to allow the formation of a 3D cuboid structure. (c) "Knobs-into-holes" stacking of 2D layers along the c-axis. Each Val residue is inserted into a cavity formed by four S-CPAAV molecules in the adjacent layer. (d, e) An offset of ~5 Å between neighboring layers in both a- and b-directions so that the layers could be interdigitated.
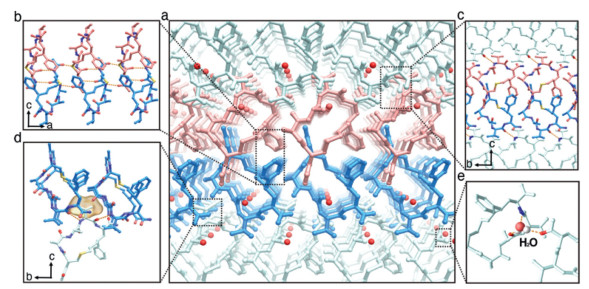
We next estimated the intermolecular interactions stabilizing the packed S-CPAAV molecules, identifying four key interfaces in the crystal lattice (Fig. 4a). (ⅰ) Along the a-axis, adjacent molecules zipped together via anti-parallel β-sheet HBs formed by the first two peptide bonds near Cys1 in its β conformation, extending into 1D column structures. An aromatic zipper, formed by the phenyl group in the staple, further stabilized this interaction (Fig. 4b). (ⅱ) Along the b-axis, HBs between the peptide bonds of Ala2 and S5-pH, along with aromatic stacking of the phenyl group in the staple, played major roles in structural stability (Fig. 4c). (ⅲ) Along the c-axis, 2D layers were primarily stabilized by "knobs-into-holes" interactions: Ala2 at the contact surface and Val4 inserted into a concave site formed by four S-CP molecules, creating strong hydrophobic interactions, while HBs between peptide bonds in adjacent layers further strengthen the structure (Fig. 4d). (ⅳ) A water molecule contributed additional stability by forming multiple HBs with surrounding peptide bonds (Fig. 4e). Overall, the combined interactions of each residue and peptide bond in different directions reinforced the packing stability of the entire system, leading to the formation of the 3D crystal.
Figure 4
Figure 4.
Intermolecular interactions in the S-CPAAV assembly involve four key interfaces (a) that stabilize the structure, including those along the a-axis (b), the b-axis (c), the c-axis (d), and the HBs formed around a water molecule (e).
Solvent molecules generally play a pivotal role in self-assembly [30-32]. Therefore, we further investigated the role of the water molecule in our case. The distance between the water molecule and the side chain carbon atoms of Ala3 was approximately 3.5 Å (Figs. S11a and b in Supporting information). To explore the impact of displacing the water molecule and disrupting the associated HBs, we substituted third alanine with the non-coding amino acid norvaline (An) to generate S-CPAAnV (Figs. S11c and S12a and b in Supporting information). CD and NMR analyses showed that the structural features of S-CPAAnV were highly conserved and consistent with those of S-CPAAV (Figs. S12c-f in Supporting information). Moreover, S-CPAAnV successfully self-assembled into 3D cubes (Fig. S13a in Supporting information). SCXRD and AFM analysis revealed that the molecular structure and packing pattern of S-CPAAnV were nearly identical to those of S-CPAAV (Figs. S11f, S13b, S14, and Table S2 in Supporting information). Notably, in the S-CPAAnV crystal, the terminal carbon atom of norvaline replaced the position originally occupied by the water molecule, with the HBs previously formed by water molecule now substituted by hydrophobic interactions from the norvaline side chain. Our results provide an example of how HBs can be replaced by hydrophobic interactions during self-assembly process, offering new insights into self-assembly design strategies.
In the assembly of S-CPAAnV, the terminal carbon atom of norvaline side chain is situated near the maximum threshold of steric hindrance from the surrounding atoms [33], suggested that further elongation of the side chain length might disrupt the current packing pattern. To confirm that, we replaced the third residue with a longer non-coding amino, norleucine (Ln), to generate S-CPALnV (Figs. S15a and b in Supporting information). CD and NMR analyses showed that the structural features of S-CPALnV were highly conserved and consistent with those of S-CPAAV and S-CPAAnV (Figs. S15c-f in Supporting information). Unexpectedly, the self-assembly experiments showed that S-CPALnV could also form cubic crystals (Fig. S16a in Supporting information). Further SCXRD results revealed that the molecule structure and the monolayer packing pattern of S-CPALnV were nearly identical to those of S-CPAAV and S-CPAAnV (Figs. S16b and S17a-d in Supporting information), but the interlayer stacking was in different arrangements (Fig. 5, and Figs. S17e-h in Supporting information).
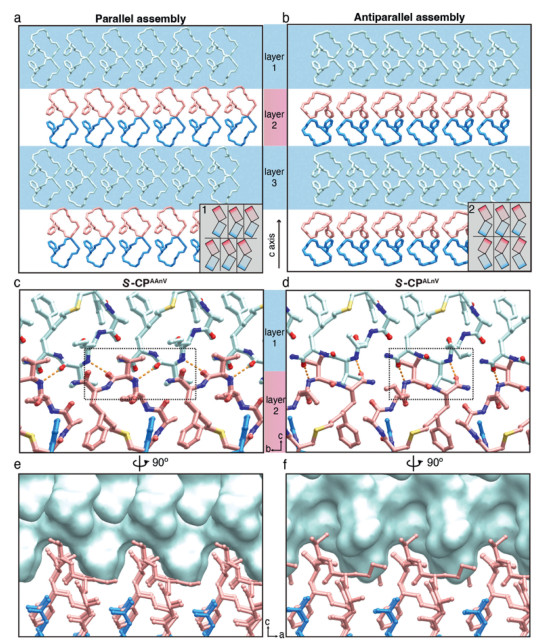
Figure 5
Figure 5.
Regulation of the cis-trans packing patterns relying on single atom switching. (a, b) Denote the two stacking modes of cis- and trans-patterns, respectively. Boxes "1" and "2" in the lower right corner of each panel are offset schematics, where the rectangle represents peptide molecule. A misalignment of the lines between the layers indicates the presence of offset, while no misalignment indicates no offset. (c, d) The interaction along the c-axis in the cis-packing mode of S-CPAAnV and the trans-packing mode of S-CPALnV. Hydrogen bonding: yellow dashed lines, hydrophobic interactions: black dashed box. (e, f) Rotating panels c and d, respectively. To enhance the intuitive observation of the spatial distribution of Val residues, we emphasized the upper layer with a cyan surface.
To describe these arrangements, we introduced the terms "cis-pattern" and "trans-pattern" to denote the groove and ridge configurations of adjacent layers that face the same and opposite directions, respectively. Thus, S-CPAAnV adopts a cis-pattern, while S-CPALnV adopts a trans-pattern. In the cis-pattern mode of S-CPALnV, we observed a ~5 Å offset between neighboring layers along both the a and b direction (Figs. 3d, e and 5a, layer 1). In contrast, in the trans-pattern mode of S-CPAAnV, the layers were in alignment (Fig. 5b, layer 2). This difference in layer arrangement resulted in a deviation of ~4.316° in β angle of the unit cell between the two crystal lattices (Tables S2 and S3 in Supporting information). While both patterns rely on HBs and hydrophobic interactions mediated by the fourth valine residues, significant disparities exist in the interlayer stacking. In the cis-pattern of S-CPALnV, the HBs formed between the peptide bonds flanking valine with angles of nearly 45° from the c axis, and the side chains of valine distributed uniformly at the layer surface to facilitate offset insertion (Figs. 5c, e, and Figs. S18a-c in Supporting information). Consequently, in the trans-pattern of S-CPALnV, the HBs formed nearly parallel to the c axis between the valine peptide bond and the C-terminal amide group, with closely spaced valine side chains in adjacent layers engaging in head-to-head hydrophobic interactions (Figs. 5d, f, and Figs. S18d-f in Supporting information). This arrangement mitigated the offset, providing ample space for the longer side chain of the third residue. Consequently, norleucine in S-CPALnV can rotate around the carbon bond to reach an optimal position, leading to two slightly different twists of the norleucine terminal carbon atoms (Fig. S17b in Supporting information).
Notably, in the assembly of S-CPALnV, the terminal carbon atom of norleucine side chain also reached the repulsive limit with surrounding atoms. We then further extended the side chain length by replaced the third residue with a non-coding amino, homomorleucine (Lh), to generate S-CPALhV (Fig. S19 in Supporting information). Although the CD results of S-CPALhV showed a typical S-CP spectrum, the peptide failed to self-assemble and instead formed an amorphous state (Fig. S20 in Supporting information). This result suggested that an overly long side chain of the third residue could lead to the collapse of the existing assembly system. Together, our investigation revealed the ability to modulate the cis-trans packing mode by adjusting the side chain length of the third amino acid. Moreover, these results highlight the versatile self-assembly stacking patterns of S-CP, offering promising strategies for future research in this field.
In conclusion, we present a cyclopentapeptide with a novel frame-like structure (S-CP), which exhibits excellent thermal stability, robustness, and remarkable self-assembly potential, as demonstrated through comprehensive computational and experimental analyses. Unlike traditional α-helices and β-sheets, S-CP lacks intramolecular interactions, allowing full exposure of the backbone, enhancing spatial orientation specificity, and providing versatile options for tuning self-assembly. In contrast to cyclic peptides with modified backbones [20], such as the introduction of alt (d, l)- and β-amino acids, S-CP is entirely composed of α-L-type amino acids, providing an additional design choice for the self-assembling basic unit. Molecular chirality is generally essential for governing structure and function in peptide self-assembly [34-36]. In our case, chirality plays a crucial role as S-CP maintains conformational stability through its in-tether chiral center, allowing precise control over assembly properties. Addtionally, S-CP possesses unique CD and NMR spectra, which provide a convenient way to identify.
Through crystal structure determination and auxiliary methods such as AFM, we uncovered the packing mode and layer-by-layer stacking mechanism of S-CP. Our analysis of residue interactions along different axes, combined with atomic-level insights, allowed for precise system tuning. Interestingly, we achieved the substitution of HBs by introducing hydrophobic interactions, which allowed the replacement of water molecules in the crystal lattice. Nonetheless, this does not imply that solvent molecules are insignificant. On the contrary, solvents play a critical role in modulating intermolecular interactions, including hydrogen bonding, van der Waals forces, and hydrophobic interactions, which can either promote or inhibit specific self-assembly behaviors. Additionally, different S-CPs under various pH conditions and in different solvents with unique polarity, hydrogen bond donor/acceptor capabilities, can alter molecular alignment and assembly patterns, potentially leading to distinct self-assembled forms, such as hydrogels [37] or peptide glasses [38, 39]. Given these effects, further exploration of solvent interactions may enable precise control over self-assembly, enhancing the versatility of S-CPs for applications across industrial, pharmaceutical, and biomedical fields. Future research will focus on understanding these solvent effects under varying environmental conditions, potentially broadening the practical applications of this peptide scaffold.
Furthermore, we showed that the cis-trans arrangement of the assembly could be modulated through single-atom changes in specific residues, a rarely reported finding [40]. Notably, the trans arrangement reduces interlayer overlap and creates additional cavities, offering new possibilities for designing layered and porous nanomaterials with finely tuned structures and properties. Altogether, this work serves as an exciting example of developing geometrically structured basic units and opens new avenues for bottom-up peptide design and self-assembly.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by the National Basic Research Program of China 973 Program (Nos. 2021YFA0910803, 2021YFC2103900), the National Natural Science Foundation of China (No. 21977011), the Natural Science Foundation of Guangdong Province (Nos. 2022A1515010996 and 2020A1515011544), the Shenzhen Science and Technology Innovation Committee (Nos. RCJC20200714114433053, JCYJ20180507181527112 and JCYJ20200109140406047), the Shenzhen-Hong Kong Institute of Brain Science-Shenzhen Fundamental Research Institutions (No. 2019SHIBS0004), the Shenzhen Fundamental Research Program (No. GXWD20201231165807007–20200827170132001), Tian Fu Jin Cheng Laboratory (Advanced Medical Center) Group Racing Project (No. TFJC2023010008).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.110785.
[1]
J. Zhang, Y. Wang, B.J. Rodriguez, et al., Chem. Soc. Rev. 51 (2022) 6936–6947. doi: 10.1039/d2cs00122e
S. Xuan, X. Jiang, R.K. Spencer, et al., Proc. Nat. Acad. Sci. U. S. A. 116 (2019) 22491–22499. doi: 10.1073/pnas.1909992116
Figure 1
Self-assembly exploration of S-CP. (a) Chemical structure of S-CP scaffold. (b) CD spectra of S-CPAAV and S-LPAAV in deionized water. (c) SEM image of the S-CPAAV cuboid assemblies (scale bar: 20 µm). (d) AFM image of the S-CPAAV cuboid assemblies (scale bar: 1 µm). (e) The height measurement of layers depicted in panel d by different colors. Six independent measurements were conducted. The mean layer height was calculated to be 1.721 µm with a standard deviation of 0.175 µm.
Figure 2
Structure characteristics of S-CP. (a) Crystal structure of S-CPAAV. Off-white: C, white: H, dark blue: N, red: O. The residues, the Ac group, the phenyl and the chiral center are labeled. (b) Ramachandran plot of the residues in S-CPAAV crystal structure. (c) ROESY map of S-CPAAV. (d) Variable temperature CD of S-CPAAV. (e) The high possibility (at least > 80%) of S-CP scaffold peptides preferring a predominant conformational state. (f-h) Structure robustness of the S-CP scaffold. The substitutions for R2, R3, and R4 are shown from left to right, and the main structures of the corresponding mutants are compared with S-CPAAV.
Figure 3
Hierarchical assembly rules of S-CPAAV. (a) One-dimension column structure formation. Two adjacent molecules alternate in the a-axis to form a 1D column represented by full or only the frame structure in stick or surface model. The distinct groove and ridge are indicated by the red dash curves. (b) Two-dimension layer and three-dimension cuboid structure formation. The 1D columns form an ab 2D plane along the b-axis by nesting groove and ridge with a thickness of ~17 Å, and multiple 2D layers are further stacked in the c-axis to allow the formation of a 3D cuboid structure. (c) "Knobs-into-holes" stacking of 2D layers along the c-axis. Each Val residue is inserted into a cavity formed by four S-CPAAV molecules in the adjacent layer. (d, e) An offset of ~5 Å between neighboring layers in both a- and b-directions so that the layers could be interdigitated.
Figure 4
Intermolecular interactions in the S-CPAAV assembly involve four key interfaces (a) that stabilize the structure, including those along the a-axis (b), the b-axis (c), the c-axis (d), and the HBs formed around a water molecule (e).
Figure 5
Regulation of the cis-trans packing patterns relying on single atom switching. (a, b) Denote the two stacking modes of cis- and trans-patterns, respectively. Boxes "1" and "2" in the lower right corner of each panel are offset schematics, where the rectangle represents peptide molecule. A misalignment of the lines between the layers indicates the presence of offset, while no misalignment indicates no offset. (c, d) The interaction along the c-axis in the cis-packing mode of S-CPAAnV and the trans-packing mode of S-CPALnV. Hydrogen bonding: yellow dashed lines, hydrophobic interactions: black dashed box. (e, f) Rotating panels c and d, respectively. To enhance the intuitive observation of the spatial distribution of Val residues, we emphasized the upper layer with a cyan surface.

 DownLoad:
DownLoad:

 下载:
下载:






 下载:
下载:
