Citation:
Yinghao Zhang, Ke Shao, Yihang Zhu, Haokun Zhang, Yinuo Zhuo, Huihui Bao, Yeye Ai, Yongguang Li. Unanticipated optical properties of π-conjugated cyclometalated Pt(Ⅱ) complexes for advanced information storage and anti-counterfeiting materials[J]. Chinese Chemical Letters,
2025, 36(9): 110735.
doi:
10.1016/j.cclet.2024.110735
Unanticipated optical properties of π-conjugated cyclometalated Pt(Ⅱ) complexes for advanced information storage and anti-counterfeiting materials
English
Unanticipated optical properties of π-conjugated cyclometalated Pt(Ⅱ) complexes for advanced information storage and anti-counterfeiting materials
College of Material, Chemistry and Chemical Engineering, Key Laboratory of Organosilicon Chemistry and Material Technology Ministry of Education, Hangzhou Normal University, Hangzhou 311121, China
b.
Jafron Biomedical Co., Ltd., Zhuhai 519000, China
c.
Hangzhou Institute of Quality & Metrology, Hangzhou 310019, China
Received Date:
20 September 2024 Accepted Date:
06 December 2024 Revised Date:
02 December 2024 Available Online:
15 September 2025
Abstract:
In this work, we synthesize two luminescent Pt(Ⅱ) complexes using different π-conjugated bidentate ligands. Both complexes are assembled into three-dimensional (3D) networks through non-classical intermolecular interactions in the crystal state. Unexpectedly, substituting pyridine with the more extensively π-conjugated quinoline significantly increases the dihedral angles between the phenyl and quinolyl groups of the bidentate ligands. This alteration disrupts the π-π interactions between molecules, resulting in distinct optical properties upon exposure to external stimuli. By integrating these complexes into polymers, we fabricate electrospun films containing luminescent nanofibers that exhibit reversible optical changes. These findings have paved the way for the development of high-performance optical encryption and anti-counterfeiting materials, achieved through the employment of simple chromophores.
Optical materials that respond to environmental stimuli [1-8] are crucial in industry and daily life. These smart materials offer extensive potential in information storage, detection, sensing, and memory devices [9-27]. Responsive optical materials can be categorized into several types based on their photophysical mechanisms: (1) covalent bond breakage, usually irreversible, (2) molecular switches with reversible configurations, (3) noncovalent interactions between chromophores, and (4) reversible molecular conformations [7,28]. These changes lead to alterations in electronic orbitals, resulting in changes of apparent and emission colors.
Among these, force-responsive materials represent a key category. For instance, pressure can shorten inter- and intramolecular distances under anisotropic mechanical milling and isotropic hydrostatic pressure. In most cases, optical materials exhibit similar pressure-induced optical behavior, with minimal differences in wavelength red-shifts or blue-shifts [29-31]. To date, various pressure-responsive organic compounds, halide chalcogenides and carbon dots show promising applications in sensing, scintillators, information storage and anti-counterfeiting [32-35]. Luminescent complexes have recently attracted attention for exploration because of their well-defined molecular structures and packing arrangements that facilitate structure-property relationship studies [36,37].
Platinum(Ⅱ) [Pt(Ⅱ)] complexes with square planar geometry, offer abundant photophysical properties, attainable through molecular design and intermolecular regulation [13,25,38]. Ligands such as bidentate, tridentate, and tetradentate, when coordinated with Pt(Ⅱ), have been extensively investigated for use in phosphorescent light-emitting diodes (PHOLEDs), biosensors, and other optical materials because of their good stability, long wavelength emission and high quantum yield [10,39-48]. By modifying the ligands, it is possible to fine-tune the highest occupied molecular orbital (HOMO) to lowest unoccupied molecular orbital (LUMO) energy gap of Pt(Ⅱ) complexes, adjusting their photo-physical properties [13,49]. These modifications also endow Pt(Ⅱ) complexes with varied conformations and configurations, involving intermolecular interactions, including π-π, metal-metal, and van der Waals interactions, resulting in distinct assembled optical properties. We have recently designed and synthesized a series of Pt(Ⅱ) molecular hinges that demonstrate varied luminescent responses to mechanical grinding and isotropic hydrostatic pressure by altering the alkyl chains on the regulatory axis [3]. This demonstrates that subtle changes in molecular structure can lead to significant alterations in molecular stacking and optical properties.
In the present work, Pt(Ⅱ) complexes modified with different π-surface cyclometalated ligands exhibit distinct optical properties that are being investigated for applications in optical encryption and anti-counterfeiting applications. Remarkably, expanding the π-surface area does not enhance the intermolecular π-π interactions as initially hypothesized; instead, it results in a significant increase in the dihedral angle between the phenyl and quinolyl groups. The two complexes exhibit distinct optical properties under external stimuli. This difference in optical properties is used to prepare luminescent electrospun films with polyurethane acrylate (PUA) as a dopant, which displays reversible optical changes. These films are further utilized in the development of encryption and anti-counterfeiting materials with distinct optical characteristics.
Cyclometalated N^C-Pt(Ⅱ) complexes 1 and 2 (Scheme S1 in Supporting information) are synthesized by a three-step procedure according to the previous literature [42]. The complexes are characterized by 1H nuclear magnetic resonance (1H NMR) spectrum and 13C NMR spectrum (Figs. S1–S4 in Supporting information), matrix-assisted laser desorption ionization time-of-flight mass spectrometry (MALDI-TOF), elemental analysis and X-ray single crystal. The details can be found in the Supporting information.
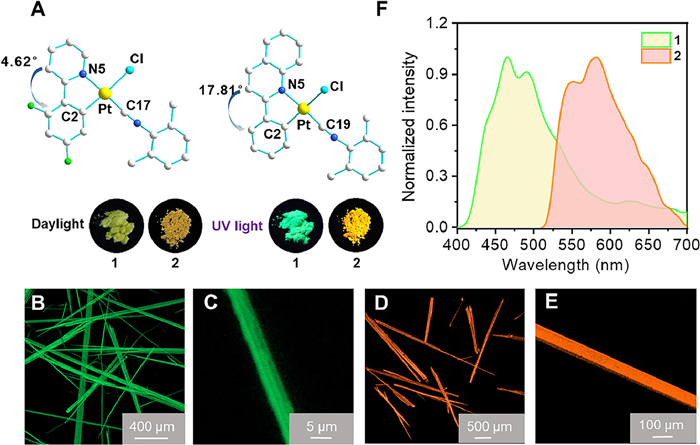
Needle-shaped crystals of 1 and 2 are successfully obtained by diffusion of diethyl ether into dichloromethane solutions. These crystals are subsequently analyzed using X-ray single crystal diffraction and confocal laser scanning microscopy (CLSM) (Figs. 1A–E; Figs. S5, S6 and Table S3 in Supporting information). The molecules of 1 are arranged in a head-to-tail configuration, forming dimers through nonclassical intermolecular C—H…Cl interactions with a distance of 2.8 Å These dimers further associate into one-dimensional (1D) chains through intermolecular C—H…F interactions (2.6 Å) (Fig. S7 in Supporting information). The 1D chains then extend into a three-dimensional (3D) network by aligning head-to-head laterally. This arrangement is facilitated by π-π interactions between the 2-isocyano-1,3-dimethylbenzene auxiliary ligands, with a π[Ph]-π[Ph] distance of 3.4 Å, and between the 2-isocyano-1,3-dimethylbenzene and the isocyano triple bond, with a π[C-π[Ph] distance of 3.4 Å. Additionally, intermolecular C—H…π interactions (2.9 Å) and C—H…F interactions (2.4 Å) contribute to the overall 3D structure (Fig. S7). Molecule 2, characterized by a significantly larger π-conjugated ligand, exhibits a distorted planar structure. This is evident from the larger dihedral angle of 17.81° between the phenyl and quinoline groups, and an angle of 12.2° between the 2-phenylquinoline Pt(Ⅱ) center and the plane formed by the isocyanide and chloride ligands through Pt(Ⅱ) center. The molecules of 2 adopt a head-to-head fashion with the formation of 1D chains under π-π interactions between the phenylquinoline and isocyano triple bond (3.4 Å, π[C-π[Ph] distance), and intermolecular C—H…π interactions (3.0 Å) (Figs. S8 and S9 in Supporting information). Respective C—H…π hydrogen bonds between the 2-phenylquinoline ligands and 2-isocyano-1,3-dimethylbenzene auxiliary ligands connect the 1D chain into the 3D structure (Fig. S9 in Supporting information). The two crystals display different optical properties in response to external stimuli, particularly 2, which exhibits distinct optical behaviors under mechanical milling and hydrostatic pressure.
Figure 1
Figure 1.
(A) Molecular structures of 1 and 2 (inset: photographs under daylight and 365 nm UV light). Confocal laser scanning microscope (CSLM) images of crystals 1 (B, C) and 2 (D, E). (F) Emission spectra of 1 and 2 (1.0 × 10–5 mol/L) in degassed dichloromethane solution.
1 and 2 display similar ultraviolet–visible (UV–vis) absorption bands when dissolve in dichloromethane (1.0 × 10–5 mol/L) (Fig. S10 in Supporting information). The strong and high-energy intense absorption bands at 255–300 and 300–350 nm are attributed to intra-ligand (IL) (π→π*) transitions, while the weak and low-energy absorption bands in the range of 365–400 nm are assigned as a mixture of ligand-to-ligand charge transfer (LLCT) [π(phenyl)→π*(N^C)] and metal-to-ligand charge transfer (MLCT) [dπ(Pt)→π(N^C)] transitions. In degassed dichloromethane solution, 1 and 2 show green and yellow emission, respectively. These Pt(Ⅱ) complexes exhibit emissions with the observed lifetime prolonged to 1.38–1.85 µs in the CH2Cl2 solutions (Figs. S11 and S12 in Supporting information). The much longer lifetimes and large Stokes shifts imply that these low-energy emissions are of triplet parentage. The emissions originate from the Pt(Ⅱ) metal-perturbed 3IL (π→π*) excited state with some mixing of 3MLCT [dπ(Pt)→π(N^C)] excited state (Fig. 1F). Due to the narrower HOMO-LUMO gap of the 2-phenylquinoline ligand, both the absorption and emission bands of 2 exhibit a red shift compared to 1. Both 1 and 2 are weakly emissive in fluid solution at room temperature, probably due to the intramolecular rotation and structural distortion. The coordination skeleton around the Pt center has been distorted from the planar configuration in the ground state to the quasi-tetrahedron configuration in the excited state. The emission bands of 1 and 2 remain unchanged across different solvents, indicating no interaction between the solvent molecules and the Pt(Ⅱ) moiety (Figs. S13 and S14 in Supporting information). This observation aligns with the understanding that the emission primarily originates from the 3IL (π→π*) exited state, which is minimally influenced by the solvent environment.
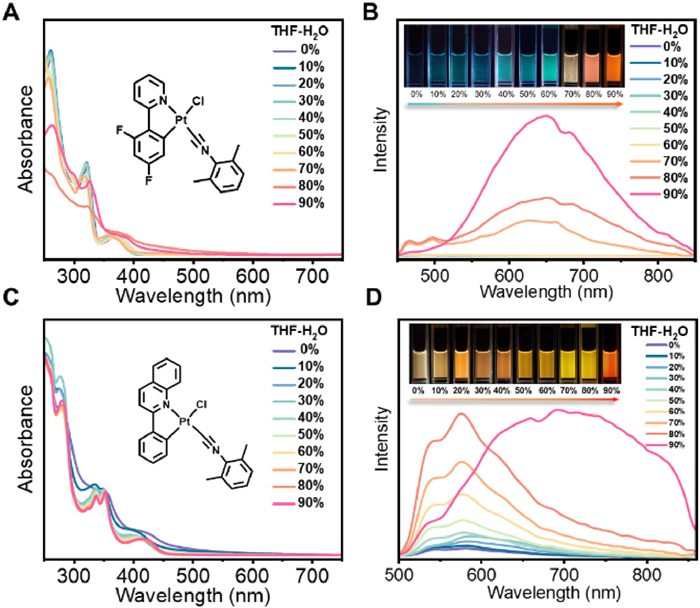
Both 1 and 2 exhibit aggregation induced phosphorescence enhancement (AIPE) behavior upon aggregate formation in tetrahydrofuran (THF)/H2O mixtures at a concentration of 1 × 10–4 mol/L. Upon photoexcitation, 1 shows a rather weak well-resolved vibronic-structured 3IL emission band in THF (Fig. 2). The emission intensity is gradually increased accompanying with the appearance of aggregated emission band at 623 nm as the increase of the H2O fraction. When the H2O portion reaches 70%, both the monomeric and aggregated emission intensities are obviously enhanced. The aggregated emission intensity boosts further when the water content reaches 90% with the observation of nanofibers (Fig. 2 and Fig. S15 in Supporting information). Due to the varying ratios of enhanced intensity of the dual emission bands, the perceived emissive colors gradually change from green to red (Fig. S16 in Supporting information). On the other hand, 2 shows a gradual increase in monomer emission intensity with increasing water content, culminating in the formation of an aggregated emission peak at 713 nm when the water content reaches 90%. In the aggregated state, the rigidification of the molecules would lead to a restricted motion, which slows down the nonradiative decay rate, thereby enhancing the emission intensity. The aggregates form under the influence of π-π and/or Pt…Pt interactions, resulting in a color change from yellow to red. Despite possessing more π-conjugated ligands, 2 exhibits a slower accumulation process, likely due to its more distorted planar configuration (Figs. 1A and 2).
Figure 2
Figure 2.
Absorption spectral changes of 1 (A) and 2 (C) (1.0 × 10–4 mol/L) upon increasing the H2O content in THF at 298 K and emission spectral changes of 1 (B) and 2 (D) (1.0 × 10–4 mol/L) upon increasing the H2O content in THF at 298 K (λex = 365 nm) (Insets: corresponding emission photos under 365 nm UV irradiation).
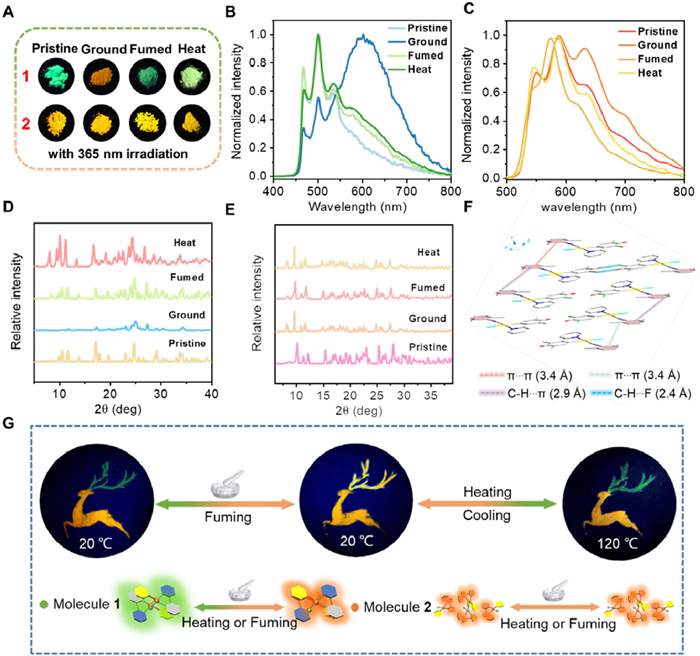
In the solid state, 1 and 2 are highly emissive mainly because of the physical restriction of the coordination skeletal deformation, manifesting their excellent aggregation-induced emission (AIE) properties. In addition, the emission lifetimes of solid powder are prolonged to 2.22–6.50 µs, unambiguously demonstrating their triplet parentage (Figs. S17–S20 in Supporting information). 1 and 2 exhibit distinct responses to mechanical stress induced by grinding in a mortar. Initially, 1 shows a pale green apparent color, which changes to yellow upon grinding, as well as emission color changes from green to orange (Figs. 3A and B). The intensity of the original green emission peak weakens, and a new orange peak emerges at 633 nm. The excitation spectrum of the emission band at λ = 633 nm (ground state) shows the presence of ground-state aggregation compared with that of the emission band at λ = 633 nm (pristine state) due to the increase in the compactness of the molecular packing and enhanced intermolecular interactions such as π−π stacking and Pt…Pt interactions in the ground state as opposed to the monomeric 3IL emission of the pristine state (Fig. S21 in Supporting information). Both emissive and apparent colors can be recovered upon CH2Cl2 vapor or heating stimuli (Figs. 3A and B). In contrast, both apparent and emissive colors of 2 almost remain unchanged (Figs. 3A and C). The differentiated results are further investigated by powder X-ray diffraction (PXRD) experiments (Figs. 3D and E). Both 1 and 2 initially display intense and sharp diffraction peaks, indicative of their crystalline nature. After grinding, 1 lead to a decrease in the intensity of these diffraction peaks, suggesting a transition from a crystalline state to a more amorphous state. Notably, for 1, the observation of a broad peak appears at 2θ = 25.038 with a d-spacing of 3.5 Å suggests the formation of stronger π-π and/or Pt…Pt interactions and alteration of the electronic structure, which do not appear of ground 2 (Figs. 3D–F). Upon heating to 120 ℃ or exposing to CH2Cl2 vapor, the diffraction patterns of 1 and 2 can be recovered (Figs. 3B and C). This recovery indicates a recrystallization process, where the materials revert to a more crystalline state after being subjected to thermal energy or solvent vapor treatment.
Figure 3
Figure 3.
(A) Photographs of 1 and 2 prepared from the pristine, ground, fumed and heat states under 365 nm UV light. Emission spectrum of 1 (B) and 2 (C) in different states. XRD spectrum of 1 (D) and 2 (E) in different states. (F) Crystal packing diagrams of 1 showing the presence of non-classical C—H…F, C—H…π hydrogen bonds and π-π interactions. (G) Emission patterns of 1 and 2 for patterning applications at 365 nm ultraviolet light.
Further supports have been revealed by the investigation using in situ attenuated total reflection infrared (ATR-IR) and Raman experiments. As depicted in Fig. S22 (Supporting information), the C≡N stretch (2198 cm–1) of complex 1 shows a shift to a lower frequency (2183 cm–1) upon grinding. Subsequently, the initial spectra could be recovered upon fuming with dichloromethane vapor or heat treatment. The recorded Raman spectra at ambient and ground state are shown in Fig. S23 (Supporting information). The Raman peak of 1 at 2187 cm–1 from the stretching vibration v(C≡N) of 2-isocyano-1,3-dimethylbenzene auxiliary ligand upshifts to low frequency (2182 cm–1), which indicates that grinding can regulate the molecular packing mode. Upon fuming with dichloromethane vapor or heat treatment, the stretching vibration is recovered to the initial peak, indicating the restoration to the more crystalline state. For 2, due to its more distorted planar configuration, the ATR-IR and Raman measurements remain consistent across the pristine, ground, fumed and heat states (Figs. S24 and S25 in Supporting information).
The unique optical properties of Pt(Ⅱ) complexes provide promising opportunities for information storage and anti-counterfeiting applications. By strategically utilizing 1 and 2, a fawn pattern can be created (Fig. 3G, Figs. S26 and S27 in Supporting information). The antlers are crafted from 1, while the body of the deer is constructed from 2. Initially, the pattern displays green luminescent antlers paired with an orange luminescent deer body. When mechanical grinding is applied, the antlers’ green emission transforms to an orange hue. Further treatments, such as fuming with dichloromethane or heating to 120 ℃, can restore the antlers’ green emission (Fig. S28 in Supporting information). However, this reversion is temporary; as the temperature decreases, the antlers revert to orange luminescence. In contrast, the antlers treated with dichloromethane vapor maintain their green glow. Importantly, throughout these processes, the color of the deer's body remains consistent and unaltered. These dynamic variations in emission under specific conditions enable the pattern to encode multiple pieces of information, thereby demonstrating its potential applications in information storage and anti-counterfeiting.
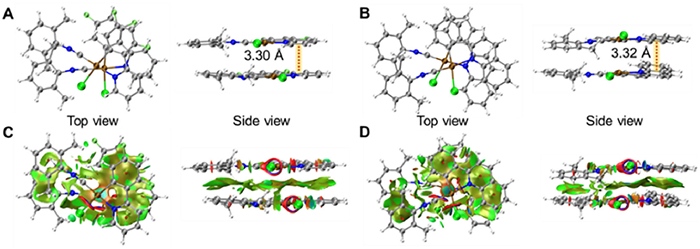
Apart from the photophysical investigations, the ground-state geometries of the dimers of 1 and 2 have been optimized and shown in Figs. 4A and B. In the optimized geometries, the cyclometalated Pt(Ⅱ) units adopt a head-to-head orientation that are nearly parallel to each other. The Pt…Pt distances within the dimers of 1 and 2 are computed to be 3.33 and 3.38 Å, suggesting the presence of Pt…Pt interactions in aggregate formation. Furthermore, the interplanar distances between the coordination planes are determined to be 3.30 and 3.32 Å, indicating the existence of π–π interactions in the dimers. The shorter Pt…Pt separations observed in both 1 and 2 align with our emission studies, where the 3MMLCT emission is found at lower energy levels. Noncovalent interaction (NCI) analysis has been conducted on the dimers. In the NCI plots, the green surfaces on the cyclometalated ligands and Pt(Ⅱ) centers between the complexes indicate the presence of Pt…Pt and π–π stacking interactions, which play important roles in enhancing the intermolecular interactions (Figs. 4C and D).
Figure 4
Figure 4.
Optimized ground-state geometries of the dimers of (A) 1 and (B) 2. Isosurfaces of the noncovalent interaction for (C) 1 and (D) 2.
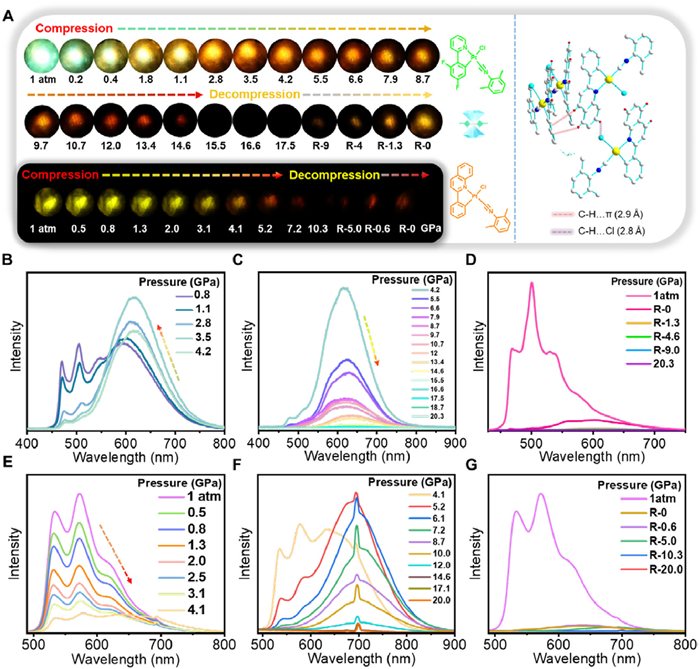
The use of hydrostatic pressure via a diamond anvil cell (DAC) induces an isotropic force, differing from the shear force exerted during mechanical grinding (Fig. 5). For 1, as hydrostatic pressure increases, a new aggregated emission band emerges at 596 nm. The emission intensity gradually strengthens as the monomeric emission diminishes (Figs. 5A and B, Fig. S29 in Supporting information). At 1.1 GPa, the aggregated emission intensity surpasses that of the monomers, reaching its maximum at 4.2 GPa. Further pressurization causes the emission band to decrease in intensity and progressively shift towards 653 nm (Fig. 5C). Emission images reveal a color evolution from green to orange-yellow, and then to red, suggesting a reduction in the π-π and/or Pt…Pt distances, leading to stronger intermolecular interactions (Fig. 5A). Notably, this emission band displays partial reversibility upon pressure release (Fig. 5D). For 2, the emission intensity at 573 nm weakens with increased hydrostatic pressure (Fig. 5E). At 4.1 GPa, a new aggregated emission appears at 631 nm, shifting towards 700 nm with further pressurization (Figs. 5E and F). The aggregated emission intensity reaches its maximum at 5.2 GPa. Both complexes exhibit isotropic pressure-induced luminescence color changes, showing more significant redshifts compared to anisotropic mechanical grinding. This emission band also displays partial reversibility upon pressure release (Fig. 5G). Moreover, the emission of 2 shows a notably larger redshift than 1. This may be due to the higher isotropic static water pressure, which promotes a more planar conformation in the initially distorted plane, enhancing intermolecular π-π and/or Pt…Pt interactions.
Figure 5
Figure 5.
(A) In situ emission micrographs of the powder form of 1 and 2 under different pressures (inset: crystal packing diagrams of 2 showing the non-classical hydrogen bonding interactions of C—H…Cl and C—H…π). (B–D) Emission spectra of 1 under different pressures. (E–G) Emission spectra of 2 under different pressures.
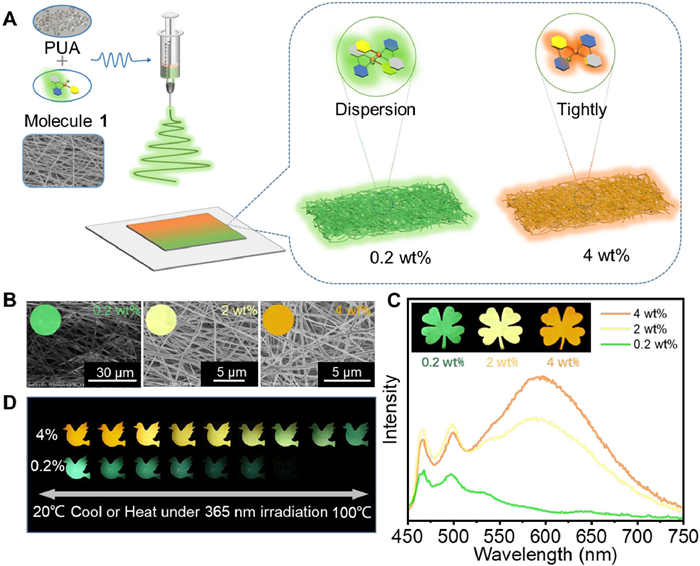
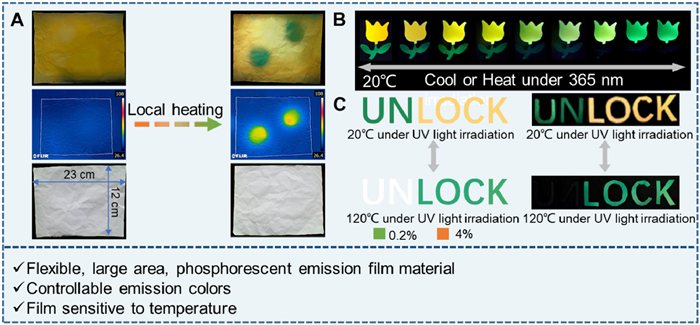
Since 1 exhibits significant temperature sensitivity, large-scale flexible films with good fiber structure have been fabricated on a decimeter scale using the electrospinning method with PUA as a dopant (Figs. 6A and B, Fig. S30 in Supporting information). For instance, leaves display green, yellow and orange emissive colors as the doping concentration in PUA increases from 0.2% to 4% in quality percent (Figs. 6B and C, Fig. S31 in Supporting information). As the temperature rises, the green emissive color of 0.2% quality fraction doping film fades, while the orange emissive color of 4% quality fraction doping film turns to green (Figs. S32 and S33 in Supporting information). These color changes are reversible with temperature regulation (Fig. 6D and Fig. S34 in Supporting information). Localized temperature increases can be detected by infrared thermometers through changes in emissive color (Fig. 7A). As shown in Fig. 7B, at a temperature of 70 ℃, the green emission of leaves made with a 0.2% doping film disappears, and the orange emission of a flower made with a 4% doping film shifts to yellow and then green as the temperature further increases. This temperature-dependent luminescent film is further developed as an optical material for anti-counterfeiting purpose. Under UV light irradiation, confusing messages like, “UNLOCK” or “LOCK” are obtained. However, upon heating, only the real message “LOCK” becomes visible. Cooling down the temperature, the confusing message immediately restores and the whole process remains invisible under daylight (Fig. 7C). Thus, a temperature-responsive cryptographic material is realized using simple complex 1, showcasing the potential of temperature-sensitive luminescent materials for advanced anti-counterfeiting techniques where authenticity can be verified through temperature-induced luminescence changes.
Figure 6
Figure 6.
(A) Constructing electrospun fiber films with different emissions by adjusting the loadings of 1. (B) Scanning electron microscope (SEM) images of electrospun fiber films with different loadings of 1 (inset: corresponding photographs under 365 nm UV light). (C) Emission spectra of electrospun fiber films with different loadings of 1 (inset: leaf patterns of different emissions with different loadings of 1). (D) Emission patterns of electrospun fiber films with different loadings of 1 at different temperatures.
Figure 7.
(A) Large-scale electrospun fiber film for temperature detection. (B) Emission patterns of electrospun fiber films with different loadings of 1 at different temperatures. (C) Constructing advanced information encryption materials based on electrospun fiber films responsive to temperature.
In conclusion, two luminescent Pt(Ⅱ) complexes featuring different π-conjugated bidentate ligands are synthesized, which exhibit planar and twisted molecular conformations, respectively. Both complexes are assembled into three-dimensional (3D) networks with non-classical intermolecular interactions in the crystal state. Differences in molecular structure and conformation lead to varied responses under mechanical grinding and hydrostatic pressure, resulting in distinct changes in emission behavior. These optical behavior changes are reversibly induced by external heat and vapor stimuli. The responsiveness of these materials to diverse external stimuli enables the creation of advanced security features that can be applied across multiple domains. The authenticity of documents or products can be verified through luminescence changes triggered by external stimuli, providing a high level of security due to the complexity of the chromophores and their responses to various stimuli.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work is supported by the National Natural Science Foundation of China (Nos. 22201057 and 22472044), Zhejiang Provincial Natural Science Foundation of China (Nos. LR22B010001 and LQ23B010001). Dr. Z. Ni in the College of Material, Chemistry and Chemical Engineering at Hangzhou Normal University is gratefully acknowledged for his kind assistance in theoretical calculation and helpful discussion.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.110735.
[1]
T. Xiao, D. Ren, L. Tang, et al., J. Mater. Chem. A 11 (2023) 18419–18425. doi: 10.1039/D3TA03498D
[2]
J.R. Otaegui, D. Ruiz-Molina, J. Hernando, et al., Adv. Fun. Mater. 34 (2024) 2402510. doi: 10.1002/adfm.202402510
[3]
Y. Ai, Y. Li, M.H.Y. Chan, et al., J. Am. Chem. Soc. 143 (2021) 10659–10667. doi: 10.1021/jacs.1c04200
[4]
D. Cui, F. Bai, L. Zhang, et al., Angew. Chem. Int. Ed. 63 (2024) e202319815. doi: 10.1002/anie.202319815
J. Li, K. Chen, J. Wei, et al., J. Am. Chem. Soc. 143 (2021) 18317–18324. doi: 10.1021/jacs.1c09272
Figure 1
(A) Molecular structures of 1 and 2 (inset: photographs under daylight and 365 nm UV light). Confocal laser scanning microscope (CSLM) images of crystals 1 (B, C) and 2 (D, E). (F) Emission spectra of 1 and 2 (1.0 × 10–5 mol/L) in degassed dichloromethane solution.
Figure 2
Absorption spectral changes of 1 (A) and 2 (C) (1.0 × 10–4 mol/L) upon increasing the H2O content in THF at 298 K and emission spectral changes of 1 (B) and 2 (D) (1.0 × 10–4 mol/L) upon increasing the H2O content in THF at 298 K (λex = 365 nm) (Insets: corresponding emission photos under 365 nm UV irradiation).
Figure 3
(A) Photographs of 1 and 2 prepared from the pristine, ground, fumed and heat states under 365 nm UV light. Emission spectrum of 1 (B) and 2 (C) in different states. XRD spectrum of 1 (D) and 2 (E) in different states. (F) Crystal packing diagrams of 1 showing the presence of non-classical C—H…F, C—H…π hydrogen bonds and π-π interactions. (G) Emission patterns of 1 and 2 for patterning applications at 365 nm ultraviolet light.
Figure 5
(A) In situ emission micrographs of the powder form of 1 and 2 under different pressures (inset: crystal packing diagrams of 2 showing the non-classical hydrogen bonding interactions of C—H…Cl and C—H…π). (B–D) Emission spectra of 1 under different pressures. (E–G) Emission spectra of 2 under different pressures.
Figure 6
(A) Constructing electrospun fiber films with different emissions by adjusting the loadings of 1. (B) Scanning electron microscope (SEM) images of electrospun fiber films with different loadings of 1 (inset: corresponding photographs under 365 nm UV light). (C) Emission spectra of electrospun fiber films with different loadings of 1 (inset: leaf patterns of different emissions with different loadings of 1). (D) Emission patterns of electrospun fiber films with different loadings of 1 at different temperatures.
Figure 7
(A) Large-scale electrospun fiber film for temperature detection. (B) Emission patterns of electrospun fiber films with different loadings of 1 at different temperatures. (C) Constructing advanced information encryption materials based on electrospun fiber films responsive to temperature.

 DownLoad:
DownLoad:

 下载:
下载:








 下载:
下载:
