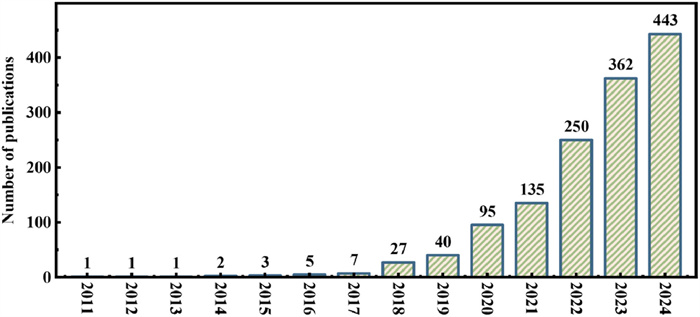
Figure 1.
The annual publications of ML in materials/catalysts research for environmental remediation from 2011 to 2024.
Application of machine learning for material prediction and design in the environmental remediation
Yunzhe Zheng , Si Sun , Jiali Liu , Qingyu Zhao , Heng Zhang , Jing Zhang , Peng Zhou , Zhaokun Xiong , Chuan-Shu He , Bo Lai

Developing more efficient materials and discovering new materials have always been important goals for researchers. This helps accelerate reaction rates, allow for cleaner and more energy-efficient chemical reactions, and address increasingly severe environmental issues more effectively [1-4]. Researching new materials typically involves a lengthy process of seven stages, discovery, development, performance optimization, system design and integration, validation, manufacturing, and deployment, which can span over a decade from initial research first to use [5]. Currently, researchers largely rely on empirical knowledge and trial-and-error experiments for materials development, which entails significant experimental costs, time, and a degree of luck. Therefore, density functional theory (DFT), as a precise and reliable theoretical computational method, assists researchers in predicting material properties, exploring new materials, and understanding material behavior, playing a crucial role in material design and development [6]. However, limitations such as high computational costs, restricted predictive capabilities, and insufficient accuracy in systems with strong electron correlation effects (such as transition metal compounds and heavy element systems) hinder its further advancement [7]. Therefore, it is necessary to develop new methods to assist in material design and development. The rise of artificial intelligence (AI) technology has had a transformative impact on today's data-driven scientific research [8]. Machine learning (ML), as a significant branch of AI, has been widely applied in various fields such as disease diagnosis [9], drug discovery [10], climate prediction [11], cybersecurity [12], image and speech recognition [13], and financial market analysis [14] due to its capabilities in handling large-scale data, discovering new scientific patterns, and low operational thresholds. At the same time, ML technology aids in integrating knowledge and data from multiple disciplinary fields, promoting interdisciplinary research and innovation [15, 16]. Recently, ML has been widely applied in the environmental field to simulate, predict, and design materials to find more efficient, cost-effective, and environmentally friendly materials [17, 18]. Taking biochar (BC) materials as an example, Wang et al. predicted the reaction rate constants for pollutant degradation in the BC/PMS system by combining ML with the characteristics of BC materials, ultimately expanding the application scenarios of the BC/PMS system in advanced oxidation processes (AOPs) [19]. Palansooriya et al. used ML to predict the efficiency of BC in fixing heavy metals in biochar-amended soils, where nitrogen content and BC application rate are two significant features affecting metal immobilization [20]. Meanwhile, Wang et al. applied ML techniques to guide the rational design of BC, discovering that high surface area and oxygen content can significantly enhance the contribution of non-radicals, targeting the enhancement of BC's non-radical degradation pathway for pollutants [21].
In this manuscript, we focus on reviewing the application of ML to the prediction and design of materials in the environmental domain and discuss its development in detail. It is hoped that this review will open new horizons for the design and development of materials in the environmental domain. The framework of this review is listed as follows. Section 2 introduces the basic definition of ML, categorizes the commonly used ML classifications into three categories, and summarizes in detail the basic process of ML applied to materials design and development. Section 3 discusses the three main applications of ML in materials science, namely predicting material properties, designing and developing new materials, and analyzing and interpreting materials characterization, and introduces the current research status of these applications. Section 4 summarizes the current state of research on ML in different common environmental material categories. Finally, Section 5, provides an outlook of this review, pointing out the main current challenges and future research trends in ML applications and materials science of environmental remediation.
ML is a method that enables computer systems to learn and analyze large amounts of data to discover and extract patterns, rules, and structures that are difficult for the human eye to discern, thereby achieving intelligent task completion [22]. In the early development of ML in the mid-20th century, due to limitations in computing resources, foundational models such as perceptrons, K-Nearest Neighbor algorithms, and decision trees were explored, along with foundational work in pattern recognition [23-25]. With advancements in computer hardware and the widespread application of statistical methods, ML began to be applied in practice. Over the past decade, with the resurgence of deep learning and breakthroughs in fields such as image recognition [26], natural language processing [27], and games (such as AlphaGo [28]), deep learning has propelled ML research to the forefront. With the combined push of theoretical development, algorithm improvements, and the availability of massive amounts of data, ML has become more adept at solving complex problems and has greatly facilitated interdisciplinary and cross-domain interactive development, driving innovation and progress in the era. It can be observed in Fig. 1 that the annual publications on the application of ML in materials and catalysts research for environmental remediation have been growing exponentially from 2011 to 2024, using the keywords "Machine Learning, " "Environment, " "Material, " and "Catalyst" in the X-mol literature search engine.
ML can be mainly divided into three categories based on the learning method: supervised learning, unsupervised learning, and semi-supervised learning. Supervised learning algorithms model and analyze the mapping relationship from input data to output labels in known datasets, enabling accurate predictions when faced with new unlabeled data [29]. For example, predicting the decontamination effect of new materials in the aqueous environment or optimizing reaction conditions based on predicted reaction outcomes [30-33]. Common models used in supervised learning include linear regression (LR) [34], decision trees (DT) [35], random forests (RF) [36], and neural networks (NN) [37], which have been maturely applied in environmental material science. In addition, supervised learning models with high interpretability, such as LR and RF, facilitate the understanding of the relationship between material properties and environmental response. In contrast to supervised learning, unsupervised learning targets unlabeled datasets to discover underlying structures or patterns in the data [38]. For instance, classifying environmental materials through clustering techniques, revealing potential correlations between catalyst performances, or uncovering hidden patterns in catalyst design [39]. Common models in unsupervised learning include clustering algorithms [39], dimensionality reduction algorithms [40], and association rule learning [41]. Semi-supervised learning is a method that combines supervised and unsupervised learning, using a small amount of labeled data and a large amount of unlabeled data for training. Its goal is to enhance the model's generalization ability and performance with limited labeled data by utilizing a large amount of unlabeled data when labeling data takes a lot of time and costs [42]. For example, in the design of environmental remediation single-atom catalysts (SACs), an initial model is built with a small amount of experimental data, and then training is continued using unlabeled data to predict more potentially efficient catalysts [43]. Common models in semi-supervised learning are self-encoders, semi-supervised support vector machines, semi-supervised clustering, and so on [42].
The general workflow of ML in environmental research are shown in Fig. 2, including data collection, data preprocessing, feature engineering, model selection, training, validation, which will be discussed in detail in this subsection.
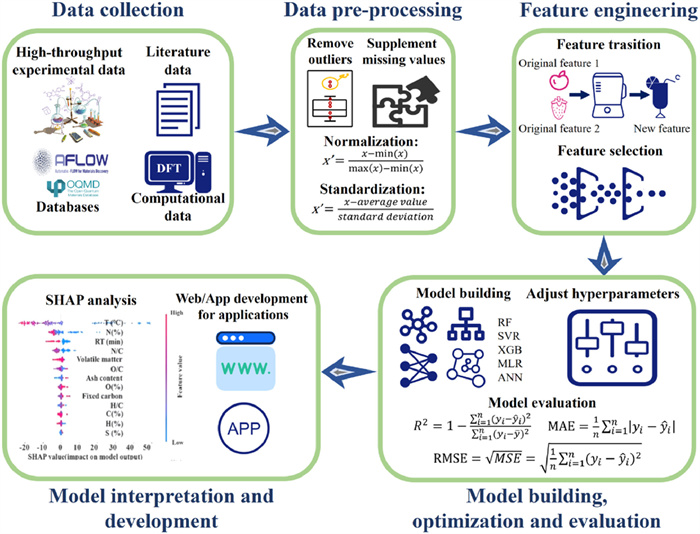
Data collection is the most important first step in ML, which directly affects the performance of the model and the ultimate prediction results [44, 45]. Typically, the sources of data in ML materials are mainly as follows:
(1) High-throughput experimental data. The material property data is obtained from the process of synthesizing and testing material samples by researchers, but the data volume is relatively small and usually used for model validation rather than establishment.
(2) Databases. Many research institutions and universities have established many material performance databases by aggregating experimental data from multiple individuals or teams. These databas provide a large amount of reliable and accurate data. Some common material databases information is organized in Table 1. However, relying entirely on databases may lead to overfitting of the model due to data bias [46].
 DownLoad:
CSV
DownLoad:
CSV
| Database name | Web address | Web functions |
| The materials project | Platform for providing material properties and computational data. | |
| OQMD | Open quantum materials database for large-scale computation and data management of materials structures and properties. | |
| NIST materials data | Provides standardized datasets and material property databases in the field of materials science. | |
| PubChem | Provides a large-scale database of chemical molecules and their properties to support the retrieval and analysis of chemical information. | |
| NOMAD | Provides a platform for storing and analyzing computational data in materials science, supporting large-scale data archiving and access. | |
| Magpie | Provides machine learning models and tools for material property prediction to support material discovery and design. | |
| AFLOW | Provides a materials database for automated first-principles calculations, including information on crystal structures, band gaps, etc. | |
| AiiDA | Provides a database for the management and archiving of material science computing workflows, supporting reproducible and verifiable scientific computing. | |
| Citrination | Provides a database of material properties and experimental data to support data-driven material design and discovery. | |
| Materials cloud | Provides a collaborative platform for computational data and tools for materials science to support data sharing and scientific research. | |
| ChemSpider | Database providing information on chemical substances, covering compound structures, properties and literature information. | |
| MPContribs | Provide a community-contributed data platform for materials projects to support user submission and access to materials computation data. |
(3) Literature data. In recent years, the annual publication volume of papers has grown exponentially, and a large amount of data points, diversified experimental conditions and the latest material data have been included. However, the reliability and usability of data in literature deserve attention. The data from different sources, different experimental conditions, and some easily ignored variables (such as stirring speed, air humidity.) may be incompatible, and researchers need to spend a lot of time screening the data. Therefore, data preprocessing is crucial for literature data.
(4) Computational chemistry data. Computational chemistry combines theoretical chemistry and computer science to provide complete physical and chemical information about materials through computer simulation of the real situation of materials. DFT calculations can provide data such as materials band structure, electron density, lattice constants and band gaps. Through molecular dynamics simulations, some dynamic behavior and thermodynamic properties of materials can be obtained. These computational methods provide rich and high-quality data but require huge computing resources, high computational costs and a long time.
Each type of data has its unique value and application scenarios. Current data in the materials field suffers from data quality issues such as wide sources and small samples, and it is difficult to achieve excellent predictions from models with data collected by a single method [47]. Therefore, data collection often comes from multiple approaches. High-quality and strongly correlated data can significantly improve the accuracy and efficiency of the model. However, material data tends to be high-dimensional, small-sample, and noisy, making it difficult to obey a particular data distribution. For collecting strongly correlated data, researchers' domain knowledge is crucially important, requiring both an understanding of materials and certain programming skills [48]. In plain terms, if the initially selected input and output data are theoretically unrelated, no matter how the model is later established and optimized, it would be difficult to achieve good predictive results. In addition to requiring real, reliable and reproducible data for collecting high-quality data, data preprocessing is also crucial.
Data from different sources often have different requirements in terms of pre-processing strategies. For example, different data may differ in format, unit, scale, which need to be unified according to the specific situation before data merging. In addition, high-throughput experimental data may contain a lot of noise, which requires complex denoising, and literature data may require text mining and information extraction. Preprocessing the data separately allows the most appropriate processing to be selected based on the specifics of each type of data. However, if the data exhibit high consistency in terms of source and format, or if the preprocessing steps are very similar and straightforward, then a unified preprocessing approach after merging the data can enhance efficiency, especially when handling large datasets. Data preprocessing is the process of cleaning, organizing and transforming raw data into a format that ML algorithms can understand and use, improving data quality and ensuring that raw data can be effectively used for analysis and model building [49]. For data cleaning, first of all, it is necessary to remove data that appears repeatedly or contains little effective information to ensure data validity. Outliers and abnormal values in the data can also be removed using statistical methods such as boxplots. Secondly, it is necessary to handle missing values in the data. Methods such as mean or median imputation can continue to be used as long as the missing value rate is less than 10% of the total data and it is considered reliable [50]. Data scaling is also necessary to ensure that different types of data can be compared on the same scale. For example, normalization (scaling data to between 0 and 1 while retaining the original distribution shape of the data) and standardization (transforming data to have a mean of 0 and standard deviation of 1, making the data conform to the standard normal distribution).
After data collection and preprocessing are complete, there are a large number of datasets that can be used for ML modeling. However, it does not mean that model performance will be higher with more input data. The features may contain a lot of redundant, irrelevant and meaningless features, thus interfering with and affecting the model learning. Through appropriate feature engineering, the risk of overfitting the training data can be reduced, making the model's generalization ability stronger on new data, reducing the complexity of the model and making it easier to explain and maintain [51]. Additionally, feature selection can reduce the dimensionality of the data and reduce computational resource consumption, speeding up training [52]. Feature engineering generally includes the following steps: (1) Feature creation. Generate new features from existing data, such as extracting month or day of week from dates, or calculating sales growth rates. (2) Feature selection. The process of identifying and retaining the features that are most valuable for the model's predictions. For example, all input feature data can be linearly correlated with the output data through Pearson's correlation coefficient analysis to preliminarily filter out lowly correlated features [53]. (3) Feature extraction. Extract useful information from the raw data, usually achieved through dimensionality reduction techniques such as principal component analysis [54] or linear discriminant analysis [55]. These technologies can help reduce the number of features while retaining important information, thereby enhancing the efficiency of the model [56]. When conducting feature engineering, the researcher's domain knowledge can help identify potential features, features that significantly impact the model, and possible transformation methods to ensure that the created features have practical meanings. Recently, several new feature selection methods have greatly facilitated researchers in efficiently identifying useful features. NCOR-FS, by embedding domain knowledge in materials science, reduces the correlation between features, thereby enhancing the predictive accuracy and interpretability of machine learning models [57]. DML-FSdek, as a data-driven multi-layer feature selection method, also incorporates expert knowledge and automates the selection of feature subsets without manual tuning of hyperparameters, reducing the risk of critical features being removed [58]. In summary, the integration of domain knowledge in feature engineering through methods like NCOR-FS and DML-FSdek has significantly improved the efficiency and effectiveness of feature selection.
After data preprocessing, and feature engineering filtering to ensure high-quality inputs, the preparatory work for ML is completed. The actual ML work is model selection, optimization, and evaluation. It is crucial to select a model that is suitable for the research research tasks from the many available models. For tasks with clear data labels, supervised learning models should be adopted. Supervised learning is commonly used to process regression and classification tasks. If the model output is a continuous variable, it is a regression process. If the output is discrete, it is a classification process. In unsupervised learning, clustering analysis methods can group material samples to help identify potential categories and similarities in material data and thus discover new promising materials.
Even after selecting a suitable model according to the task type, the model may not immediately show excellent predictive capabilities. Each ML algorithm has its specific hyperparameters that affect the model's learning ability and performance. Grid search can be used to tune hyperparameters, which involves setting a group of candidate values for each hyperparameter and testing every combination to select the combination with the best performance [59]. This requires massive computational resources, so initially, hyperparameters can be tuned within a reasonable range based on the understanding of the model. Taking the number of decision trees (N_estimators) in a random forest model as an example, for a relatively small dataset it can be set within 50-100, and for a larger dataset, the number of decision trees should be increased to 100-500. Researchers can also determine the potential optimal hyperparameter range more efficiently by manually inputting a small number of hyperparameters. Additionally, Bayesian optimization algorithms can find good hyperparameter combinations with relatively few iterations [60].
To select an optimal model, model evaluation is necessary to allow researchers to compare model performance. Commonly used evaluation metrics for classification tasks include Accuracy, Precision, Recall, F1 score and area under the receiver operating characteristic curve (AUC-ROC). Commonly used evaluation metrics for regression tasks include coefficient of determination (R2), mean squared error (MSE), root mean squared error (RMSE), mean absolute error (MAE), with the formulas shown in (1), (2), (3), (4). The dataset can be split into two parts according to different proportions for training and testing, with the training set used to establish the model and test set to validate real-world applicability. Overfitting occurs when model performance on the training set is significantly higher than on the test set, meaning the model has excessively learned random noise and specific patterns in the training data rather than truly learned the potential patterns in the data. Underfitting occurs when test set performance is significantly higher than training set performance [61]. A good model should not only learn good patterns on the training set but also show strong generalization capabilities on the test set, akin to a methodology moving from theory to practice. Cross-validation can be used to validate model stability by excluding accidental effects of a specific data split resulting in good performance [62]. It is a statistical method that involves partitioning the dataset into several subsets, with some used for training and some for testing model predictive capabilities. This approach not only helps evaluate model accuracy but also effectively prevents overfitting. Commonly, K-fold cross-validation randomly splits the dataset into K subsets. The model is trained K times using K-1 subsets for training and the remaining one for testing. The final performance evaluation is the average of these K test results, excluding accidental effects of the data partition.
|
|
(1) |
|
|
(2) |
|
|
(3) |
|
|
(4) |
where yi and
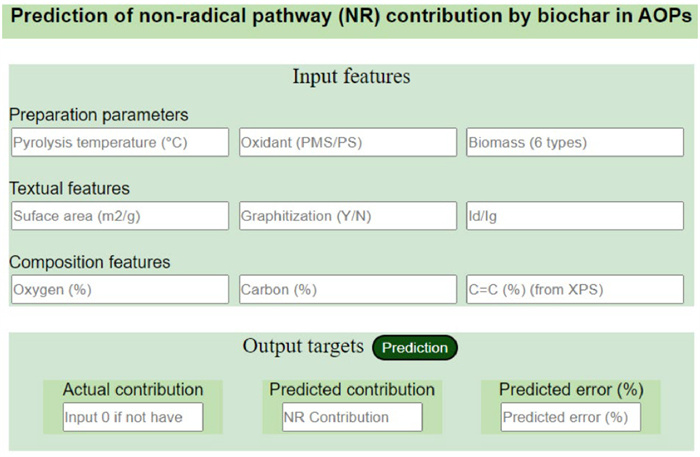
As ML models increase in complexity, improving model transparency and interpretability has become increasingly important. In the past, researchers only trained models through input and output without understanding the internal mechanisms, known as the "black box" problem. Due to the lack of transparency in black box models, researchers may face difficulties when analyzing model results. This issue is particularly prominent in fields like healthcare, finance, and research that require reliable explanations and validation of model decisions [63]. Now more and more model explanation methods are being widely adopted, mainly categorized into global interpretability (evaluating the contribution of all features to model output) and local interpretability (focusing on how specific features affect the final outcome, aiming to explain the behavior of complex models around particular data points) [64]. Shapley Additive exPlanations (SHAP) is the most commonly used model explanation method. Derived from the Shapley value concept in game theory to fairly distribute rewards among collaborative players, SHAP analyzes the weighted averages of different arrangements of feature subsets to determine each feature's contribution to the prediction. Through SHAP analysis, ML model predictions can be explained intuitively by understanding the influence of different features, promoting improvement in model explainability and understandability. This helps address the "black box" problem by enhancing transparency especially for high-stake domains like healthcare and finance that require validation and accountability in model decisions [65, 66]. Finally, to enhance the application capabilities of the model and increase the impact of the literature work, some researchers will establish websites to make the models available to the public. Wang et al. developed a Graphic User Interface in the webpage by integrating Python (version 3.7) and Flask (version 2.1.3) [21]. This webpage can predict the non-basic contribution in the BC-AOP system as demonstrated in Fig. 3, and its development is based on a well-trained eXtreme Gradient Boosting (XGB) model (online address:
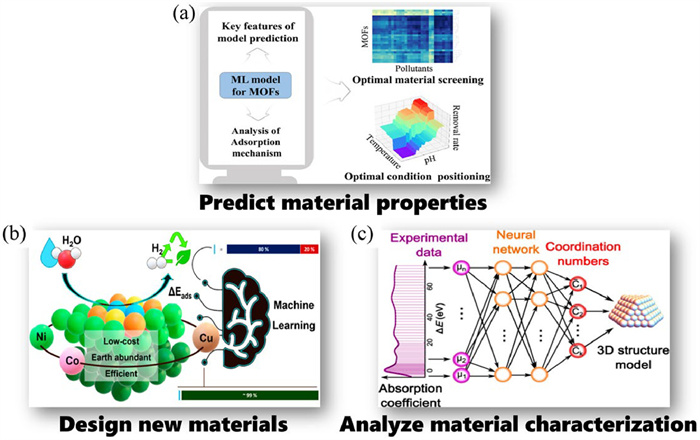
ML is widely used in environmental materials research, aiming to design and develop new materials in a cost-effective, time- and labor-saving manner, and to promote the development of materials science. In the application of ML, thorough exploration and appropriate representation of knowledge are crucial for enhancing model performance and interpretability. In materials science, knowledge can encompass physical laws, chemical structures, electronic properties, etc., and this knowledge can be represented in various ways, such as logical rules, graph structures. These representation schemes not only aid in constructing more accurate ML models but also enhance the interpretability of the models, making the prediction results easier for domain experts to understand and validate [67]. Furthermore, knowledge representation schemes have a significant impact on interpreting material data. Within the ML framework, combining material properties and performance with knowledge representation schemes can lead to a better understanding and prediction of material behaviors. For instance, by integrating physical and chemical knowledge of materials, ML models can more accurately predict the performance of battery materials, thus expediting the discovery and design process of new materials [68]. This approach not only improves the accuracy of model predictions but also enhances the generalization capability of the models, enabling them to handle a broader range of material data. ML can be applied to materials research in three main ways: predict material properties, design new materials, and analyze material characterization. ML technology can provide performance prediction of physicochemical properties, electronic structure (e.g., band gap, electron density), thermodynamic and mechanical properties (e.g., melting point, thermal conductivity, modulus of elasticity), for example, predicting the pollutant adsorption capacity of MOFs materials (Fig. 4a) [69]. ML can also be used to screen out new materials with potential applications or materials that meet specific performance requirements by simulating and predicting possible combinations of structures and compositions, such as identifying the most active HER catalysts in nickel-cobalt alloy bases by combining DFT and ML methods (Fig. 4b) [70]. For the analysis and interpretation of material characterization, ML can detect and identify various types of defects in the characterization image, and also make the interpretation and analysis of spectral data (e.g., photoelectron spectra, Raman spectra, infrared spectra) faster and more efficient. For instance, utilizing ML techniques to uncover hidden relationships between X-ray absorption near-edge structure (XANES) spectral features and catalyst geometric structures as shown in Fig. 4c [71]. Further details will be discussed in the following three subsections.
The properties of materials (including physical and chemical properties, electronic structural properties, thermodynamics, and mechanical properties) are crucial factors in determining their application potential and performance. Traditionally, the detection of these properties relies on experimental testing and physics-based computational methods such as DFT. However, these methods are often time-consuming, labor-intensive, and may not be sufficiently accurate or applicable when dealing with complex material systems. Due to its unique advantages in handling large datasets and discovering underlying patterns, ML has emerged as an innovative approach in predicting material properties. Predicting adsorption capacity is a significant application of ML in material property prediction, such as predicting the adsorption of carbon molecular sieves and activated carbon for N2, O2, and N2O [72], predicting the adsorption capacity of activated carbon for hydrophobic organic compounds [73], and for Congo red [74]. These predictions not only enhance the efficiency of materials design, but also drive the development of more efficient adsorbents in environmental applications. In the environment, there exist numerous pollutants that are difficult to degrade and detect, such as perfluorooctanoic acid (PFOA), which exhibits significant stability due to its unique structure. Utilizing ML algorithms can not only help understand the relationship between pollutant degradation and material properties but also provide a more convenient means for preliminary assessment of new materials' performance in future studies. Recently, Alnaimat et al. developed an RF model with an RMSE of 7.7 and an R2 of 0.965 based on variables in an electrochemical system to predict the efficiency of electro-oxidation for removing PFOA [75]. This model offers a reliable ML tool for environmental remediation work. Additionally, ML models predicting the degradation of various recalcitrant organic pollutants such as levofloxacin [76], norfloxacin [76], and tetracycline [77] have also been developed in recent years. Exploring the structure-property relationship is an important research direction in materials science. By delving into the relationship between the microstructure of materials and their macroscopic properties, scientists can better understand the characteristics of materials, thereby providing more effective guidance for material design, preparation, and application. When predicting a specific property, researchers often use experimental parameters as part of the input features, conducting feature analysis while optimizing parameters, and replacing the originally time-consuming parameter optimization experiments. The ML algorithm identifies patterns and dependencies in the operational parameters, which guides the optimization process to focus on the most influential factors. Li et al. accurately predicted the removal rates of different MOFs for typical environmental pollutants in aqueous environments using the XGB model. They also predicted the optimal Metal-Organic Frameworks and their best adsorption conditions for specific pollutants (such as pollutant concentration, pH value, solid-liquid ratio, and temperature), validating their accuracy through experiments [69].
Reverse material design refers to the targeted design of new materials by understanding the specific performance requirements of the desired material, and then using ML techniques to analyze and predict the correlation between the structure and properties of materials. This method helps address the high costs and time consumption associated with traditional trial-and-error methods, enabling the faster development of material solutions with specific functionalities and performances. In order to design and develop catalysts that simultaneously catalyze chlorobenzene and nitrogen oxides, Wu et al. collected data such as metal ion acidity and standard reduction potential. They built an ML model to find an analytical formula for metal ion acidity through symbolic regression and evaluated its redox capability using an ML regression model. Ultimately, they developed a bifunctional catalyst that catalyzes both chlorobenzene and nitrogen oxides, confirming its superiority in guiding the synthesis of materials through experiments [78]. To develop low-capital cost catalytic technologies in the natural gas field, catalysts combining high catalytic activity, mass transfer intensity, low hydraulic resistance, and high effective thermal conductivity were designed. Motaev et al. used ML methods to analyze a small amount of experimental data to determine the optimal yield conditions for light hydrocarbon fractions, highlighting the critical roles of Brunauer-Emmett-Teller (BET) surface area, pressure, and temperature. They successfully designed porous Co3O4 with a small admixture of Al2O3 particles, Co2O3 through the classical calcination of quartz sand impregnated with a cobalt nitrate solution, and a catalyst using a hydroxyapatite support material [79]. One of the major challenges in catalyst design is the development of a general descriptor model that accurately captures the intricate interactions between geometric and electronic structures [80]. Due to the vast number of variables in high-dimensional catalytic systems, a unified low-cost descriptor that optimizes various reactions and simultaneously resolves experimental and theoretical results has yet to emerge. Recently, Lin et al. unified the activity and selectivity predictions for multiple electrocatalytic reactions (O2 reduction reaction (ORR), O2 evolution reaction (OER), CO2 reduction reaction (CO2RR), and nitrogen reduction reaction (NRR)) by combining simple and readily accessible descriptors such as d-band theory and frontier orbitals [81]. This model, based on fewer than 4,500 DFT calculations, allows for the rapid prediction of highly active dual-atom sites for various reactions and products, replacing the need for over 50,000 high-throughput calculations. Using this model, the best ORR/OER bifunctional catalyst, Co-Co/Ir-Qv3, was predicted and synthesized. This catalyst demonstrated a high ORR half-wave potential of 0.941/0.937 V and a low OER overpotential of only 330/340 mV at 10 mA/cm2. These studies highlight the transformative impact of ML in material design, offering pathways to overcome traditional challenges. Future research can further refine these techniques, addressing current limitations such as data quality and computational demands, to provide more sophisticated solutions for environmental remediation.
Materials characterization is crucial in materials science as it involves detailed descriptions of a material's structure, composition, properties, and other characteristics using various technical methods. With the rapid increase in data volume and the continuous development of characterization techniques, traditional analytical methods are gradually proving to be inadequate. Due to its unique advantages in handling complex datasets and extracting useful information, ML has become a primary tool in materials characterization analysis and interpretation. This section will explore the applications of ML in materials characterization analysis and interpretation, including aspects such as image analysis [82], spectroscopic analysis [83], defect detection [84], and more.
Materials characterization often involves the use of microscopy techniques (such as scanning electron microscopy (SEM) and transmission electron microscopy (TEM)) to obtain nano-scale image information. ML has shown great potential in image processing and analysis. In a study analyzing SEM images, Hajilounezhad et al. developed a deep learning framework called CNTNet to predict the mechanical properties of carbon nanotube (CNT) forests in simulated images [82]. The research team generated images of CNT forests using physics-based simulation methods and trained deep learning classifiers on these images to predict their stiffness and buckling load performance, achieving a classifier accuracy of over 91%. The CNT-Net framework paves the way for high-throughput materials discovery using SEM images. Spectroscopic techniques (such as photoelectron spectroscopy [85], Raman spectroscopy [83]) are another important class of materials characterization tools used to analyze material composition and structural information. Through ML, the interpretation and analysis of spectroscopic data become faster and more efficient. X-ray absorption spectroscopy (XAS) is sensitive to the local atomic structure surrounding the absorbing metal species and offers the possibility to monitor in situ material transformations, making it one of the few experimental methods that can detect and analyze the structure-property relationship of metal nanoparticles, such as catalytic activity. Timoshenko et al. successfully unraveled the hidden relationships between gold and catalyst structures using supervised machine learning techniques applied to XANES spectroscopy, including average size, shape, and morphology of platinum nanoparticles [86]. This method is not only applicable to determining nanoparticle structures in real-time studies but can also be extended to other nanoscale systems. Additionally, this method allows real-time XANES analysis and is a promising approach for high-throughput and time-resolved studies. Tiny defects in materials (such as voids, microcracks) significantly impact material performance. Manual detection of these defects is often time-consuming and subject to large subjective biases. ML can automatically detect and identify defects in images through training, significantly improving efficiency and accuracy. Kajendirarajah et al. demonstrated an effective method by combining artificial neural networks (ANNs) with tip-enhanced Raman spectroscopy to analyze defects in CNTs based on the ID/IG ratio analysis generated from hyperspectral maps [84]. This research highlights the potential of combining ML with nanoscale research techniques to improve characterization and analysis methods.
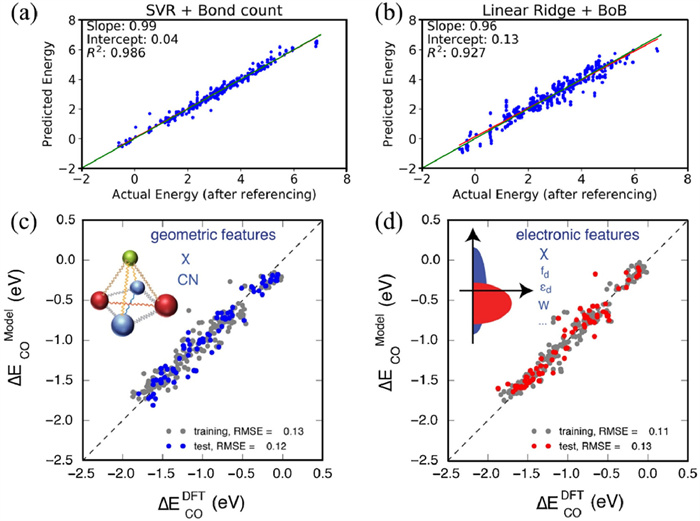
Evaluating the interaction between the catalyst surface and adsorbate molecules is a key aspect of the rational design of catalysts. Traditional calculations for the energy of various adsorptions and transition states using DFT are costly [87], while ML models can predict these by learning patterns within data, thus reducing computational costs. Chowdhury et al. analyzed the adsorption energy database of surface species involved in the decarboxylation and decarbonylation of acetic acid on eight single metal transition metal catalysts (Ni, Pt, Pd, Ru, Rh, Re, Cu, Ag) using ML techniques, finding that ML models can accurately predict the adsorption energies of various species on other metal surfaces, especially in dealing with complex chemical reactions and multiple metal surfaces. The authors also found that modeling with simple coordinate-free species descriptors (e.g., bond counts C-C, C-H) (R2 = 0.986, intercept = 0.04) (Fig. 5a) achieved a better fitting effect compared to complex coordinate-based descriptors (bag-of-bonds (BoB) techniques) (R2 = 0.9827, intercept = 0.13) (Fig. 5b) [30]. Li et al. developed an ML chemical adsorption model based on ANNs and similarly found that using more readily accessible geometric features (including local electro-negativity and effective coordination number of adsorption sites) to predict the adsorption energy of CO on metal surfaces can demonstrate better performance, with a prediction error of 0.12 eV (Fig. 5, Fig. 5) [31]. This work can aid in screening multi-metal alloys for CO2 electro-reduction and guide the synthesis of promising candidate alloys with lower overpotentials. In Wang et al.'s work, they found that the electric dipole moment, as a convenient and accurate descriptor, is key for rapidly and accurately predicting molecular adsorption energies and charge transfers [33]. Additionally, descriptors rooted in the d-band chemisorption theory and its recent developments, including sp-band and d-band characteristics, have been used to predict the adsorption energies of metal surfaces [88].
Metal catalyst surfaces consist of various surface sites, with their catalytic activity typically dominated by a few specific active sites. Identifying these active sites is a challenging task, and designing them is crucial for achieving high-performance heterogeneous catalysts. Jinnouchi et al. proposed a universal ML approach using local similarity kernels, allowing for the exploration of catalytic activity based on local atomic configurations. They then applied this to the direct NO decomposition on Rh-Au alloy nanoparticles [89]. This method can effectively predict the energetics of catalytic reactions on nanoparticles, providing detailed information about the structure, size, and composition-related catalytic activity of active sites. The structure sensitivity of metal catalysts depends on molecules and reactions; finding inherent active sites for catalysis and the structure sensitivity for rational catalyst design remains a significant challenge [90-92]. Shu et al. employed interpretable ML methods and a strategy combining first-principles calculations to reveal the structure sensitivity of metal catalysts. By constructing a two-dimensional descriptor containing topological structure descriptors, combined with reaction energy terms, they successfully predicted reaction barriers for various chemical bonds in metal catalysts [93]. This method not only exhibits high accuracy but also generalizes well for datasets containing different symmetries, bond orders, and steric hindrances among various chemical bonds.
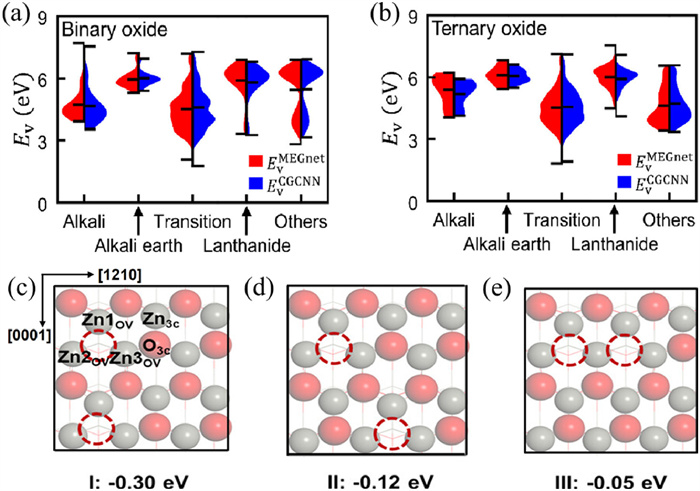
High-valent metal oxides (such as Fe(Ⅳ) [94, 95], Fe(Ⅴ) [96], Mn(Ⅳ) [97, 98], and Mn(Ⅴ) [99]) are considered as key in selective partial oxidation. Computational screening can help explain which chemical environments can lead to stable metal oxides, and high-throughput screening can go beyond a few compounds in experiments to design spaces across the entire periodic table. However, in isolated, uncoordinated metal sites, linear scaling relationships are often distorted or broken, weakening the predictive power of linear scaling relationships [100]. Therefore, non-linear ML models are the most promising to accelerate high-throughput screening [101]. To overcome the reactivity variations of stable metal-oxides with changing metal spin states and to complicate the development of structure-property relationships, Nandy et al. successfully predicted the formation energies of metal-oxide intermediates under a range of first-row metal, oxidation state, and spin states using ANN ML models. Through importance analysis of features, they found that non-local electronic ligand properties dominate compared to other transition metal complex properties (such as spin state or ionization potential) [101]. Due to the lack of detailed computations, understanding the surface coverage under reaction conditions and the presence of numerous possible reaction sites for O* and HO* on metal oxide materials is much more complex than studying transition metal catalysts [102]. Back et al. used ML to develop an automated and high-throughput method to predict the O2 evolution reaction overpotential on any oxide surface. They found that several active sites and surface ratios of low-index IrO2 and IrO3 are more active than the most stable rutile (110), except for rutile (110). They identified promising active sites in IrO2 and IrO3 with theoretical overpotentials 0.2 V higher than rutile (110) [103]. Oxygen vacancies (VO) are fundamental and inherent defects in metal oxides, whose formation is influenced by growth conditions and annealing processes. The concentration and distribution of oxygen vacancies determine the catalytic activity of metal oxides. Park et al. used Materials Graph Network and Crystal Graph Convolutional Neural Networks to establish an ML model for predicting the oxygen vacancy formation energy, finding that most of the binary oxides (Fig. 6a) and ternary oxides (Fig. 6b) composed of pure lanthanide metals were found to have large eV values (> 5 eV) [104]. Han et al. conducted more detailed work and found that in the presence of VOs, the surface structure transitions from the wurtzite phase to the body-centered-tetragonal phase, undergoing structural changes. Due to the additional electron enrichment near VO from the departure of oxygen, leading to structural deformation, the activity of new VO reaction sites gradually increases. Through calculations, it was found that OV mainly exists in three arrangements: (i) Clustered along the lower energy [0001] direction (Fig. 6c); (ii) located in isolated positions (Fig. 6d); (iii) clustered along the [12̅10] direction (Fig. 6e). Different arrangements will affect the dissociation barrier of the C-O bond, significantly influencing the reactivity of the cleavage reaction [105]. Ji et al. established an ML model using a support vector machine (SVM) and found that doping transition metals promotes the formation of oxygen vacancies, where Au and Ta can effectively lower the energy of intermediates and are considered the most effective dopants [106].
In the field of environmental remediation, ML has significantly optimized the design and screening process of metal catalysts and its oxides. Through ML models, researchers can accurately predict key data such as adsorption energies and formation energies, reducing the high costs associated with traditional computational methods. Furthermore, ML methods have helped reveal the impact of various physical and chemical properties on catalytic activity and stability, identifying active sites of efficient catalysts and oxygen vacancy distributions, aiding in the development of more efficient catalysts. Overall, ML demonstrates enormous potential in improving the accuracy of catalyst performance predictions and promoting the design of novel catalytic materials.
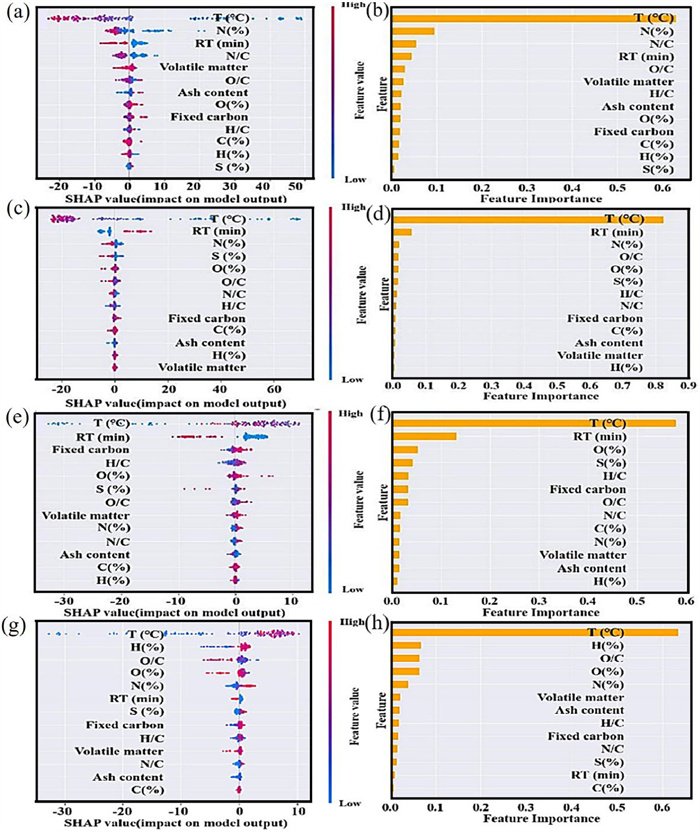
Carbon-based materials, including BC, CNT, and graphene, have garnered significant attention in the field of environmental applications due to their advantages of metal-free character, acid and alkali resistance, biocompatibility, environmental friendliness, and multi-functionality. These materials are extensively utilized in carbon sequestration, soil remediation, climate change mitigation, catalytic reactions, wastewater treatment, and energy storage, making them ideal candidates to address a variety of environmental challenges [107]. ML has exhibited great potential in the rational design of carbon-based materials. BC, an emerging carbonaceous material, is characterized by its large specific surface area, rich functional groups, and low economic cost. Its performance is mainly influenced by internal and external factors. Internal factors refer to the physical and chemical properties of the BC itself. In recent years, ML methods have been widely applied to predict the specific surface area [108], total pore volume [108], adsorption capacity [109], and composition [110] of BC, successfully aiding in the synthesis of high-performance BC catalysts. External conditions are the experimental conditions and parameters that can enable BC to exhibit excellent performance. For instance, Wang et al. used an XGB model to discover through experiments that simultaneously adjusting the calcination temperature and biomass precursor can effectively enhance non-radical degradation pathways [21]. Leng et al. utilized SHAP analysis to interpret and analyze the model, discovering that pyrolysis temperature is the most significant factor in predicting nitrogen recovery (char-nitrogen yield) in BC (Figs. 7a and b), amine-N (Fig. 7, Fig. 7), pyrrolic-N (Fig. 7, Fig. 7), and pyridinic-N (Fig. 7, Fig. 7). Similarly, CNTs, with their high specific surface area and strong adsorption capabilities, are used in pollutant adsorption, sensing, and monitoring [111, 112]. To achieve low-cost and controllable growth of CNTs, Chu et al. synthesized CNT arrays with different morphologies and performances by altering roughness, immersion time, and preheating temperature. They then utilized ML techniques to develop a mathematical model optimizing both tube diameter and dispersity, providing new insights into the large-scale and continuous production of CNT arrays [113]. Understanding and controlling the influence of structure and experimental conditions on properties has been a longstanding barrier to the widespread adoption of CNTs in many applications. The successful integration of ML has overcome this obstacle, guiding the design of CNTs with higher stiffness, greater buckling load [82], and lower equivalent sheet resistance [114]. Graphene, the most explored representative of two-dimensional materials, benefits significantly from the integration with ML methods. Due to the inherent chemical inertness of graphene limiting its application scope, heteroatom doping can significantly enhance its properties [115, 116]. Addressing the complexity brought by structural and chemical freedom in catalyst design, Dieb et al. employed ML in combination with atomic simulation to discover stable structures of B-graphene with a boron concentration as high as 31.25%. They also found that B-graphene exhibited semiconductor behavior at a doping concentration of 25% [116]. Rational control of defects in graphene is crucial, as defects can modulate graphene's physicochemical properties, significantly altering its electronic, optical, thermal, and mechanical performances [117, 118]. To deeply understand the impact of defects, Motevalli et al. leveraged ML based on over 20,000 electronic structure simulations and 220 features, revealing that the presence of C-C bond defects in graphene is decisively influenced by the presence of oxygen, while the concentration and distribution of hydrogen atoms determine the frequency of bond breakage [119].
ML has greatly propelled the design and optimization of non-metal catalysts in environmental remediation, particularly carbon-based materials. Through ML methods, researchers can predict and control the physical and chemical properties of carbon-based materials such as BC, CNTs, and graphene, enhancing their performance in areas like pollutant adsorption, energy storage, and catalytic reactions. These studies not only optimize material fabrication processes but also improve the accuracy of performance predictions, fostering the widespread application of carbon-based materials.
SACs have gained widespread attention in the field of environmental remediation in recent years due to their unique catalytic properties and high atomic efficiency. Compared to traditional multi-atom catalysts, SACs offer higher utilization of active sites, saving the use of expensive metal resources. Additionally, because the single atoms are dispersed on a carrier, interactions between atoms are reduced, providing SACs with higher stability in catalytic reactions. These outstanding characteristics demonstrate significant potential for the application of SACs in environmental protection and pollution control. In the realm of atmospheric pollution control, SACs can efficiently catalyze the conversion of volatile organic compounds (VOCs), nitrogen oxides (NOx), and sulfur dioxide (SO2), significantly reducing pollutant concentrations in the air. In the field of water treatment, SACs exhibit excellent catalytic performance, capable of efficiently degrading organic pollutants and heavy metal ions in water bodies, thereby enhancing water purification efficiency. In recent years, ML technology has shown significant potential in the design and development of SACs, bringing revolutionary changes to traditional empirical and experimental catalyst design methods and significantly promoting the application and development of SACs in the ORR [120], OER [121], CO2RR [122], NRR [17], and hydrogen evolution reaction (HER) [123].
ORR is a crucial electrochemical reaction and a key step in many energy conversion and storage technologies. To better understand the formation and stability mechanisms of SACs and design more economically efficient ORR-SACs, Wu et al. proposed rational design guidelines for the ORR activity of transition metal single atoms on defective carbon surfaces. Utilizing ML to predict the ORR activity of all transition metal SACs is 130,000 times faster than DFT calculations with an error rate of only 8.33%. They found that the dispersion of metal nanoparticles (NPs) into stable single-atom arrays on defective carbon surfaces is controlled by the NP decomposition energy barrier and the diffusion barrier of single atoms on the carbon surface [124]. Rechargeable metal-air batteries are characterized by high energy density, low self-discharge rates, and low emissions, holding significant potential to reduce reliance on fossil fuels [125]. ORR plays a discharge role in these batteries, while OER plays an equally crucial charging role. To find efficient and stable OER catalysts, Fu et al. based on computational data from 75 SACs, utilized a Gradient Boosting Regression (GBR) model to capture the potential relationship between physical properties of SACs and overpotentials, revealing the impact of features such as atomic radius, d/f orbital electron count, and Bader electronegativity on activity. They ultimately designed four ORR catalysts, nine OER catalysts, and five ORR/OER bifunctional electrocatalysts, comprehensively verifying their stability [126]. Converting CO2 into useful chemicals or fuels (e.g., CO2RR) is an effective method to address the greenhouse effect, bringing significant environmental and economic benefits [127]. However, a major challenge of CO2RR is the competition with the accompanying HER, leading to reduced efficiency and selectivity [128]. To design SACs that are efficient and suppress the HER process, Zhang et al. employed a combined ML and DFT approach, utilizing multiple effective indicators such as catalyst stability, hydrogen evolution inhibition capability, and CO adsorption strength to screen out two efficient CO2RR-SACs [127]. Among them, Mn-NC2 has the lowest reaction barrier and the best catalytic activity, making it an ideal candidate for high-activity CO2RR, while Pt-NC2 exhibits high selectivity and catalytic activity for *CHO formation, with the best inhibition effect on HER. Developing low-energy, environmentally friendly NRR technologies is crucial for reducing fossil fuel usage and greenhouse gas emissions. Similar to the situation with CO2RR, the NRR process is also affected by competition from HER, leading to decreased efficiency [129]. Zafari et al. used a deep neural network (DNN) to predict the adsorption and free energy of boron (B) doped graphene SACs in the NRR process and identified CrB3C1 as an efficient electrocatalyst with a low overpotential of 0.13V and high selectivity for NRR [130]. Hydrogen is a clean, renewable energy source, and HER in water electrolysis is a method for producing clean hydrogen using electrical energy. Liu et al. demonstrated through ML regression that the electronic affinity and first ionization energy of SACs are closely related to the HER process, providing theoretical guidance for HER-SACs [131].
ML has demonstrated immense potential in the design and development of SACs, significantly enhancing the efficiency and stability of catalysts. ML methods enable rapid prediction and optimization of SACs' performance in various reactions, including ORR, OER, CO2RR, NRR, and HER, providing efficient and cost-effective catalytic solutions for atmospheric pollution control and water treatment. In conclusion, ML technology has greatly facilitated the application and advancement of SACs in environmental protection and pollution control.
We mainly focused on the application of ML in the research and development process of common materials in the environmental field. So far, the application of ML technology in the field of material design has demonstrated its enormous potential. By extracting complex nonlinear relationships from large amounts of data, ML has not only improved the accuracy and efficiency of material performance prediction, achieving high-throughput screening and feature importance analysis, but also provided innovative methods and tools for the discovery and design of new materials. However, there are still many challenges and shortcomings that researchers need to address. Here, we summarize some of the current shortcomings of ML-guided material design and possible solutions, hoping to bring some new breakthroughs to the application of ML:
(1) High-quality and accurately labeled data are key to training effective models. However, in many cases, experimental data may contain noise, or high-quality datasets may be scarce. Therefore, how to effectively preprocess data, improve data quality, and extract useful information from limited data is an urgent issue that needs to be addressed. At the same time, data sharing and open data policies help promote research and development of ML algorithms, reducing the difficulty of data acquisition. Furthermore, the effective collection of thousands of continuously updated literature data is challenging. Recently, the development of text mining, natural language processing techniques [48], and the cDA-DK model [132] has enabled the automatic and accurate extraction of data information from the literature. However, further development of data mining models that cater to different data needs and types will help maximize the collection and utilization of existing literature data in the future.
(2) Model interpretability is crucial for ensuring the reliability and controllability of material design. Many deep learning models are considered "black box" models due to their complex structures, making it difficult to explain their underlying mechanisms. Material science research requires interpretable models to reveal the fundamental reasons for material behavior. Addressing the challenge of model interpretability will continue to be an important research direction in future material design studies.
(3) Multiscale modeling aims to integrate information from different scales to more comprehensively describe the structure, properties, and behavior of materials. Multiscale modeling often requires the integration of data from different scales, including atomic-level structural information, lattice-level properties, and macroscopic experimental data. How to effectively integrate this data to establish a comprehensive model is a challenge. Multiscale modeling often requires large computational resources and time, especially when involving atomic-level simulations and quantum mechanical calculations. Efficient multiscale modeling to reduce computational costs is a challenge that researchers need to address.
(4) The emergence of large-scale generative artificial intelligence, such as Generative Adversarial Networks (GANs) [133] and Variational Autoencoders (VAEs) [134], enables the generation of novel material structures by learning from existing data, thus exploring vast design spaces beyond human intuition. Liu et al. found that large language models like ChatGPT can effectively address materials-related issues, such as generating new materials, solving differential equations, and answering common queries [135]. However, due to the current lack of specialized knowledge in the materials domain, the application of these models is still limited to basic solutions and requires continuous optimization in question-answer dialogues to meet specific needs. In the future, collaborative efforts among environmental domain experts, materials scientists, and artificial intelligence specialists are needed to develop large-scale generative artificial intelligence models that integrate domain-specific knowledge. This collaboration aims to propel the development of environmental remediation materials into a new phase.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yunzhe Zheng: Writing – original draft, Visualization, Validation, Conceptualization. Si Sun: Investigation, Formal analysis. Jiali Liu: Formal analysis. Qingyu Zhao: Investigation. Heng Zhang: Project administration. Jing Zhang: Conceptualization. Peng Zhou: Project administration. Zhaokun Xiong: Supervision. Chuan-Shu He: Writing – review & editing, Supervision, Project administration. Bo Lai: Writing – review & editing, Supervision, Resources.
The authors wish to thank the National Natural Science Foundation of China (Nos. 52370083 and 52170088), Sichuan Science and Technology Program (No. 2024NSFTD0014) and Key R & D Program of Heilongjiang Province (No. 2023ZX02C01) for financial support.
Y. Zheng, X. Wang, Y. Dong, et al., Appl. Catal. B: Environ. Energy 361 (2025) 124580. doi: 10.1016/j.apcatb.2024.124580
H. Zhao, X. Tan, H. Chai, et al., Chin. Chem. Lett. (2024) 110571. DOI: https://doi.org/ 10.1016/j.cclet.2024.110571.
S. Sun, S. Song, S. Yang, et al., Chin. Chem. Lett. 35 (2024) 109242. doi: 10.1016/j.cclet.2023.109242
M.J. Wang, J. Yang, L. Peng, et al., Chin. Chem. Lett. (2024) 110573. DOI: https://doi.org/ 10.1016/j.cclet.2024.110573.
Y. Liu, T. Zhao, W. Ju, et al., J. Materiomics. 3 (2017) 159-177. doi: 10.4142/jvs.2017.18.2.159
Y. Sun, P. Zhou, M. Sun, et al., ACS Catal. 14 (2024) 6525-6534. doi: 10.1021/acscatal.4c00048
G.R. Schleder, A.C.M. Padilha, C.M. Acosta, et al., J. Phys. : Mater. 2 (2019) 032001. doi: 10.1088/2515-7639/ab084b
M. Wu, D.C. Kozanoglu, C. Min, et al., Adv. Eng. Inform. 50 (2021) 101368. doi: 10.1016/j.aei.2021.101368
M.M. Ahsan, S.A. Luna, Z. Siddique, Healthcare 10 (2022) 541. doi: 10.3390/healthcare10030541
S. Dara, S. Dhamercherla, S.S. Jadav, et al., Artif. Intell. Rev. 55 (2022) 1947-1999. doi: 10.1007/s10462-021-10058-4
R. Yang, J. Hu, Z. Li, et al., Atmos. Environ. 338 (2024) 120797. doi: 10.1016/j.atmosenv.2024.120797
I.H. Sarker, A.S.M. Kayes, S. Badsha, et al., J. Big Data 7 (2020) 41. doi: 10.1186/s40537-020-00318-5
K. Noda, Y. Yamaguchi, K. Nakadai, et al., Appl. Intell. 42 (2015) 722-737. doi: 10.1007/s10489-014-0629-7
B.M. Henrique, V.A. Sobreiro, H. Kimura, Expert Syst. Appl. 124 (2019) 226-251. doi: 10.1016/j.eswa.2019.01.012
X. Zhao, L. Wang, Y. Pei, J. Phys. Chem. C 125 (2021) 22513-22521. doi: 10.1021/acs.jpcc.1c05734
Z. Wang, H. Zhang, J. Li, Nano Energy 81 (2021) 105665. doi: 10.1016/j.nanoen.2020.105665
Z.W. Chen, Z. Lu, L.X. Chen, et al., Chem. Catalysis 1 (2021) 183-195.
J. Feng, Y. Ji, Y. Li, J. Mater. Chem. A 11 (2023) 14195-14203. doi: 10.1039/d3ta01883k
R. Wang, H. Chen, Z. He, et al., Environ. Sci. Technol. 58 (2024) 16867-16876. doi: 10.1021/acsnano.4c02631
K.N. Palansooriya, J. Li, P.D. Dissanayake, et al., Environ. Sci. Technol. 56 (2022) 4187-4198. doi: 10.1021/acs.est.1c08302
R. Wang, S. Zhang, H. Chen, et al., Environ. Sci. Technol. 57 (2023) 4050-4059. doi: 10.1021/acs.est.2c07073
L. Zhou, S. Pan, J. Wang, et al., Neurocomputing 237 (2017) 350-361. doi: 10.1016/j.neucom.2017.01.026
J. Orbach, Arch. Gen. Psychiatry 7 (1962) 218-219. doi: 10.1001/archpsyc.1962.01720030064010
T. Cover, P. Hart, IEEE Trans. Inf. Theory 13 (1967) 21-27. doi: 10.1109/TIT.1967.1053964
D. Beattie, Int. J. Man-Mach. Stud. 1 (1969) 331-332. doi: 10.1016/S0020-7373(69)80028-3
L. Liu, Y. Wang, W. Chi, IEEE Access (2020) 1-1. DOI:http://doi.org/10.1109/ACCESS.2020.3021590.
D.W. Otter, J.R. Medina, J.K. Kalita, IEEE Trans. Neural Netw. Learn. Syst. 32 (2021) 604-624. doi: 10.1109/tnnls.2020.2979670
J.X. Chen, Comput. Sci. Eng. 18 (2016) 4-7.
A.M. Żurański, J.I. Martinez Alvarado, B.J. Shields, et al., Acc. Chem. Res. 54 (2021) 1856-1865. doi: 10.1021/acs.accounts.0c00770
A.J. Chowdhury, W. Yang, E. Walker, et al., J. Phys. Chem. C 122 (2018) 28142-28150. doi: 10.1021/acs.jpcc.8b09284
Z. Li, X. Ma, H. Xin, Catal. Today 280 (2017) 232-238. doi: 10.1016/j.cattod.2016.04.013
X. Ma, Z. Li, L.E. Achenie, et al., J. Phys. Chem. Lett. 6 (2015) 3528-3533. doi: 10.1021/acs.jpclett.5b01660
X. Wang, S. Ye, W. Hu, et al., J. Am. Chem. Soc. 142 (2020) 7737-7743. doi: 10.1021/jacs.0c01825
N. Artrith, Z. Lin, J.G. Chen, ACS Catal. 10 (2020) 9438-9444. doi: 10.1021/acscatal.0c02089
J. Park, I. Chung, H. Jeong, et al., Appl. Catal. B: Environ. Energy 361 (2025) 124622. doi: 10.1016/j.apcatb.2024.124622
M. Tamtaji, H. Gao, M.D. Hossain, et al., J. Mater. Chem. A 10 (2022) 15309-15331. doi: 10.1039/d2ta02039d
M.E. Günay, R. Yıldırım, Chem. Eng. J. 140 (2008) 324-331. doi: 10.1016/j.cej.2007.09.047
H.B. Barlow, Neural Comput. 1 (1989) 295-311. doi: 10.1162/neco.1989.1.3.295
Z. -Z. Shi, W. -P. Li, W. -J. Kang, et al., ACS Catal. 13 (2023) 9181-9189. doi: 10.1021/acscatal.3c01400
M.L. Raymer, W.F. Punch, E.D. Goodman, et al., IEEE Trans. Evol. Comput. 4 (2000) 164-171. doi: 10.1109/4235.850656
K. Vougas, T. Sakellaropoulos, A. Kotsinas, et al., Pharmacol. Ther. 203 (2019) 107395. doi: 10.1016/j.pharmthera.2019.107395
J.E. van Engelen, H.H. Hoos, Mach. Learn. 109 (2020) 373-440. doi: 10.1007/s10994-019-05855-6
S. Wang, C. Qian, S. Zhou, ACS Appl. Mater. Interfaces 15 (2023) 40656-40664. doi: 10.1021/acsami.3c08535
Y. Roh, G. Heo, S.E. Whang, IEEE Trans. Knowl. Data Eng. 33 (2021) 1328-1347. doi: 10.1109/tkde.2019.2946162
S. Shi, S. Sun, S. Ma, et al., J. Inorg. Mater. 37 (2022) 1311-1325. doi: 10.15541/jim20220149
C. -Y. Chang, M. -T. Hsu, E.X. Esposito, et al., J. Chem Inf. Model. 53 (2013) 958-971. doi: 10.1021/ci4000536
Y. Liu, S. Ma, Z. Yang, et al., J. Chin. Ceram. Soc. 51 (2023) 427-437. doi: 10.1007/s00404-023-07044-2
Y. Liu, X. Zou, Z. Yang, et al., J. Chin. Ceram. Soc. 50 (2022) 863.
S. García, S. Ramírez-Gallego, J. Luengo, et al., Big. Data. Anal. 1 (2016) 9. doi: 10.1186/s41044-016-0014-0
S.J. Hadeed, M.K. O'Rourke, J.L. Burgess, et al., Sci. Total Environ. 730 (2020) 139140. doi: 10.1016/j.scitotenv.2020.139140
T. Verdonck, B. Baesens, M. Óskarsdóttir, et al., Mach. Learn. 113 (2024) 3917-3928. doi: 10.1007/s10994-021-06042-2
V. Bolón-Canedo, N. Sánchez-Maroño, A. Alonso-Betanzos, Prog. Artif. Intell. 5 (2016) 65-75. doi: 10.1007/s13748-015-0080-y
M. Tamtaji, S. Chen, Z. Hu, et al., J. Phys. Chem. C 127 (2023) 9992-10000. doi: 10.1021/acs.jpcc.3c00765
N.M. Sirakov, T. Shahnewaz, A. Nakhmani, Electronics 13 (2024) 282. doi: 10.3390/electronics13020282
A. Tharwat, T. Gaber, A. Ibrahim, et al., AI Commun. 30 (2017) 169-190. doi: 10.3233/AIC-170729
B. Conroy, R. Nayak, A.L.R. Hidalgo, et al., Microporous Mesoporous Mater. 335 (2022) 111802. doi: 10.1016/j.micromeso.2022.111802
Y. Liu, X. Zou, S. Ma, et al., Acta Mater. 238 (2022) 118195. doi: 10.1016/j.actamat.2022.118195
Y. Liu, J. -M. Wu, M. Avdeev, et al., Adv. Theory Simul. 3 (2020) 1900215. doi: 10.1002/adts.201900215
M.Y. Shams, A.M. Elshewey, E. -S.M. El-kenawy, et al., Multimed. Tools Appl. 83 (2024) 35307-35334.
D.P. Kuttichira, S. Gupta, D. Nguyen, et al., Knowl. -Based Syst. 241 (2022) 108238. doi: 10.1016/j.knosys.2022.108238
S. Pothuganti, Int. J. Adv. Res. Electr. Electron. Instrum. Eng 7 (2018) 3692-3695.
I. Tougui, A. Jilbab, J. El Mhamdi, Healthc. Inform. Res. 27 (2021) 189-199. doi: 10.4258/hir.2021.27.3.189
N. Khan, M. Nauman, A.S. Almadhor, et al., IEEE Access 12 (2024) 90299-90316. doi: 10.1109/access.2024.3420415
H. Hakkoum, A. Idri, I. Abnane, Eng. Appl. Artif. Intell. 131 (2024) 107829. doi: 10.1016/j.engappai.2023.107829
M. Lu, F. Gao, Y. Tan, et al., ACS Appl. Mater. Interfaces 16 (2024) 3593-3604. doi: 10.1021/acsami.3c18490
Q. Fu, T. Xu, D. Wang, et al., Carbon 223 (2024) 119045. doi: 10.1016/j.carbon.2024.119045
L. v. Rueden, S. Mayer, K. Beckh, et al., IEEE Trans. Knowl. Data Eng. 35 (2023) 614-633.
Y. Liu, B. Guo, X. Zou, et al., Energy Storage Mater. 31 (2020) 434-450. doi: 10.3390/ijerph17020434
J. Li, J. Wang, H. Mu, et al., ACS ES & T Eng. 3 (2023) 1258-1266.
N.K. Pandit, D. Roy, S.C. Mandal, et al., J. Phys. Chem. Lett. 13 (2022) 7583-7593. doi: 10.1021/acs.jpclett.2c01401
J. Timoshenko, D. Lu, Y. Lin, et al., J. Phys. Chem. Lett. 8 (2017) 5091-5098. doi: 10.1021/acs.jpclett.7b02364
H. Mashhadimoslem, A. Ghaemi, Environ. Sci. Pollut. Res. 30 (2023) 4166-4186. doi: 10.1007/s11356-022-22508-9
H. Lee, Y. Choi, Chemosphere 350 (2024). Doi: https://doi.org/ 10.1016/j.chemosphere.2023.141003.
R. Ahmad Aftab, S. Zaidi, A. Aslam Parwaz Khan, et al., Alex. Eng. J. 71 (2023) 355-369. doi: 10.1016/j.aej.2023.03.055
S. Alnaimat, O. Mohsen, H. Elnakar, J. Environ. Manage. 370 (2024) 122857. doi: 10.1016/j.jenvman.2024.122857
S. Jiang, W. Xu, Q. Xia, et al., J. Hazard. Mater. 471 (2024) 134309. doi: 10.1016/j.jhazmat.2024.134309
N. Huang, K. Gao, W. Yang, et al., Bioresour. Technol. 361 (2022) 127710. doi: 10.1016/j.biortech.2022.127710
J. Wu, J. Zhang, G. Qian, et al., Appl. Catal. A: Gen. 668 (2023) 119487. doi: 10.1016/j.apcata.2023.119487
K. Motaev, M. Molokeev, B. Sultanov, et al., Ind. Eng. Chem. Res. 62 (2023) 20658-20666. doi: 10.1021/acs.iecr.3c03147
X. Chang, Z. -J. Zhao, Z. Lu, et al., Nat. Nanotechnol. 18 (2023) 611-616. doi: 10.1038/s41565-023-01344-z
X. Lin, X. Du, S. Wu, et al., Nat. Commun. 15 (2024) 8169. doi: 10.1038/s41467-024-52519-8
T. Hajilounezhad, R. Bao, K. Palaniappan, et al., npj Comput. Mater. 7 (2021) 134. doi: 10.1038/s41524-021-00603-8
P. Solís-Fernández, H. Ago, ACS Appl. Nano Mater. 5 (2022) 1356-1366. doi: 10.1021/acsanm.1c03928
U. Kajendirarajah, M. Olivia Aviles, F. Lagugne-Labarthet, Phys. Chem. Chem. Phys. 22 (2020) 17857-17866. doi: 10.1039/d0cp02950e
S. Xiang, P. Huang, J. Li, et al., Phys. Chem. Chem. Phys. 24 (2022) 5116-5124. doi: 10.1039/d1cp05513e
J. Timoshenko, D. Lu, Y. Lin, et al., J. Phys. Chem. Lett. 8 (2017) 5091-5098. doi: 10.1021/acs.jpclett.7b02364
D. -Y. Xing, X. -D. Zhao, C. -S. He, et al., Chin. Chem. Lett. 35 (2024) 109436. doi: 10.1016/j.cclet.2023.109436
Z. Li, S. Wang, W.S. Chin, et al., J. Mater. Chem. A 5 (2017) 24131-24138. doi: 10.1039/C7TA01812F
R. Jinnouchi, R. Asahi, J. Phys. Chem. Lett. 8 (2017) 4279-4283. doi: 10.1021/acs.jpclett.7b02010
M.T.M. Koper, Nanoscale 3 (2011) 2054-2073. doi: 10.1039/c0nr00857e
J. Lim, C. -Y. Liu, J. Park, et al., ACS Catal. 11 (2021) 7568-7577. doi: 10.1021/acscatal.1c01413
H. Zhang, M. Jin, Y. Xiong, et al., Acc. Chem. Res. 46 (2013) 1783-1794. doi: 10.1021/ar300209w
W. Shu, J. Li, J.X. Liu, et al., J. Am. Chem. Soc. 146 (2024) 8737-8745. doi: 10.1021/jacs.4c01524
T. Chantarojsiri, J.W. Ziller, J.Y. Yang, Chem. Sci. 9 (2018) 2567-2574. doi: 10.1039/c7sc04486k
X. Dai, T. Liu, Y. Du, et al., Chin. Chem. Lett. (2024) 110548. DOI: https://doi.org/ 10.1016/j.cclet.2024.110548.
F.T. de Oliveira, A. Chanda, D. Banerjee, et al., Science 315 (2007) 835-838. doi: 10.1126/science.1133417
T.H. Parsell, R.K. Behan, M.T. Green, et al., J. Am. Chem. Soc. 128 (2006) 8728-8729. doi: 10.1021/ja062332v
H. Wang, M. Luo, Y. Wang, et al., Chin. Chem. Lett. (2024) 110348. DOI: https://doi.org/ 10.1016/j.cclet.2024.110348.
R. Gupta, T. Taguchi, B. Lassalle-Kaiser, et al., PNAS Nexus 112 (2015) 5319-5324. doi: 10.1073/pnas.1422800112
T.Z.H. Gani, H.J. Kulik, ACS Catal. 8 (2018) 975-986. doi: 10.1021/acscatal.7b03597
A. Nandy, J. Zhu, J.P. Janet, et al., ACS Catal. 9 (2019) 8243-8255. doi: 10.1021/acscatal.9b02165
M. Bajdich, M. García-Mota, A. Vojvodic, et al., J. Am. Chem. Soc. 135 (2013) 13521-13530. doi: 10.1021/ja405997s
S. Back, K. Tran, Z.W. Ulissi, ACS Catal. 9 (2019) 7651-7659. doi: 10.1021/acscatal.9b02416
S. Park, N. Lee, J.O. Park, et al., ACS Mater. Lett. 6 (2023) 66-72. doi: 10.54912/jci.2022.0022
Y. Han, J. Xu, W. Xie, et al., ACS Catal. 13 (2023) 5104-5113. doi: 10.1021/acscatal.3c00658
Y. Ji, P. Liu, Y. Huang, Phys. Chem. Chem. Phys. 25 (2023) 5827-5835. doi: 10.1039/d2cp04635k
S. Mallakpour, M. Lormahdiabadi, Langmuir 38 (2022) 4065-4076. doi: 10.1021/acs.langmuir.2c00091
H. Li, Z. Ai, L. Yang, et al., Bioresour. Technol. 369 (2023) 128417. doi: 10.1016/j.biortech.2022.128417
W. Zhang, R. Chen, J. Li, et al., Biochar 5 (2023) 25. doi: 10.1007/s42773-023-00225-x
Y. Li, R. Gupta, S. You, Bioresour. Technol. 359 (2022) 127511. doi: 10.1016/j.biortech.2022.127511
L. Leng, X. Lei, N. Abdullah Al-Dhabi, et al., Chem. Eng. J. 485 (2024) 149862. doi: 10.1016/j.cej.2024.149862
J. Peng, Y. He, C. Zhou, et al., Chin. Chem. Lett. 32 (2021) 1626-1636. doi: 10.1016/j.cclet.2020.10.026
D. Chu, Z. Ji, X. Zhang, et al., N. J. Chem. 47 (2023) 21883-21896. doi: 10.1039/d3nj04124g
E.M. Khabushev, D.V. Krasnikov, O.T. Zaremba, et al., J. Phys. Chem. Lett. 10 (2019) 6962-6966. doi: 10.1021/acs.jpclett.9b02777
A. Eftekhari, H. Garcia, Mater. Today Chem. 4 (2017) 1-16. doi: 10.1016/j.mtchem.2017.02.003
M.D. T, Z. Hou, K. Tsuda, J. Chem. Phys. 148 (2018) 241716. doi: 10.1063/1.5018065
A. Cortijo, M.A.H. Vozmediano, Nucl. Phys. B 763 (2007) 293-308. doi: 10.1016/j.nuclphysb.2006.10.031
F. Banhart, J. Kotakoski, A.V. Krasheninnikov, ACS Nano 5 (2011) 26-41. doi: 10.1021/nn102598m
B. Motevalli, B. Sun, A.S. Barnard, J. Phys. Chem. C 124 (2020) 7404-7413. doi: 10.1021/acs.jpcc.9b10615
X. Zhang, J. Liu, R. Li, et al., J. Colloid Interface Sci. 645 (2023) 956-963. doi: 10.1016/j.jcis.2023.05.011
S. Lin, H. Xu, Y. Wang, et al., J. Mater. Chem. A 8 (2020) 5663-5670. doi: 10.1039/c9ta13404b
X. Wan, Z. Zhang, H. Niu, et al., J. Phys. Chem. Lett. 12 (2021) 6111-6118. doi: 10.1021/acs.jpclett.1c01526
L. Wu, T. Guo, T. Li, iScience 24 (2021) 102398. doi: 10.1016/j.isci.2021.102398
L. Wu, T. Guo, T. Li, J. Mater. Chem. A 8 (2020) 19290-19299. doi: 10.1039/d0ta06207c
N.K. Wagh, S.S. Shinde, C.H. Lee, et al., Nano-Micro Lett. 14 (2022) 190. doi: 10.1007/s40820-022-00927-0
Q. Fu, T. Xu, C. He, et al., Langmuir 40 (2024) 10726-10736. doi: 10.1021/acs.langmuir.4c00803
Q.M. Zhang, Z.Y. Wang, H. Zhang, et al., Phys. Chem. Chem. Phys. 26 (2024) 11037-11047. doi: 10.1039/d4cp00325j
Y. -J. Zhang, V. Sethuraman, R. Michalsky, et al., ACS Catal. 4 (2014) 3742-3748. doi: 10.1021/cs5012298
B.H.R. Suryanto, C.S.M. Kang, D. Wang, et al., ACS Energy Lett. 3 (2018) 1219-1224. doi: 10.1021/acsenergylett.8b00487
M. Zafari, D. Kumar, M. Umer, et al., J. Mater. Chem. A 8 (2020) 5209-5216. doi: 10.1039/c9ta12608b
T. Liu, X. Zhao, X. Liu, et al., J. Energy Chem. 81 (2023) 93-100. doi: 10.1016/j.jechem.2023.02.018
Y. Liu, X. Ge, Z. Yang, et al., J. Power Sources 545 (2022) 231946. doi: 10.1016/j.jpowsour.2022.231946
I. Goodfellow, J. Pouget-Abadie, M. Mirza, et al., Commun. ACM 63 (2020) 139–144. doi: 10.1145/3422622
X. Luo, Z. Wang, P. Gao, et al., npj Comput. Mater. 10 (2024) 254. doi: 10.1007/978-3-031-60441-6_17
Y. Liu, Z. Yang, Z. Yu, et al., J. Materiom. 9 (2023) 798-816. doi: 10.1016/j.jmat.2023.05.001

Figure 1 The annual publications of ML in materials/catalysts research for environmental remediation from 2011 to 2024.

Figure 3 A graphic user Interface for predicting non-basic contribution in the BC-AOP system. Reprinted with permission [21]. Copyright 2023, American Chemical Society.

Figure 4 Three main ways to ML applications in environmental materials research: (a) Predict material properties. Reprinted with permission [69]. Copyright 2023, American Chemical Society. (b) Design new materials. Reprinted with permission [70]. Copyright 2022, American Chemical Society. (c) Analyze material characterization. Reprinted with permission [71]. Copyright 2017, American Chemical Society.

Figure 5 (a) The results of Support Vector Regression (SVR) using simple coordinate-free species descriptors, bond counts (C-C, C-O, C-H, O-H, C-M, and O-M counts), and (b) the results of a linear regression model using complex coordinate-based descriptors BOB, in predicting the various metals. Reprinted with permission [30]. Copyright 2018, American Chemical Society. Comparison of the model performance using the (c) the geometry based, and (d) electronic structured based primary features for CO adsorption energy on metal surfaces. Reprinted with permission [31]. Copyright 2017, Elsevier.

Figure 6 EVMEGnet and EVCGCNN distribution of (a) binary and (b) ternary oxides in the latent space composed of solely alkali, alkali earth, transition, lanthanide, or other metals. In parts a–c, red and blue areas represent the distribution of EVMEGnet and EVCGCNN, respectively. Reprinted with permission [104]. Copyright 2023, American Chemical Society. The distribution of OVs on the surface along the [0001] direction (c), forming at isolated locations away from each other (d) and along the [12̅10] direction (e). Reprinted with permission [105]. Copyright 2023, American Chemical Society.

Figure 7 (a) SHAP analysis and (b) model interpretation based on feature analysis for char-N yield prediction. (c) SHAP analysis and (d) model interpretation based on feature analysis for amine-N prediction. (e) SHAP analysis and (f) model interpretation based on feature analysis for pyrrolic-N prediction. (g) SHAP analysis and (h) model interpretation based on feature analysis for pyridinic-N prediction. Reprinted with permission [111]. Copyright 2024, Elsevier.
Table 1. Common material databases information.
| Database name | Web address | Web functions |
| The materials project | Platform for providing material properties and computational data. | |
| OQMD | Open quantum materials database for large-scale computation and data management of materials structures and properties. | |
| NIST materials data | Provides standardized datasets and material property databases in the field of materials science. | |
| PubChem | Provides a large-scale database of chemical molecules and their properties to support the retrieval and analysis of chemical information. | |
| NOMAD | Provides a platform for storing and analyzing computational data in materials science, supporting large-scale data archiving and access. | |
| Magpie | Provides machine learning models and tools for material property prediction to support material discovery and design. | |
| AFLOW | Provides a materials database for automated first-principles calculations, including information on crystal structures, band gaps, etc. | |
| AiiDA | Provides a database for the management and archiving of material science computing workflows, supporting reproducible and verifiable scientific computing. | |
| Citrination | Provides a database of material properties and experimental data to support data-driven material design and discovery. | |
| Materials cloud | Provides a collaborative platform for computational data and tools for materials science to support data sharing and scientific research. | |
| ChemSpider | Database providing information on chemical substances, covering compound structures, properties and literature information. | |
| MPContribs | Provide a community-contributed data platform for materials projects to support user submission and access to materials computation data. |
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: