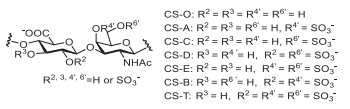
Figure 1.
CS repeating units and some CS subtypes.
Semisynthesis of rare chondroitin sulfate B and T oligosaccharides
Kai Zhou , Ao Sun , Yuchao Wang , Hang Dong , Chenkai Bai , Yidian Mo , Xuyang Ding , Xiangbao Meng , Zhongtang Li , Zhongjun Li

Chondroitin sulfate (CS) is a linear, sulfated polysaccharide belonging to the glycosaminoglycan (GAG) superfamily, which is present on the surface and in the extracellular matrix [1] of cells including in nerve and cartilage tissue, playing an important role in many biological processes such as cell recognition [2], cancer metastasis [3], parasitic infections [4], viral infections [5], and neuron growth regulation [6]. CS contains the repeating disaccharide units of ᴅ-glucuronic acid (GlcA) and N-acetyl-ᴅ-galactosamine (GalNAc) linked by β-(1→3) bonds, and the repeating disaccharide units are connected by β-(1→4) bonds [7]. Sulfate groups may be attached to the C-2, C-3 positions of GlcA and the C-4′, C-6′ positions of GalNAc, which can derive sixteen CS subtypes at most (Fig. 1) [8]. Notably, almost all the sulfate groups are located at the C-4′, C-6′ positions of GalNAc for CS subtypes from terrestrial animals [9], while for those from marine species, the sulfate groups are located at the C-2 [10, 11] or C-3 [12-14] positions of GlcA and very rarely at both C-2 and C-3 positions [15]. The sulfate groups at different positions of CS lead to structural diversity, which may be important for the various biological functions of CS [16-19].
As shown in Fig. 1, CS could be divided into five common subtypes including CS-O, CS-A, CS-C, CS-D, CS-E according to the specific sulfation position on GlcA and GalNAc [20-22]. There are four subtypes of CS (CS-A, CS-C, CS-D, CS-E) in the human body, which are extensively investigated for their synthetic methods and biological activities [23-27]. Whereas, CS-B and CS-T found in marine species are rare CS subtypes and have been rarely studied [8, 10]. Apart from that CS-B and CS-T are less abundant in nature, it is difficult to prepare CS-B and CS-T by chemical synthetic methods because of their sulfate groups at the C-2 positions of GlcA and C-4′ or both C-4′ and C-6′ positions of GalNAc. Therefore, it is necessary and challenging to develop simple and efficient synthetic methods of CS-B and CS-T for their functional study.
Traditionally, CS oligosaccharides are prepared by degrading natural CS and isolating depolymerized products, which usually affords a complex mixture with low yields. Given the scarcity of CS-B and CS-T in nature, it is impracticable to obtain CS-B and CS-T oligosaccharides from nature resource by such a strategy. Additionally, CS oligosaccharides can also be accessed via enzymatic synthesis, chemical synthesis and semisynthesis. Liu et al. and Huang et al. have reported some excellent progresses in the field of enzymatic synthesis of CS oligosaccharides [28-30]. But no enzymatic synthesis of CS-B and CS-T oligosaccharides has been reported so far, mainly due to the narrow substrate applicability and high cost of the enzymes. Chemical synthesis of CS oligosaccharides mainly refers to the splicing strategy based on monosaccharide blocks. Suda et al. have reported a synthetic method to obtain CS-B and CS-T disaccharides with a reaction route of 23 steps based on the monosaccharide splicing strategy [31], which had low efficiency (Fig. 2).
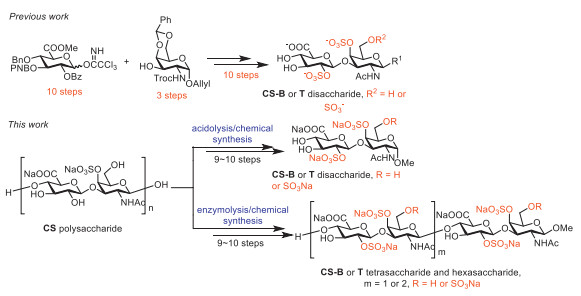
Previously, our research group reported a semisynthetic method of CS-A, CS-C and CS-E oligosaccharides based on enzymatic degradation of natural chondroitin (CH) polysaccharides [32]. Based on our works in enzymatic hydrolysis of CH and hyaluronic acid (HA) [23, 32, 33], we chose to prepare size-defined CS oligosaccharides without sulfates by enzymatic degradation of natural CH polysaccharides for the subsequent synthesis of CS-B and CS-T oligosaccharides.
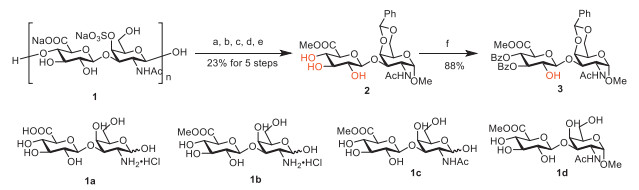
We aimed to develop a semisynthetic method to prepare CS-B and CS-T disaccharides from the common CH disaccharide. Compared with acquiring CH disaccharide blocks by enzymatic degradation of natural CH polysaccharides, acidolysis is more efficient and economical for the preparation of CH disaccharide blocks [34]. We chose CS-A polysaccharides as the starting materials for preparing CH disaccharide blocks due to its abundance and low cost. CS-A polysaccharides 1 was treated with concentrated hydrochloric acid to afford compound 1a (Scheme 1). 1a underwent methyl esterification of the carboxyl group to give 1b. It is interesting that the hydroxyl at C-1′ position of GalNAc of 1a did not undergo methyl glycosylation when the amino group of GalNAc of 1a was deprotected. Subsequent acetylation of amino and methyl glycosylation afforded 1d. Compound 1d was treated with benzaldehyde dimethyl acetal in the presence of p-toluenesulfonic acid to afford 2. To achieve regioselective sulfation at C-2 position of GlcA of CS-B and CS-T disaccharides, a regioselective protection reaction was performed with benzoyl cyanide [35-38] to give 3, which has a free hydroxyl at C-2 position of GlcA, in an isolated yield of 88%. The reaction condition of 3 is presented in Table 1. The structural confirmation of 3 is presented in the supplement materials.
 DownLoad:
CSV
DownLoad:
CSV
 |
||||
| Entry | Solvent | T (℃) | 2:2b: 2c | Yield (%) |
| 1 | CHCl3 | 0 | 1:2:0.2 | < 20 |
| 2 | CHCl3 | 0→rt | 1:2:0.2 | 40b |
| 3 | CHCl3 | 0→rt | 1:6:0.2 | 0c |
| 4 | CHCl3 | 0→rt | 1:4:0.2 | < 20c |
| 5 | CHCl3 | 0→rt | 1:3:0.2 | 42b |
| 6 | DCM | 0→rt | 1:2.2:0.2 | 38c |
| 7 | DCM | −40 | 1:2.2:0.2 | 78c |
| 8 | DCM | −78 | 1:2.2:0.2 | 44b |
| 9 | DCM | −50 | 1:2.2:0.2 | 82c |
| 10 | DCM | −60 | 1:2:0.2 | 88 |
| a Unless otherwise noted, all reactions were carried out on a 0.2 mmol scale under argon atmosphere, 2.0 mL dry solvent, 25 mg/mL 4 Å MS. b Isolated yield, main by-product was compound that had only 1 benzoyl group (at C-3 position of GlcA). c Isolated yield, main by-product was compound that had 3 benzoyl groups. |
||||
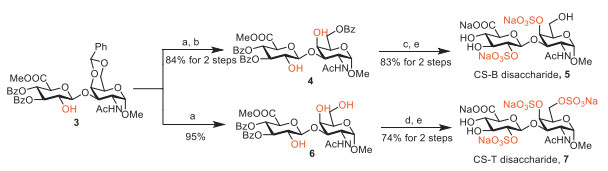
To achieve regioselective sulfations at C-4′ or both C-4′ and C-6′ positions of GalNAc of CS-B and CS-T disaccharides, these hydroxyls need to be selectively deprotected. Benzylidene group of 3 was easily removed to give 6 in acidic condition. The high reactivity of hydroxyl at C-6′ position of GalNAc allowed its regioselective protection with benzoyl cyanide to afford 4. Compound 4 was treated with triethylamine sulfur trioxide, followed by saponification with lithium hydroperoxide and sodium hydroxide [39] to afford the CS-B disaccharide 5. Treatment of compound 6 with triethylamine sulfur trioxide failed to produce the desired product 7. However, treatment of 6 with chlorosulfonic acid, followed by saponification with lithium hydroperoxide and sodium hydroxide afforded the CS-T disaccharide 7 smoothly (Scheme 2).
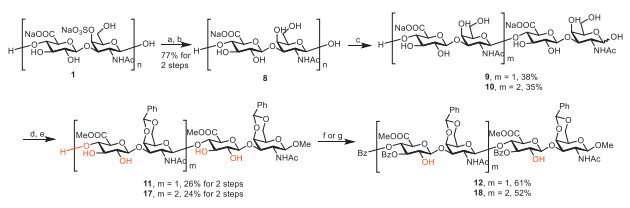
After completing the efficient synthesis of CS-B and CS-T disaccharides, we started to explore the synthesis of more complex oligosaccharides of CS-B and CS-T. CS-A polysaccharides 1 was desulfated and cleaved by enzymes to give CH tetrasaccharide block 9 and hexasaccharide block 10 (Scheme 3) [33]. Compounds 9 and 10 were treated with acetyl chloride and methanol to undergo esterification and methyl glycosylation, followed by treatment with benzaldehyde dimethyl acetal in the presence of p-toluenesulfonic acid to afford 11 and 17. The regioselective sulfation at C-2 positions of GlcA of CS-B and CS-T oligosaccharides was very challenging. Compound 2 has only three equatorial hydroxyl groups, but 11 and 17 have five and seven equatorial hydroxyls respectively. Due to the poor solubility of 11 and 17 in dichloromethane, they failed to afford the expected products under the condition for 3. After screening many solvents and solvent combinations, regioselective protection of 11 and 17 with benzoyl cyanide afforded 12 and 18 in an isolated yield of 61% and 52%, respectively, when a small portion of N, N-dimethylformamide was added to improve solubility. The reaction condition optimization of 12 and 18 are presented in Table 2, Table 3. The structural confirmation of 12 and 18 are presented in the supplement materials.
 DownLoad:
CSV
DownLoad:
CSV
 |
||||
| Entry | Solvent | T (℃) | 11:11b: 11c | Yield (%) |
| 1 | CHCl3 | 0→rt | 1:3:0.3 | < 20 |
| 2 | 2.5 mL DCM, 0.5 mL DMF | −40 | 1:3:0.3 | 44 |
| 3 | 2.5 mL DCM, 0.5 mL DMF | −50 | 1:3:0.3 | 50 |
| 4 | 2.5 mL DCM, 0.5 mL DMF | −60 | 1:3:0.3 | 42 |
| 5 | 2.5 mL DCM, 0.5 mL DMF | −55 | 1:3:0.3 | 57 |
| 6 | 2.5 mL DCM, 0.5 mL DMF | −55 | 1:3.2:0.3 | 61 |
| a Unless otherwise noted, all reactions were carried out on a 0.2 mmol scale under argon atmosphere, 2.0 mL dry solvent, 25 mg/mL 4 Å MS. | ||||
DownLoad:
CSV
 |
|||||
| Entry | Catalyst | Solvent | T (℃) | 17:17b: 17c | Yield (%) |
| 1 | PPY | 2.0 mL CHCl3 | 0→rt | 1:4:0.4 | 0 |
| 2 | PPY | 2.0 mL DMF | 0→rt | 1:4:0.4 | 0 |
| 3 | PPY | 2.0 mL CHCl3, 0.5 mL DMF | 0→rt | 1:6:0.4 | < 20 |
| 4 | PPY | 2.0 mL CHCl3, 0.5 mL DMF | −20→rt | 1:6:0.4 | < 20 |
| 5 | PPY | 2.0 mL DCM, 0.5 mL DMF | −20→rt | 1:6:0.4 | < 20 |
| 6 | PPY | 2.0 mL CHCl3, 0.5 mL DCM | 0→rt | 1:4:0.4 | < 20 |
| 7 | PPY | 2.0 mL DCM | −40 | 1:4:0.4 | < 20 |
| 8 | PPY | 2.0 mL DCM | −78 | 1:4:0.4 | < 20 |
| 9 | PPY | 2.0 mL DCM | −40→rt | 1:4:0.4 | < 20 |
| 10 | PPY | 2.5 mL DCM, 0.5 mL DMF | −40 | 1:4:0.4 | 42 |
| 11 | PPY | 2.5 mL DCM, 0.5 mL DMF | −50 | 1:4:0.4 | 50 |
| 12 | PPY | 2.5 mL DCM, 0.5 mL DMF | −60 | 1:4:0.4 | 36 |
| 13 | PPY | 2.5 mL DCM, 0.5 mL DMF | −50 | 1:4.3:0.4 | 52 |
| 14 | PPY | 2.5 mL DCM, 0.5 mL DMSO | −50 | 1:4.3:0.4 | 32 |
| 15 | PPY | 2.5 mL DCM, 0.5 mL pyridine | −50 | 1:4.3:0.4 | 50 |
| 16 | DBU | 2.5 mL DCM, 0.5 mL DMF | −50 | 1:4.3:0.4 | < 20 |
| a Unless otherwise noted, all reactions were carried out on a 0.2 mmol scale under argon atmosphere, 25 mg/mL 4 Å MS. | |||||
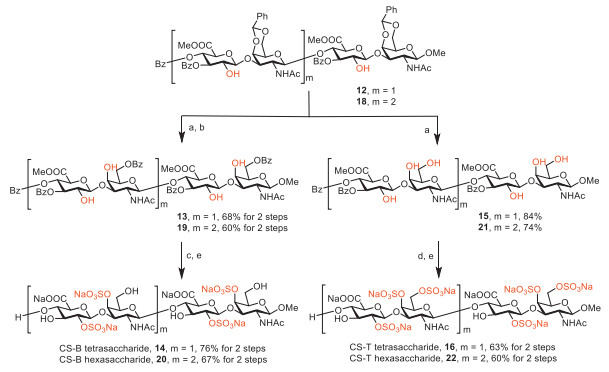
Subsequently, the benzylidene groups of 12 and 18 were deprotected under mild condition to afford 15 and 21 (Scheme 4). Regioselective protection of compound 15 and 21 with benzoyl cyanide afforded 13 and 19. Compound 13 and 19 were treated with triethylamine sulfur trioxide to undergo sulfation, and the followed ester hydrolysis with lithium hydroperoxide and sodium hydroxide afforded CS-B tetrasaccharide 14 and CS-B hexasaccharide 20. Compound 15 and 21 were treated with chlorosulfonic acid as the sulfation reagent, and the ester was hydrolyzed with lithium hydroperoxide and sodium hydroxide to afford CS-T tetrasaccharide 16 and CS-T hexasaccharide 22.
We have reported a facile semisynthesis method for synthesis of structurally homogeneous CS-B and CS-T disaccharides, tetrasaccharides and hexasaccharides in 9~10 linear steps from the commercially available natural CS-A polysaccharides. Our approach benefited from efficient enzymatic and acidic cleavage of CH polysaccharides, thus avoiding the redundant steps in constructing the disaccharide or tetrasccharide or hexasaccharide block and improving synthetic efficiency significantly. From commercially available natural CS polysaccharides to size-defined rare CS oligosaccharides, this strategy provided an excellent idea for addressing the availability of rare natural oligosaccharides. The essential regioselective protection of multiple equatorial hydroxyls made the synthetic method seem ingenious, and had significant reference value in regioselective protection research field of hydroxyls. Considering the potential application of sulfated polysaccharides in anticoagulation field, we may evaluate the anticoagulation activity of these compounds in subsequent researches.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Kai Zhou: Writing – original draft, Visualization, Validation, Supervision, Project administration, Investigation, Formal analysis, Data curation, Conceptualization. Ao Sun: Writing – review & editing, Visualization, Supervision. Yuchao Wang: Methodology. Hang Dong: Methodology. Chenkai Bai: Methodology. Yidian Mo: Methodology. Xuyang Ding: Methodology. Xiangbao Meng: Methodology. Zhongtang Li: Writing – review & editing, Supervision, Project administration, Methodology, Funding acquisition. Zhongjun Li: Writing – review & editing, Supervision, Software, Resources, Project administration, Methodology, Funding acquisition, Conceptualization.
This work was supported by the National Natural Science Foundation of China (Nos. 81930097, 82151223), and the National Key R&D Program of China (Nos. 2022YFF1203005, 2022YFC2303700).
Supplementary material associated with this article can be found, in the online version, at doi:
H. Wu, R. Zhang, B. Hu, et al., Chin. Chem. Lett. 32 (2021) 3940–3947. doi: 10.1016/j.cclet.2021.04.043
S. Jalkanen, M. Jalkanen, J. Cell. Biol. 116 (1992) 817–825. doi: 10.1083/jcb.116.3.817
S. Mizumoto, S. Yamada, K. Sugahara, Curr. Opin. Struct. Biol. 34 (2015) 35–42. doi: 10.1016/j.sbi.2015.06.004
M.W. Fried, P.E. Duffy, Vaccine 33 (2015) 7483–7488. doi: 10.1016/j.vaccine.2015.10.011
S. Yamada, K. Sugahara, Curr. Drug. Discov. Technol. 5 (2008) 289–301. doi: 10.2174/157016308786733564
J.M. Brown, J. Xia, B.Q. Zhuang, et al., Proc. Natl. Acad. Sci. U. S. A. 109 (2012) 4768–4773. doi: 10.1073/pnas.1121318109
M. Mende, C. Bednarek, M. Wawryszyn, et al., Chem. Rev. 116 (2016) 8193–8255. doi: 10.1021/acs.chemrev.6b00010
G. Vesssella, S. Traboni, D. Cimini, et al., Biomacromolecules 20 (2019) 3021–3030. doi: 10.1021/acs.biomac.9b00590
K. Wang, W. Wang, R. Zhang, et al., Food Chem. 434 (2024) 137392. doi: 10.1016/j.foodchem.2023.137392
S. Nadanka, K. Sugahara, Glycobiology 7 (1997) 253–263. doi: 10.1093/glycob/7.2.253
N. Takeda, S. Horai, J. Tamura, Carbohydr. Res. 424 (2016) 54–58. doi: 10.1016/j.carres.2016.02.006
K. Sugahara, Y. Tanaka, S. Yamada, et al., J. Biol. Chem. 271 (1996) 26745–26574. doi: 10.1074/jbc.271.43.26745
A.K. Shetty, T. Kobayashi, S. Mizumoto, et al., Carbohydr. Res. 344 (2009) 1526–1532. doi: 10.1016/j.carres.2009.02.029
K. Higashi, Y. Okamoto, A. Mukuno, et al., Carbohydr. Polym. 134 (2015) 557–565. doi: 10.1016/j.carbpol.2015.07.082
R.S. Cavalcante, A.S. Brito, L.C. Palhares, et al., Carbohydr. Polym. 183 (2018) 192–200. doi: 10.1016/j.carbpol.2017.12.018
H. Bougatef, F. Krichen, F. Capitani, et al., Carbohydr. Polym. 196 (2018) 272–278. doi: 10.1016/j.carbpol.2018.05.019
K. Kitazawa, S. Nadanaka, K. Kadomatsu, H. Kitagawa, Commun. Biol. 4 (2021) 114. doi: 10.1038/s42003-020-01618-5
S. Yang, S. Gigout, A. Molinaro, et al., Mol. Psychiatr. 26 (2021) 5658–5668. doi: 10.1038/s41380-021-01208-9
C. Spliid, A. Toledo, P. Sanderson, et al., J. Biol. Chem. 297 (2021) 101391. doi: 10.1016/j.jbc.2021.101391
G. Zhang, Q. Liu, S. Yang, et al., Chin. Chem. Lett. 30 (2019) 686–689. doi: 10.1016/j.cclet.2018.06.012
K. Wang, F. Bai, X. Zhou, et al., Int. J. Bio. Macromol. 209 (2022) 1280–1287. doi: 10.1364/oe.446015
Q. Yuan, X. Shi, H. Ma, et al., Int. J. Bio. Macromol. 262 (2024) 129969. doi: 10.1016/j.ijbiomac.2024.129969
W. Yao, Y. Zhu, X. Zhang, et al., J. Org. Chem. 83 (2018) 14069–14077. doi: 10.1021/acs.joc.8b01987
Y.S. Chng, G. Tristan, G.W. Yip, Y. Lam, Org. Lett. 21 (2019) 4559–4562. doi: 10.1021/acs.orglett.9b01457
C. Peng, Q. Wang, R. Jiao, et al., Carbohydr. Polym. 262 (2021) 117911.
A. Badri, A. Williams, A. Awofiranye, et al., Nat. Commun. 12 (2021) 1389. doi: 10.1038/s41467-021-21692-5
K. Wang, L. Qi, L. Zhao, et al., Carbohydr. Polym. 301 (2023) 120361. doi: 10.1016/j.carbpol.2022.120361
J. Li, G. Su, J. Liu, Angew. Chem. Int. Ed. 56 (2017) 11784–11787. doi: 10.1002/anie.201705638
J. Li, E.M. Sparkenbaugh, G. Su, et al., ACS Cent. Sci. 6 (2020) 1199–1207. doi: 10.1021/acscentsci.0c00712
P. Lin, Y. Xu, S. Bali, et al., Angew. Chem. Int. Ed. 2024 e202405671. doi: 10.1002/anie.202405671
M. Wakao, R. Obata, K. Miyachi, et al., Bioorg. Med. Chem. Lett. 25 (2015) 1407–1411. doi: 10.1016/j.bmcl.2015.02.054
X. Zhang, H. Liu, W. Yao, et al., J. Org. Chem. 84 (2019) 7418–7425. doi: 10.1021/acs.joc.9b00112
X. Zhang, H. Liu, L. Lin, et al., Angew. Chem. Int. Ed. 57 (2018) 12880–12885. doi: 10.1002/anie.201807546
C. Lopin, J.C. Jacquinet, Angew. Chem. Int. Ed. 45 (2006) 2574–2578. doi: 10.1002/anie.200503551
P. Peng, M. Linseis, R.F. Winter, R.R. Schmidt, J. Am. Chem. Soc. 138 (2016) 6002–6009. doi: 10.1021/jacs.6b02454
T. Li, T. Li, T. Cui, et al., Org. Lett. 20 (2018) 3862–3865. doi: 10.1021/acs.orglett.8b01446
T. Li, T. Li, M. Linseis, et al., ACS Catal. 10 (2020) 11406–11416. doi: 10.1021/acscatal.0c02112
T. Li, T. Li, Y. Sun, et al., Org. Chem. Front. 8 (2021) 260–265. doi: 10.1039/d0qo01243b
J.C. Jacquinet, C. Lopin-Bon, A. Vibert, Chem. Eur. J. 15 (2009) 9579–9595. doi: 10.1002/chem.200900741

Scheme 1 Synthesis of CH disaccharide block 3 with free C-2 hydroxyl of GlcA. Reagents and conditions: (a) 0.6 mol/L HCl, 85 ℃, 48 h; (b) 2% AcCl/MeOH, 70 ℃, 2 h; (c) NaOMe, Ac2O, MeOH, 40 ℃, 24 h; (d) 2% AcCl/MeOH, 70 ℃, 16 h; (e) p-TsOH, PhCH(OMe)2, DMF, r.t., 12 h, 23% for 5 steps; (f) BzCN, PPY, 4 Å MS, DCM, −60 ℃, 24 h, 88%.

Scheme 2 Synthesis of CS-B disaccharide 5 and CS-T disaccharide 7. Reagents and conditions: (a) AcOH (aq), 80 ℃, 30 min, 95% for 6; (b) BzCN, pyridine, 0 ℃, 12 h, 84% for 2 steps; (c) SO3·Et3N, pyridine, 60 ℃, 36 h; (d) ClSO3H, pyridine, DMF, 80 ℃, 24 h; (e) LiOH (aq)/H2O2 (aq), THF/H2O, −5 ℃ → r.t., then NaOH (aq), MeOH, 0 ℃ → r.t., 5, 83% for 2 steps, 7, 74% for 2 steps.

Scheme 3 Synthesis of CH tetrasaccharide block 12 and hexasaccharide block 18 with naked C-2 hydroxyls of GlcA. Reagents and conditions: (a) AcCl/MeOH, r.t., 7 d; (b) NaOH (aq), r.t., 24 h, 77% for 2 steps; (c) 2.5% hyaluronidase, NaOAc/NaCl buffer, pH 5.0, 37 ℃, 7 d, 38% for 9, 35% for 10; (d) 3% AcCl/MeOH, r.t., 16 h; (e) p-TsOH, PhCH(OMe)2, DMF, r.t., 12 h, 11, 26% for 2 steps, 17, 24% for 2 steps; (f) BzCN, PPY, 4 Å MS, DCM/DMF, −55 ℃, 24 h, 61% for 12; (g) BzCN, PPY, 4 Å MS, DCM/DMF, −50 ℃, 24 h, 52% for 18.

Scheme 4 Synthesis of CS-B tetrasaccharide 14, hexasaccharide 20 and CS-T tetrasaccharide 16, hexasaccharide 22. Reagents and conditions: (a) AcOH (aq), 60 ℃, 3 h, 84% for 15, 74% for 21; (b) BzCN, pyridine, 0 ℃, 12 h, 68% for 13 in 2 steps, 60% for 19 in 2 steps; (c) SO3·Et3N, pyridine, 60 ℃, 48 h; (d) ClSO3H, pyridine, DMF, 80 ℃, 24 h; (e) LiOH (aq)/H2O2 (aq), THF/H2O, −5 ℃ → r.t., then NaOH (aq), MeOH, 0 ℃ → r.t., 76% for 14 in 2 steps, 67% for 20 in 2 steps, 63% for 16 in 2 steps, 60% for 22 in 2 steps.
Table 1. Condition optimization of compound 3a.
|
||||
| Entry | Solvent | T (℃) | 2:2b: 2c | Yield (%) |
| 1 | CHCl3 | 0 | 1:2:0.2 | < 20 |
| 2 | CHCl3 | 0→rt | 1:2:0.2 | 40b |
| 3 | CHCl3 | 0→rt | 1:6:0.2 | 0c |
| 4 | CHCl3 | 0→rt | 1:4:0.2 | < 20c |
| 5 | CHCl3 | 0→rt | 1:3:0.2 | 42b |
| 6 | DCM | 0→rt | 1:2.2:0.2 | 38c |
| 7 | DCM | −40 | 1:2.2:0.2 | 78c |
| 8 | DCM | −78 | 1:2.2:0.2 | 44b |
| 9 | DCM | −50 | 1:2.2:0.2 | 82c |
| 10 | DCM | −60 | 1:2:0.2 | 88 |
| a Unless otherwise noted, all reactions were carried out on a 0.2 mmol scale under argon atmosphere, 2.0 mL dry solvent, 25 mg/mL 4 Å MS. b Isolated yield, main by-product was compound that had only 1 benzoyl group (at C-3 position of GlcA). c Isolated yield, main by-product was compound that had 3 benzoyl groups. |
||||
 下载: 导出CSV
下载: 导出CSV
Table 2. Condition optimization of compound 12a.
|
||||
| Entry | Solvent | T (℃) | 11:11b: 11c | Yield (%) |
| 1 | CHCl3 | 0→rt | 1:3:0.3 | < 20 |
| 2 | 2.5 mL DCM, 0.5 mL DMF | −40 | 1:3:0.3 | 44 |
| 3 | 2.5 mL DCM, 0.5 mL DMF | −50 | 1:3:0.3 | 50 |
| 4 | 2.5 mL DCM, 0.5 mL DMF | −60 | 1:3:0.3 | 42 |
| 5 | 2.5 mL DCM, 0.5 mL DMF | −55 | 1:3:0.3 | 57 |
| 6 | 2.5 mL DCM, 0.5 mL DMF | −55 | 1:3.2:0.3 | 61 |
| a Unless otherwise noted, all reactions were carried out on a 0.2 mmol scale under argon atmosphere, 2.0 mL dry solvent, 25 mg/mL 4 Å MS. | ||||
下载: 导出CSV
Table 3. Conditions optimization of compound 18a.
|
|||||
| Entry | Catalyst | Solvent | T (℃) | 17:17b: 17c | Yield (%) |
| 1 | PPY | 2.0 mL CHCl3 | 0→rt | 1:4:0.4 | 0 |
| 2 | PPY | 2.0 mL DMF | 0→rt | 1:4:0.4 | 0 |
| 3 | PPY | 2.0 mL CHCl3, 0.5 mL DMF | 0→rt | 1:6:0.4 | < 20 |
| 4 | PPY | 2.0 mL CHCl3, 0.5 mL DMF | −20→rt | 1:6:0.4 | < 20 |
| 5 | PPY | 2.0 mL DCM, 0.5 mL DMF | −20→rt | 1:6:0.4 | < 20 |
| 6 | PPY | 2.0 mL CHCl3, 0.5 mL DCM | 0→rt | 1:4:0.4 | < 20 |
| 7 | PPY | 2.0 mL DCM | −40 | 1:4:0.4 | < 20 |
| 8 | PPY | 2.0 mL DCM | −78 | 1:4:0.4 | < 20 |
| 9 | PPY | 2.0 mL DCM | −40→rt | 1:4:0.4 | < 20 |
| 10 | PPY | 2.5 mL DCM, 0.5 mL DMF | −40 | 1:4:0.4 | 42 |
| 11 | PPY | 2.5 mL DCM, 0.5 mL DMF | −50 | 1:4:0.4 | 50 |
| 12 | PPY | 2.5 mL DCM, 0.5 mL DMF | −60 | 1:4:0.4 | 36 |
| 13 | PPY | 2.5 mL DCM, 0.5 mL DMF | −50 | 1:4.3:0.4 | 52 |
| 14 | PPY | 2.5 mL DCM, 0.5 mL DMSO | −50 | 1:4.3:0.4 | 32 |
| 15 | PPY | 2.5 mL DCM, 0.5 mL pyridine | −50 | 1:4.3:0.4 | 50 |
| 16 | DBU | 2.5 mL DCM, 0.5 mL DMF | −50 | 1:4.3:0.4 | < 20 |
| a Unless otherwise noted, all reactions were carried out on a 0.2 mmol scale under argon atmosphere, 25 mg/mL 4 Å MS. | |||||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们


 下载:
下载: