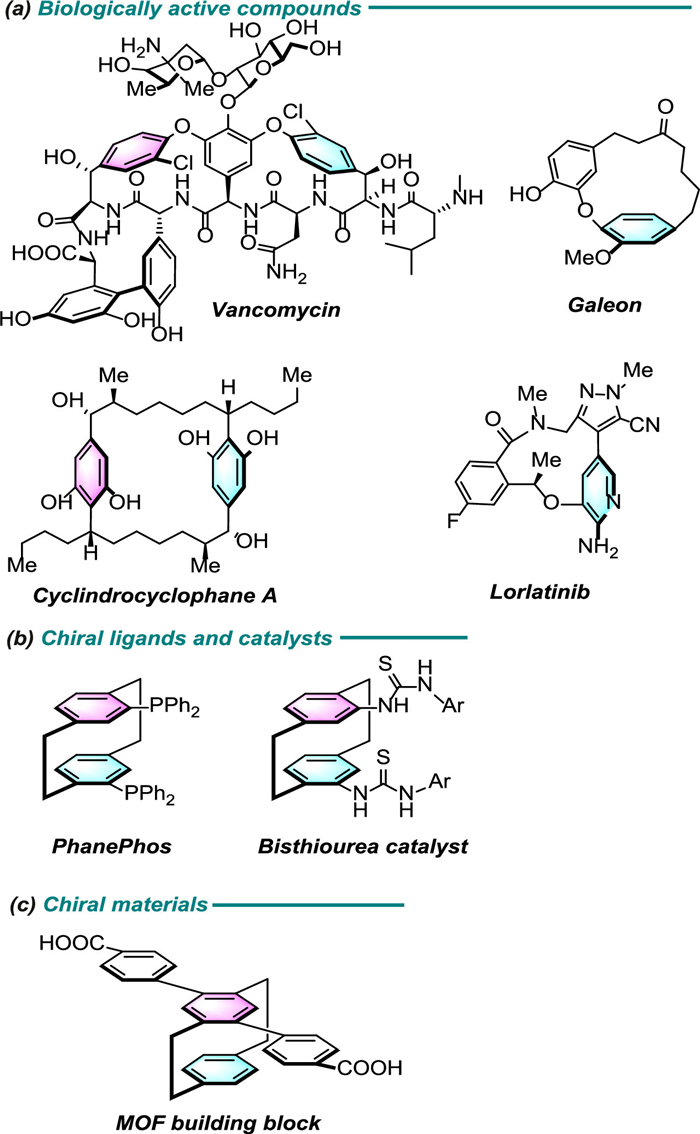
Figure 1.
Selected examples of stereo-enriched planar chiral cyclophanes.
Recent advances toward the catalytic enantioselective synthesis of planar chiral cyclophanes
Kai Zhu , Lei Yang , Yang Yang , Yanqi Wu , Fengzhi Zhang

Vancomycin, Cyclindrocyclophane A, Galeon and Lorlatinib are represented examples of biologically active compounds exhibiting a planar chiral cyclophane (Fig. 1a) [1–6]. Moreover, the substituted [2.2]paracyclophanes have found successful applications in the development of chiral ligands and catalysts (Fig. 1b) [7–11], as well as versatile chiral building blocks in functional materials (Fig. 1c) [12–15]. In the case of chiral cyclophanes, a subset of macrocycles with a skeleton in which an aromatic unit is linked by an ansa chain, their unique features resulting from their atropisomerism (or planar chirality), i.e., restricted rotation around the bond flip of the aromatic ring [16,17]. Owing to the importance and high demand of these scaffolds, methods for forming enantiomerically pure compounds containing such a stereogenic element have attracted increased attention [18–23]. However, the synthesis of planar chiral cyclophanes has been regarded as a formidable challenge due to the unfavorable entropic effects.
Historically, enantioenriched cyclophanes were prepared by the conventional chiral resolution of racemates which limited quick access to the target enantiopure backbones. Asymmetric catalysis with high efficiency and economic value now provides new insights for the assembly of such privileged scaffolds [18–23]. The expanding knowledge on asymmetric organocatalysis paved the way toward pioneering work on enantioselective construction of ansa chains. The crucial atropenrichment has also been achieved via chemoenzymatic macrocyclization. Another elegant procedure concerns chiral capsule assembly strategy i.e., the enantiocontrolled confinement of a linear substrate. In view of the remarkable advancements achieved over the past years, we feel that a general update of the most recent breakthroughs in this research is of interest. This review aims to provide an overview of the catalytic asymmetric strategies for procuring planar chiral cyclophanes with the help of representative examples. We hope that this survey will encourage the research interest of chemists for the discovery of novel methodologies in the coming years, which may be beneficial for the preparation of valuable and previously difficult-to-access chiral cyclophanes.
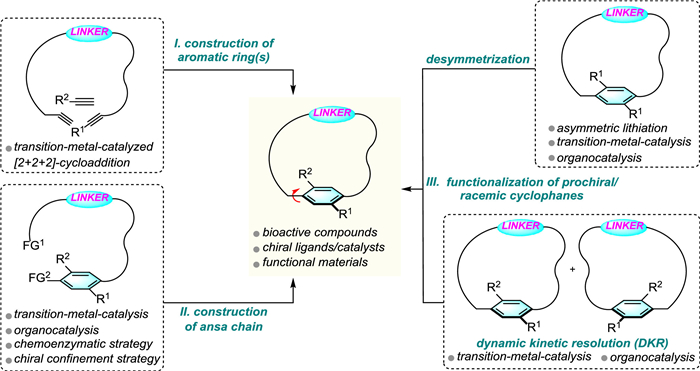
This review is divided into three main sections according to the catalytic enantioselective synthetic routes (Scheme 1): Section Ⅰ concerns the access to planar chiral cyclophanes via construction of the aromatic ring. Section Ⅱ deals with stereoselective construction of ansa chains. Section Ⅲ covers the enantioselective functionalization of prochiral/racemic cyclophanes. Methods belonging to each section are categorized according to the catalytic mode employed, namely transition-metal catalysis, organocatalysis, biocatalysis, chiral confinement strategy and so on.
Atropisomerism via construction of aromatic ring(s) is considered one of the most convenient strategies for the rapid and straightforward preparation of planar chiral cyclophanes [23]. The Rh-catalyzed asymmetric intra- and intermolecular [2 + 2 + 2] cycloaddition reactions developed by Tanaka and co-workers perfectly exemplify this strategy. Because of their convergent nature and high atom-economy, these transformations have received much attention, and remarkable advances in terms of stereo- and chemoselectivities have been realized over the past two decades.
In 2007, Tanaka and co-workers disclosed the first enantioselective catalytic [2 + 2 + 2] cycloaddition reaction toward planar chiral [7]-[10]metacyclophanes by means of Rh-catalyzed intra- and intermolecular alkyne cyclotrimerization (Scheme 2) [24,25].
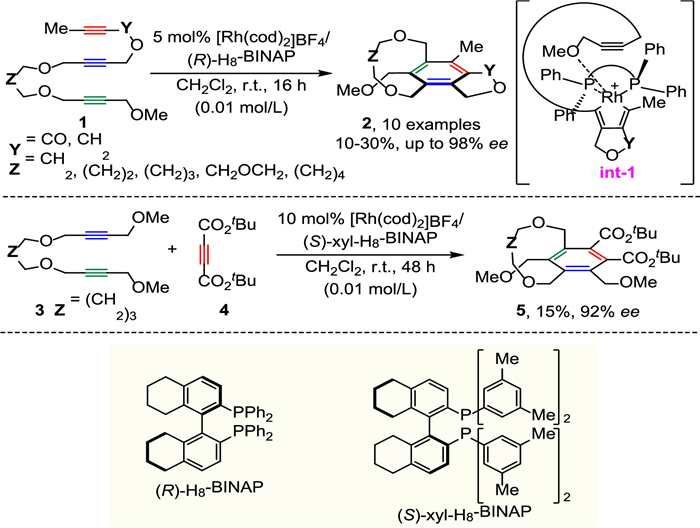
The methyl and methoxymethyl substituents at two alkyne termini in 1 are crucial to the atropostability of the product, which could increase the steric strain of the ansa chain, thus preventing ring flip to the other side from occurring. The reaction gave desired metacyclophanes 2 with high enantiomeric excess (88%−98% ee), but only 10%−30% yields were obtained, the undesired orthoacyclophanes were generated as by-products. It was anticipated that enantioselectivity was determined by preferential formation of intermediate int-1, the 1,6-diyne moiety of 1 reacted with the Rh(Ⅰ) complex, remaining alkyne inserts into the five-membered rhodacycle selectively as directed by the terminal methoxy group coordination to create robust chiral environment, the induction of planar chirality relies on the ligand chirality of (R)-H8-BINAP. Furthermore, the intermolecular [2 + 2 + 2] cycloaddition could furnish the corresponding product 5 in 15% yield with 92% ee.
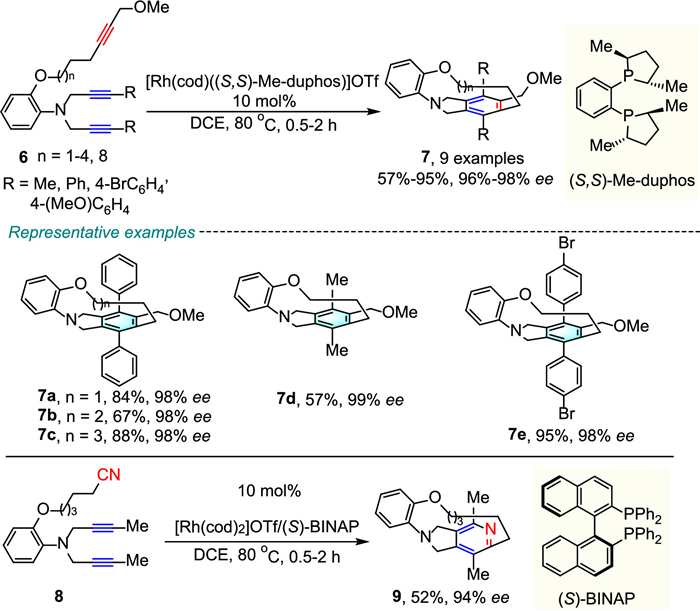
Soon after this work, Shibata's group carried out an efficient cationic Rh-catalyzed enantioselective intramolecular [2 + 2 + 2]-cycloaddition of N-branched triynes for achieving the asymmetric synthesis of atropisomeric tripodal cage compounds (Scheme 3) [26]. The successful implementation of branched substrates leads to a substantial increase in the complexity of the products. The [8]-[11] and [15]-cyclophanes 7 were obtained in good to excellent yields (57%−95%) and high enantioselectivities (96%−98% ee). When diyne-nitrile 8 was employed as substrate, the use of (S)-BINAP was necessary to achieve high enantioselectivity.
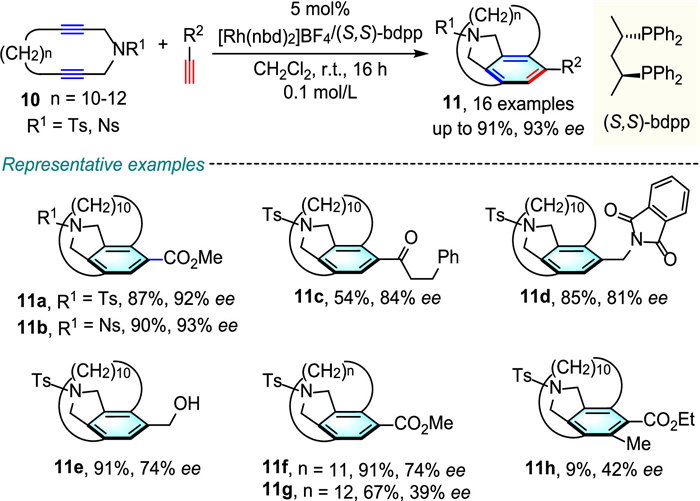
After these pioneer studies in the synthesis of planar chiral metacyclophanes, in 2013, Tanaka and co-workers further extended the application of the Rh-catalyzed [2 + 2 + 2] cycloaddition strategy to perform highly enantioselective synthesis of planar chiral carba-[10]-[12]paracyclophanes (Scheme 4) [27]. The protocol featured a broad substrate tolerance of terminal monoynes, and delivered the corresponding products 11 in moderate to good yields and stereoselectivities by using Rh(nbd)2]BF4 as catalyst and (S, S)-bdpp as ligand. However, internal monoyne (11h, 9% yield, 42% ee) was inefficient under standard conditions, probably due to the steric and/or electronic effects.
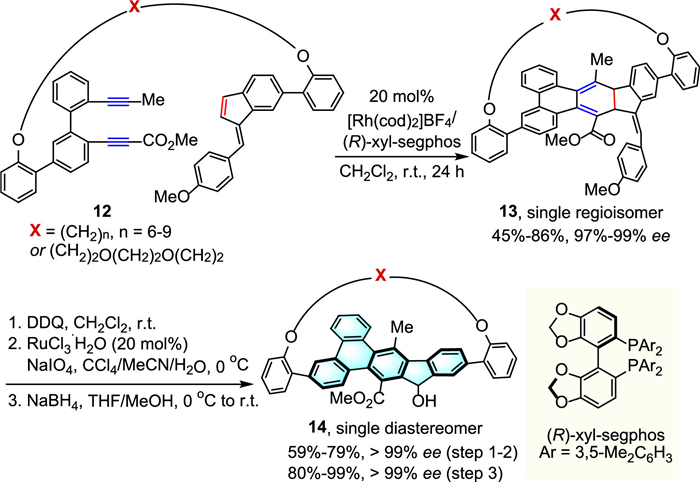
Considering the unique physical properties and potential applications of polycyclic aromatic hydrocarbon (PAH)-based chiral molecules, Tanaka's group designed an efficient intramolecular regioselective [2 + 2 + 2] cycloaddition method for the catalytic enantioselective synthesis of PAH-based planar chiral bent cyclophanes (Scheme 5) [28]. By using Rh(cod)2]BF4 as catalyst and (R)-xyl-segphos as ligand, the desired products 13 could be obtained with moderate to high yields and excellent ee values (97%−99% ee). Stepwise oxidative transformations and diastereoselective 1,2-reduction of the thus obtained 13 afforded stereochemically well-defined 9-fluorenol-based planar chiral bent cyclophanes 14 (> 99% ee), which possess higher fluorescence quantum yields than the acyclic reference molecules (78%−82% vs. 48%). Furthermore, the investigation into the impact of bending on the chiroptical properties revealed that the anisotropy factors (gabs values) for electronic circular dichroism (ECD) increased as the length of the chain became shorter.
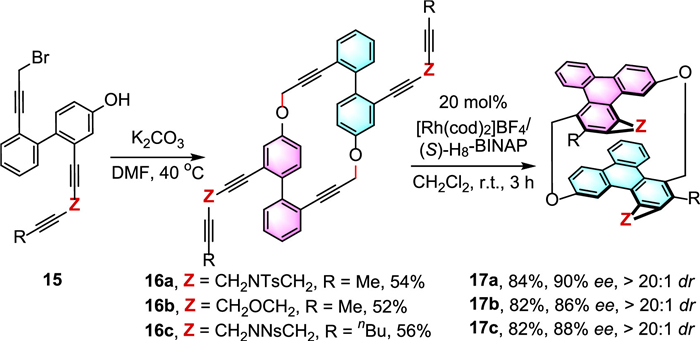
More recently, Tanaka and co-workers advanced the chemistry of atroposelective [2 + 2 + 2] cycloaddition further to derive PAH-based [2.2]cyclophanes ([2.2]triphenylenophanes) with two planar chiralities in single molecular architecture (Scheme 6) [29]. From bromo-phenol precursors 15, the addition of K2CO3 enabled the intermolecular macrocyclization, and generated cyclic hexaynes 16a-c in good yields (52%−56%). The succeeding Rh-catalyzed enantioselective intramolecular double [2 + 2 + 2] cycloaddition of 16a-c proceeded in complete diastereoselectivity (> 20:1 dr), affording 17a-c in good optical purities. Density functional theory (DFT) calculations revealed that the diastereoselectivity is kinetically governed by the second [2 + 2 + 2] cycloaddition, while the planar chirality is determined by the first [2 + 2 + 2] cycloaddition. These [2.2]triphenylenophanes showed significantly lower fluorescence quantum yields (0.6%−2.1%) than that of unsubstituted triphenylene (9%). Besides, the ECD spectra of these molecules showed a mirror-image relationship based on the distinct Cotton effects, and relatively good dissymmetry factors gabs (with maximum values of 1.9 × 10–3). The convenient preparation of these planar chiral π-conjugated [2.2]triphenylenophanes that possess attractive photophysical and chiroptical properties is projected to stimulate advancement of chiral functional materials.
An alternative pathway towards planar chiral cyclophanes consists of the construction of ansa chains as shown in route Ⅱ of Scheme 1. Among the established strategies, the direct enantioselective macrocyclization reactions revealed to be the most straightforward route and have received intensive interest due to their convergent nature and high versatility. This section illustrates contemporary advances of the field towards effective incorporation of planar chiral elements in enantioenriched cyclophanes under catalytic control.
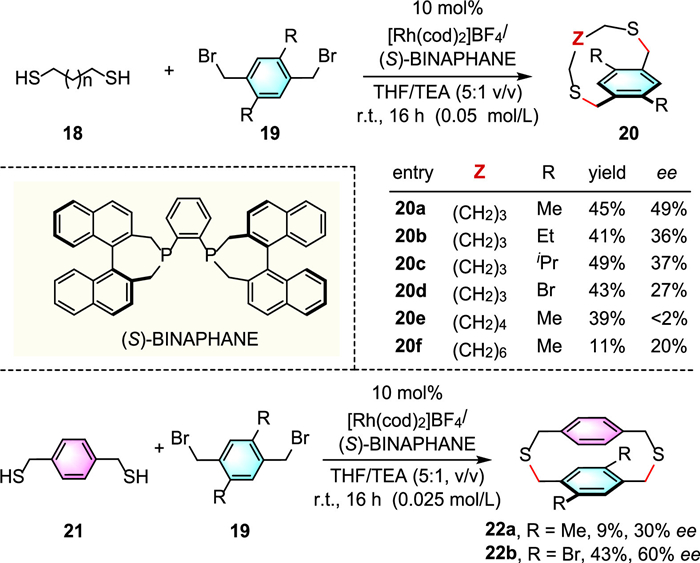
In 2007, Tanaka and colleagues reported the first example of an intermolecular catalytic enantioselective macrocyclization reaction of dithiols 18 and 1,4-bis(bromomethyl)benzenes 19 under mild conditions [30]. The desired planar-chiral dithiaparacyclophanes 20 and 22 were obtained in low to moderate yields and ee values by using the widely studied cationic Rh(Ⅰ)/(S)-binaphane catalytic system (Scheme 7). Subsequently, Tanaka and co-workers further explored the same transformations by using cationic Pd(Ⅱ) catalysis with commercially available (R)-BINAP as the cooperative ligand, and successfully assemble the corresponding products with comparable levels of yields (up to 43%) and enantiomeric excesses (up to 44%) [31].
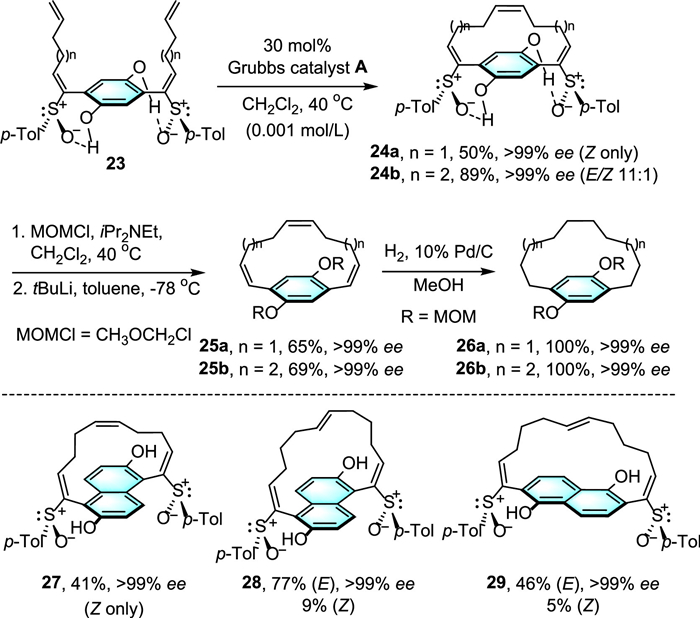
In 2009, Suzuki and co-workers employed a chiral auxiliary chemistry to achieve ruthenium-catalyzed intramolecular olefin metathesis of tetraenes 23 by including two chiral sulfoxide moieties to control the local conformation of the corresponding intermediates 24, by forming two hydrogen bonds to the phenol hydroxy groups (Scheme 8) [32]. Followed by elaborations including desulfurization and hydrogenation to obtain saturated planar chiral [10]- and [12]paracyclophanes 26 with excellent ee values (> 99% ee). On the basis of this procedure, 1,5- and 2,6-naphthalene cores also can be successfully transformed to [10]- and [12]paracyclophanes 27–29 with high ee values (> 99% ee). In another development of this chemistry, Ohmori and colleagues realized that substrates with only one chiral sulfinyl group is enough for the stereocontrol of the cyclized product, which offering efficient and general access to valuable planar chiral cyclophanes [33].
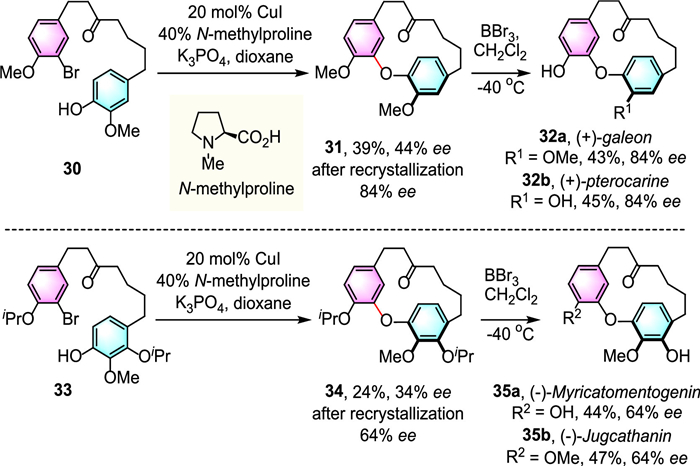
Asymmetric Ullmann reaction have attracted great interest for their widespread applications in the preparation of valuable chiral scaffolds. In 2013, Beaudry and colleagues developed an enantioselective copper-catalyzed intramolecular Ullmann cyclization of bromophenols (30, 33) for the construction of planar chiral diaryl ethers (31, 34) [34]. Among various chiral ligands, N-methyl proline was found the most efficient one for the stereocontrol. After further purified by recrystallization, cyclophanes (31, 34) can be prepared with good optical purity, which can be further converted to various diarylether heptanoid natural products, including (+)-galeon 32a, (+)-pterocarine 32b, (-)-myricatomentogenin 35a and (-)-jugcathanin 35b without the loss of enantiopurity (Scheme 9).
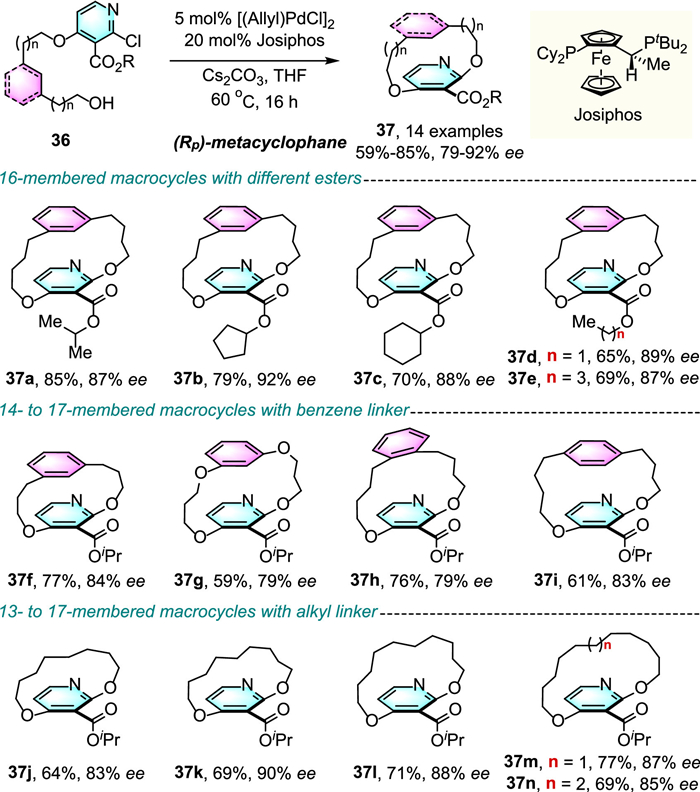
Very recently, Li and co-workers demonstrated that palladium-catalyzed enantioselective C–O cross-coupling of linear precursors 2,3,4-trisubstituted pyridines 36 via intramolecular macrocyclization could provide various planar chiral metacyclophanes in good yields and up to 92% ee by using Josiphos as a chiral ligand (Scheme 10) [35].
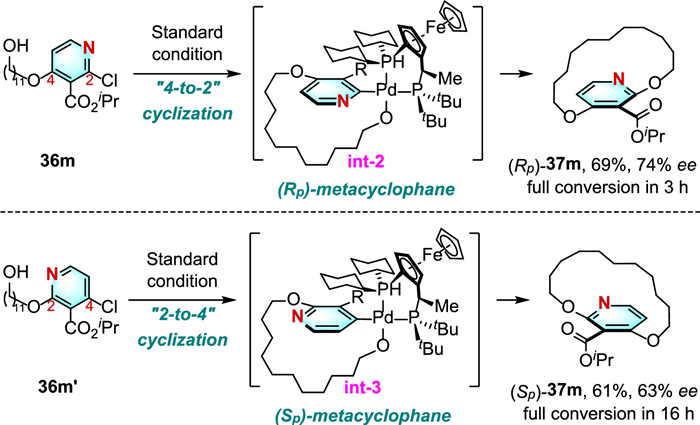
The bulky substituent at C3-position of pyridine was crucial to prevent inversion of the aromatic ring by bond rotation. Moreover, the treatment of two regioisomeric linear precursors 36 m and 36m' with the same Pd(Ⅱ)/Josiphos catalytic system enabled to prepare the two enantiomers of the metacyclophane through "4-to-2" cyclization and "2-to-4" cyclization, respectively (Scheme 11). Mechanistic studies revealed that stereochemical bias is efficiently imposed by the bulkier cyclohexyl moiety on Josiphos, permitting the oxidative addition complex int-2 and int-3 cyclization from the less crowded bottom plane of the prochiral pyridyl to avoid steric repulsion.
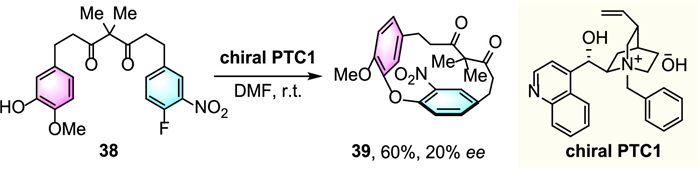
Back in 2003, seminal work on organocatalytic asymmetric cunstruction of ansa chain was exemplified by Zhu and colleagues (Scheme 12) [36]. Enantiomerically enriched diarylether cyclophane 39 was synthesized via an intramolecular nucleophilic aromatic substitution (SNAr) reaction of linear achiral diarylheptanoid 38 by using cinchonine-derived chiral phasetransfer catalyst PTC1 as the base. These results served as an encouraging proof-of-concept of atroposelective synthesis of chiral cyclophanes, albeit low enantioselectivity was obtained.
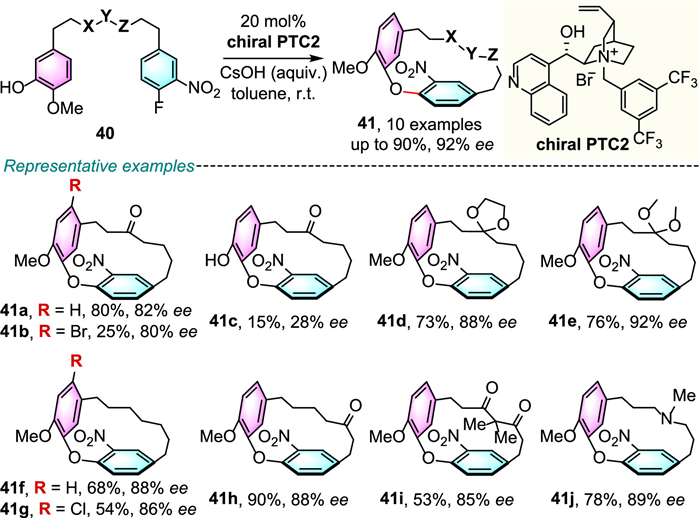
Almost 14 years later, for the same reaction, Cai and co-workers discovered that the use of base and electron-withdrawing cinchonine-derived catalyst PTC2 bearing two trifluoromethyl substituents on the aromatic ring is important for the enantiocontrol [37]. The targeted transformation, affording enantioenriched oxa[1,7]metapara-cyclophanes, was achieved under mild conditions with synthetically useful yields and good enantiomeric excess (Scheme 13). However, substrate with an additional free hydroxyl group could significantly decrease the enantioselectivity and yield (41c, 15%, 28% ee). This is reasonable since the nucleophility of the reactive phenolic anion could be weakend by the hydrogen bond, thus made it difficult to form tight ion-pairing interaction with the chiral phase-transfer catalyst for better stereoselective nucleophilic attack. The potential utility of this procedure is also investigated. The stereoenriched product 41a can be efficiently transformed into (-)-pterocarine and (-)-galeon without ee erosion.
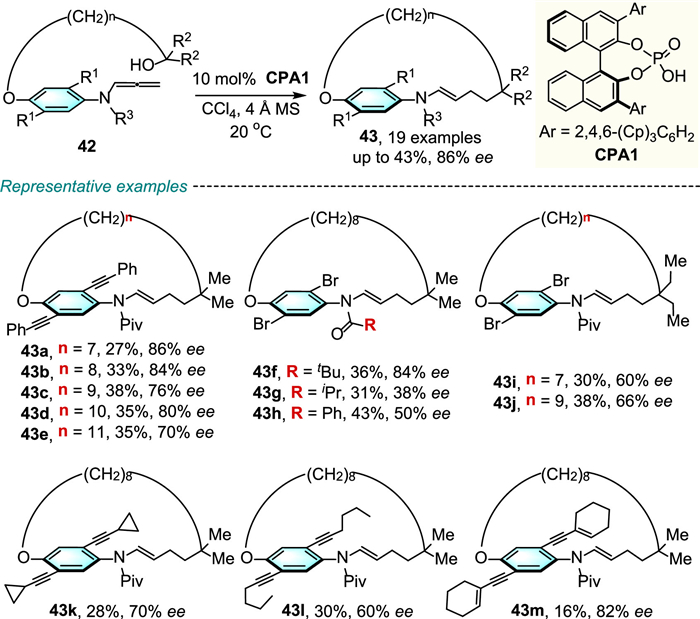
In 2022, Yang and colleagues demonstrated the innovative and successful access to planar chiral macrocycles 43 through chiral phosphoric acid (CPA)-catalyzed asymmetric intramolecular addition of the hydroxyl group into the allenamide moiety (Scheme 14) [38]. Substrates bearing di-alkynyl substitutions and di-bromo substitutions as the steric hindrance groups worked smoothly to achieve moderate yields, and good to high enantioselectivities. The configuration stability of the representative cyclophanes has been investigated by increasing temperature, no loss of enantioenrichment was observed for 43f (with n = 8) bearing di-bromo substitutions and 43d (with n = 10) bearing bulky di-phenylacetylenyl substitutions (Scheme 15). However, the 20-membered macrocycle 43n (with n = 9) bearing di-bromo substitutions rapidly racemization even at 40 ℃, which showed that the size of the ansa-bridge and substituents on the aromatic ring are critical to restrict planar rotation.
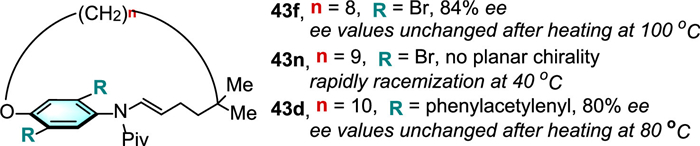
Indole-based atropisomers are widely found in bioactive molecules and chiral ligand skeletons. Recently, indole/pyrrole-based planar-chiral meta-macrocycles 45 with 10–16 membered tether-lengths were elegantly fabricated by Wang and co-workers under chiral N-heterocyclic carbene (NHC)-catalysis from intramolecular enantioselective macrocyclization of 3-carboxaldehyde indoles/pyrroles 44 in good yields and with high to excellent enantioselectivities (Scheme 16) [39].

A variety of N-substituted alkyl-alcohol chains containing ester, ether or amide group, C2-substitutions as well as C4-C7 substitutions on indole ring were well accommodated. It is noteworthy that the macrocyclic planar chirality is highly dependent on the size of the ansa chain, product with 10-membered tether-length result in decreased yield (45a, 49% yield) due to the higher strain during the formation of the ansa chain, while increase the tether-length to 18-member led to the loss of planar chirality (45i). In addition, by using the same catalytic system, atroposelective kinetic resolution of racemic rac-46 allows to access enantiopure 47, which contains both planar and axial chirality, with excellent diastereoselectivity and enantioselectivity. And recovery of optically active ent-46 with acceptable enantioselectivity was achieved (Scheme 17).
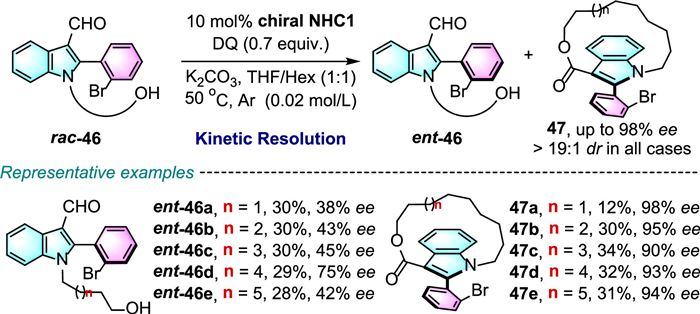
Almost at the same time, Zhao and Li reported a very similar reaction involving an intramolecular reaction of benzaldehyde substrates 48 (Scheme 18) [40].
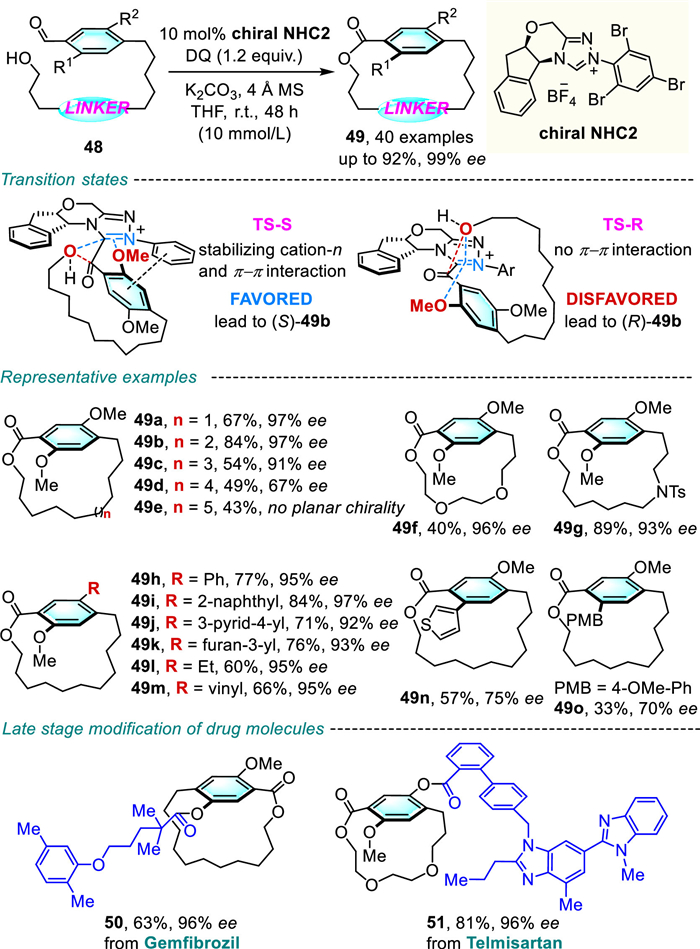
Treating 48 in the presence of chiral NHC2 initiated a stereoselective macrolactonization reaction to yield [11-14]planar chiral macrocycles in good to high yields and generally excellent enantioselectivities. DFT calculations revealed that the energy of transition state TS-S is lower than that of TS-R. In the (S)-pathway, the π-π interaction along with the weaker cation-OMe interaction are thought to efficiently stabilize the unique transition state geometry and control the stereoselectivity during the reaction. Remarkably, this powerful and reliable approach has been applied for the late-stage functionalization of several marketed drug molecules, including gemfibrozil, telmisartan, and furnished the desired drug-like atropisomers (50, 51) with excellent ee values (96% ee).
Independently, the application of NHC-catalyzed macrocyclization in the synthesis of complex planar chiral macrolactones has also been investigated recently by Chi, Wu and colleagues (Scheme 19) [41].

Under oxidative conditions, the reaction of bifunctional hydroxyl aldehyde substrate 52 with chiral NHC3 effectively form an NHC-bound acyl azolium intermediate int-4, which then reacts with the alcohol group of the substrate to complete the intramolecular lactonization process. Notably, the addition of hydrogen bond donor (HBD-1) as co-catalyst proved to be important to improve the yield and enantioselectivity, which may benefit from the hydrogen-bonding interactions provided by the thiourea moiety of HBD-1 with the acyl azolium intermediate int-4 and hydroxyl group. A library of diverse chiral macrocycles 53 with various ring sizes and substituents was obtained in high yields and stereoselectivities.
Chemoenzymatic strategy has been recognized as a powerful and versatile synthetic tool to prepare structurally diverse chiral building blocks. In 2020, Collins and colleagues showed that the reaction of di-carboxylic acids 54 (tethered with various aliphatic linkers) with ortho-substituted aromatic diols 55 can be promoted by the serine hydrolase Candida antarctica lipase B (CALB) (20 mg/0.01 mmol) to derive optically pure [12-15]paramacrolactones 56 in moderate to high yields and excellent enantioselectivities (Scheme 20) [42].
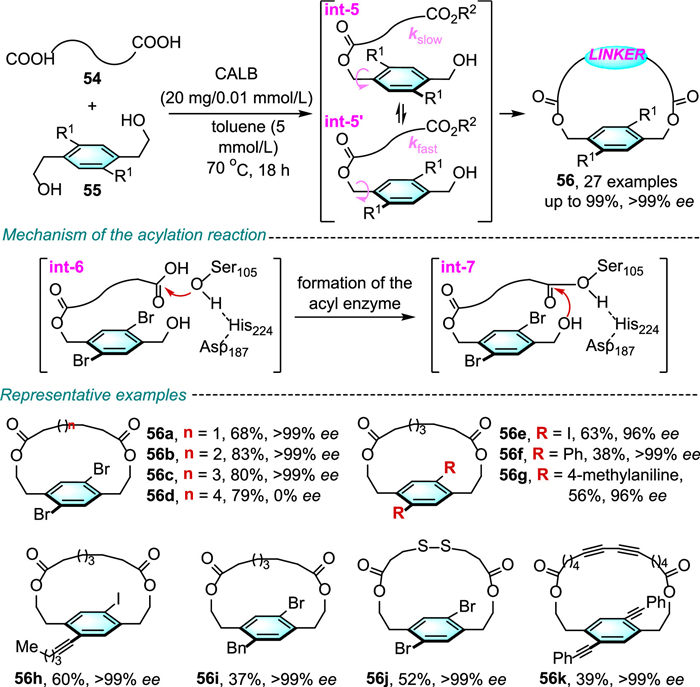
Detailed computational docking analyses revealed that, in the model structure, the catalytic serine residue (Ser105) attacks the carbonyl carbon atom of the carboxylic acid substrate 54, and the tetrahedral intermediate int-6 is subsequently generated. Then, the reaction with hydroxyl group generates the second tetrahedral intermediate int-7, with one of the bromine substituents on the aromatic ring pointing toward the exterior of the catalytic active site. Thus, CALB exclusively recognizes the major product 56 over the counter enantiomer. Collins has also shown this biocatalytic reaction to be easily scaled up to gram scale without a drop in enantioselectivity.
In 2023, Lee, Sun and colleagues published a landmark report on the use of step-by-step chiral confinement strategy to effect asymmetric intramolecular transformations (Scheme 21) [43].
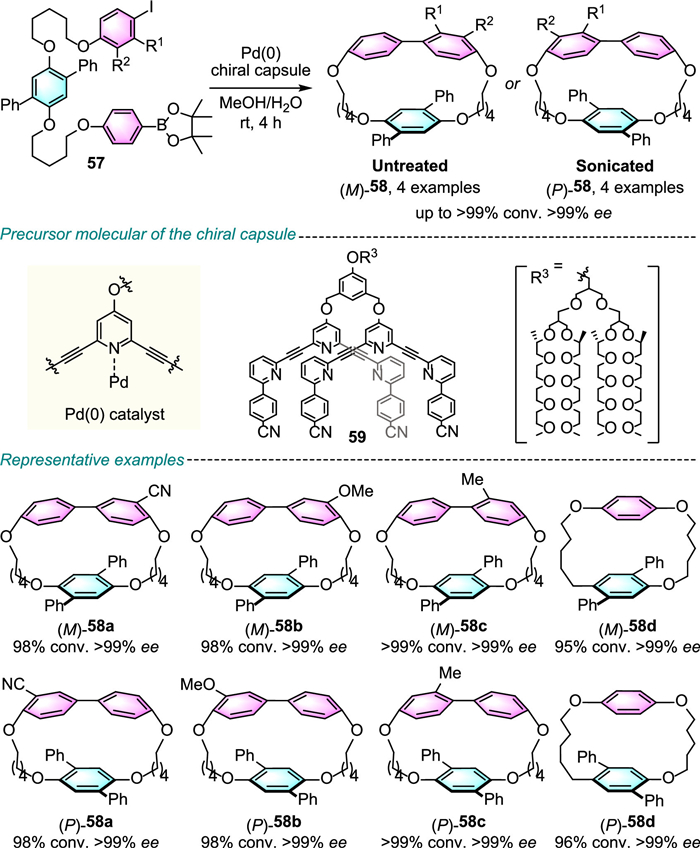
From precursor molecule 59 that comprises a conformationally flexible hydrophilic chiral dendron and an aromatic pyridine-based segment, the addition of hydrophobic linear substrate 57 can trigger 59 into a capsule-shaped chiral interstitial domain via hydrophobic interactions, and additional dipolar interactions among cyanide groups. Subsequently, the reaction under chiral-confinement generates macrocycle products (M)−58 with excellent enantioselectivities and quantitative conversions. It is noteworthy that by using sonication the chirality of the capsule assembly can be collectively switch into an opposite form with near-perfect enantioselectivity. In addition to planar chirality, the capsule assembly asymmetric macrocyclization strategy also can induce point and axial chirality with high levels of enantioselectivity.
Enantioselective functionalization of preformed (racemic or prochiral) cyclophanes constitutes another versatile strategy in catalyst-stereocontrolled synthesis of enantiomerically pure cyclophanes, which could occur through desymmetrization and dynamic kinetic resolution (DKR). In catalytic desymmetrization of meso/prochiral symmetric precursors, if the distinguishing between the reaction rates of the chiral catalyst with the two enantiotopic reactive sites is sufficiently large, enantioselectivity will be achieved. In the DKR context, benefit from the low rotational barrier of less hindered aromatic ring in the substrates, enantioenrichment of one enantiomer can be achieved quantitatively in the conversion products.
In 2010, Shibata and colleagues developed a catalytic asymmetric ortho-lithiation and dilithiation for the enantioselective synthesis of planar chiral 1,n-dioxa[n]paracyclophanes (Scheme 22) [44,45]. Here, oxygen atoms at the 1 and n positions were used as directing groups to ensure high enantioselectivity. Using stoichiometric amount of (-)-sparteine as chiral ligand and sec-butyllithium as the lithium reagent, followed by quenching with various electrophiles, such as iodine, iodomethane, benzophenone, chlorodiphenylphosphine and N,N-dimethylformamide, gave the corresponding planar chiral paracyclophanes 62–66 in good yields and excellent enantioselectivities (95%−98% ee). Further lithiation of the monolithiated paracyclophanes and trapping with electrophiles, allowing access to disubstituted planar chiral paracyclophanes 67 with almost perfect ee values. It is noteworthy that even a catalytic amount of (-)-sparteine (0.2 equiv.) gave the corresponding products in good to high yields and enantioselectivities.
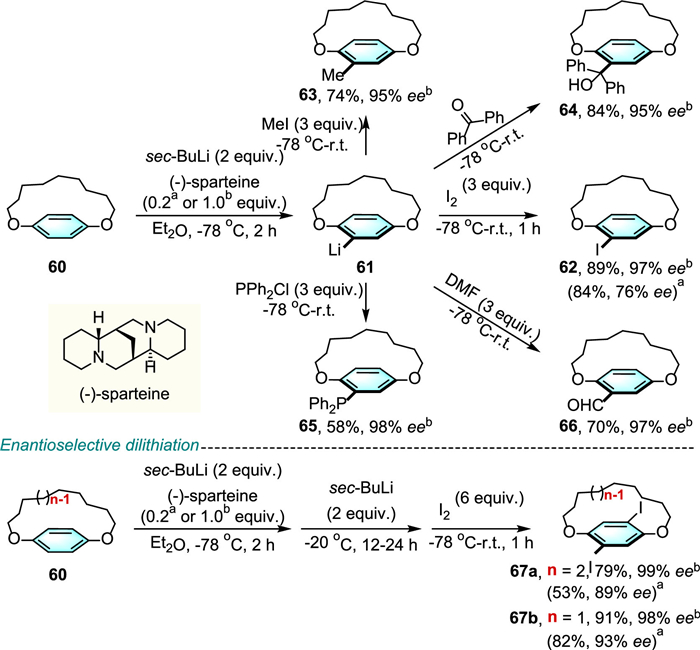
Almost at the same time, the same group reported an asymmetric Sonogashira coupling system that merges palladium catalysis and chiral ferrocene-based ligand (R, Rp)-Taniaphos for the dynamic kinetic asymmetric transformation of diiodoparacyclophanes 68 (Scheme 23) [46]. Reaction with alkynes 69 as coupling partners delivered planarly chiral dialkynylparacyclophanes 70 in good to excellent yields and up to 79% ee values.
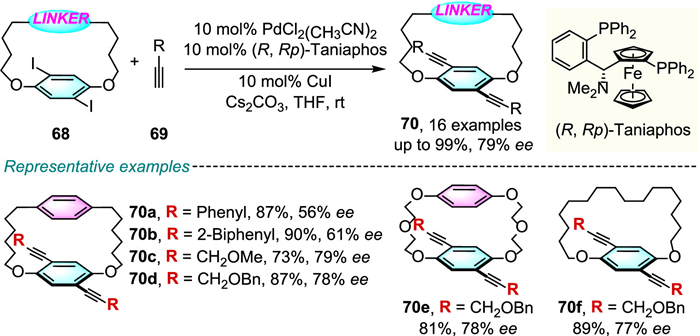
Recently, Micouin, Benedetti and colleagues described the catalytic asymmetric synthesis of precious planar chiral [2.2]paracyclophane 72 in good yields and excellent enantioselectivities through Ru-catalysed Noyori transfer hydrogenation reaction of achiral pseudo-para-diformyl[2.2]paracyclophane 71 (Scheme 24) [47,48]. The procedure was applicable even at gram scale. Mechanistic investigations of plausible hydrogenation models suggest that, the attractive CH-π interaction between [Ru(p-cymene)] complex and the π-conjugated system of [2.2]paracyclophane should favor the formation of corresponding (Rp)-isomer 72.
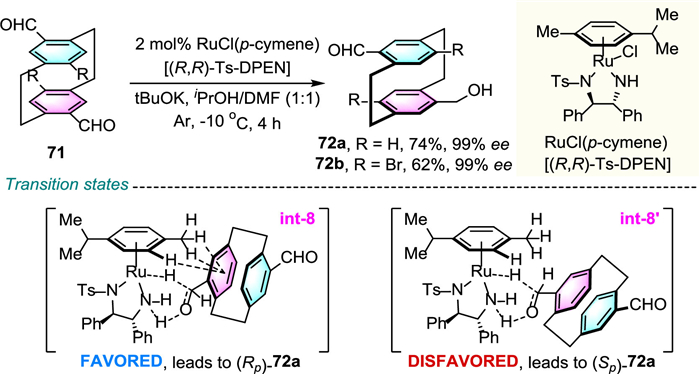
More recently, Zhao and co-workers ingeniously exemplified the DKR of pyridine-derived prochiral cyclophane 73 with palladium-catalyzed enantioselective C–H olefination toward planar-chiral cyclophane 75 (Scheme 25) [49]. Among various amino acid-derived ligands, d-pGlu-OH proved to be the most effective and allowed to reach high levels of enantioselectivity. Introduction of steric constraint substituent adjacent to ansa chain crucially bestowed the enantioselectivity and atropostability. Notably, originating from rotation of the large steric 2-pyridyl formyl group around the N–CO bond, products 75 were obtained as a mixture of cis/trans rotamers. This methodology was applicable to the modification of derivatives of marketed drugs, such as indomeyhacin and estrone, which highlight the reactions' efficacy.
In 2022, Yang, Xue and colleagues successfully accomplished the asymmetric synthesis of planar chiral paracyclophanes 80 through CPA-catalyzed enantioselective electrophilic aromatic aminations of achiral paracyclophane-type macrocycle 78 with high enantioselectivities and excellent yields (Scheme 26) [50].
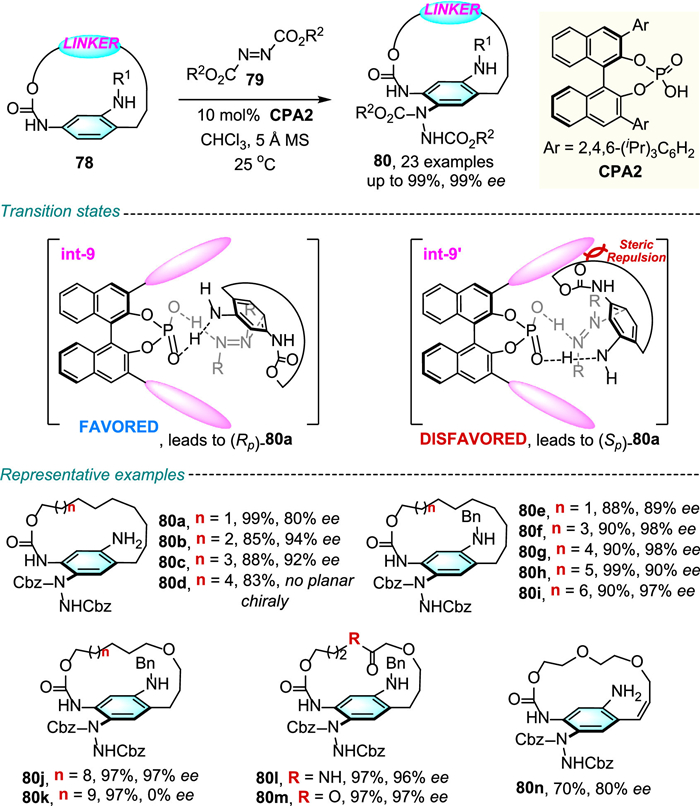
Sterically hindered substituted hydrazine was asymmetrically introduced at the ortho position of the ansa chain and blocked the rotation of the aromatic ring, leading to separation of the enantiomers. By using the more steric hindered N-Bn-substituted substrate, the macrocycle could be extended to 23-member (80j). In the proposed mechanism, the authors demonstrated that the destabilizing strong steric repulsion between the methylene group of the substrate and the isopropyl substituent of the phosphoric acid is a key factor for high enantioselectivity.
In recent years, the development of efficient catalytic kinetic resolution (KR) of functionalized [2.2]paracyclophanes has generated considerable interest [51–60]. More recently, the CPA-catalyzed electrophilic amination strategy has further been used by Yang and co-workers to perform a highly efficient catalytic enantioselective KR of substituted amido[2.2]paracyclophanes (Scheme 27) [61]. This KR approach features a broad substrate scope, both enantiomers of various pseudo-ortho-, pseudo-meta-, pseudo-para- and pseudo-geminal-substituted amido[2.2]paracyclophanes could be obtained with high to excellent ee values and KR performances (selectivity factor up to 371). Additionally, this transformation was proceeded smoothly for the enantioselective desymmetrization of pseudo-para-substituted diamido[2.2]paracyclophane 84, which provided the corresponding C–H amination product 85 with moderate yield and excellent enantioselectivity.
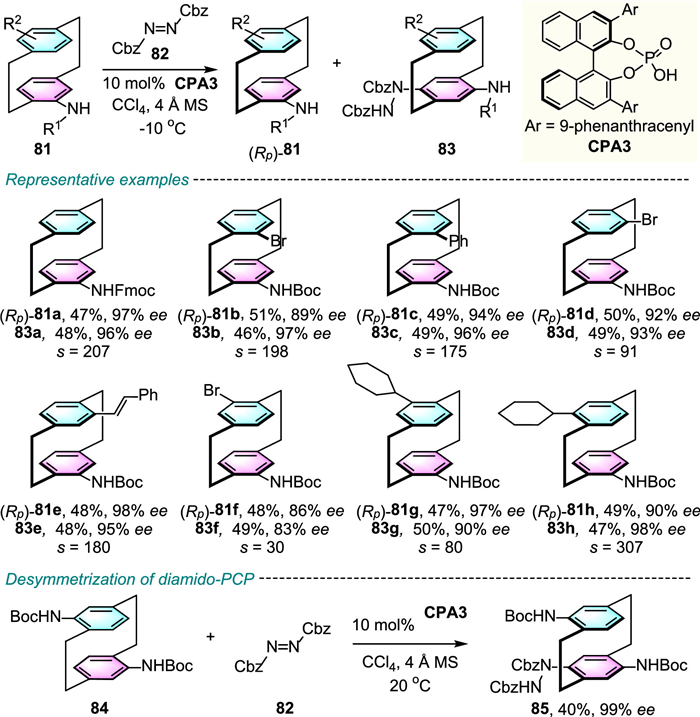
As the conformational stability of planar chiral cyclophanes is highly dependent on the ansa bridge length and aromatic ring substituent size, thus, the mutable ansa chain allows the late-stage enantioselective functionalization of aromatic ring of racemic/prochiral cyclophanes to occur through a DKR or a KR process.
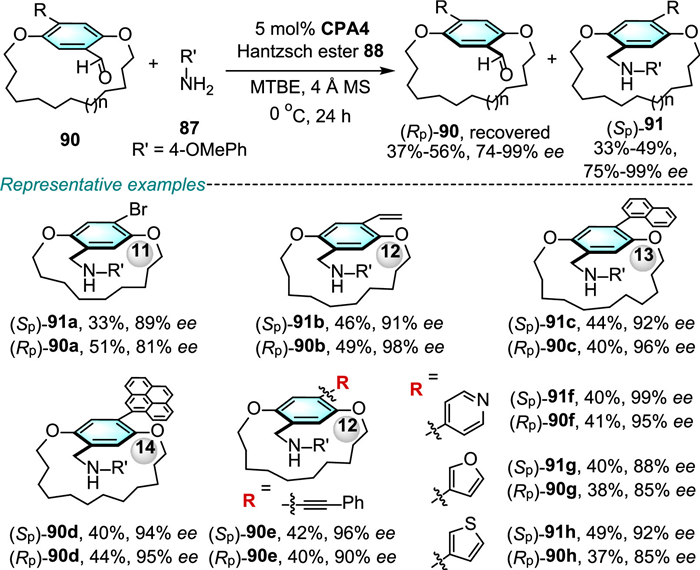
In 2023, Zhao and co-workers reported a CPA-catalyzed asymmetric transfer hydrogenation (ATH) reaction of imines with Hantzsch ester 88 for synthesis of planar chiral cyclophanes from aldehyde 86 and aromatic amines 87 (Scheme 28) [62].

The [15]-[17]cyclophanes bearing diverse substituents at the C4 position of the benzene ring undergo the DKR process, and worked well to afford the products 89 in good yields with high enantioselectivities (up to > 99% ee). Further, reducing the ansa chain length to [11]-[14]membered ansa bridge undergo the ATH reaction in a KR manner, delivering the desired products 91 with good to high enantioselectivities (75%−98% ee), while the remaining substrates 90 were recovered in acceptable yields with 74% ~ > 99% ee values (Scheme 29). Mechanism studies of the reaction revealed that the enantio-determining step is the CPA-catalyzed ATH of the imine intermediate step.
Soon after, Zhao, Li and co-workers disclosed an efficient NHC-catalyzed asymmetric oxidation esterification of diverse racemic cyclophanes 92 with alcohols 93 through DKR (Scheme 30) [63].
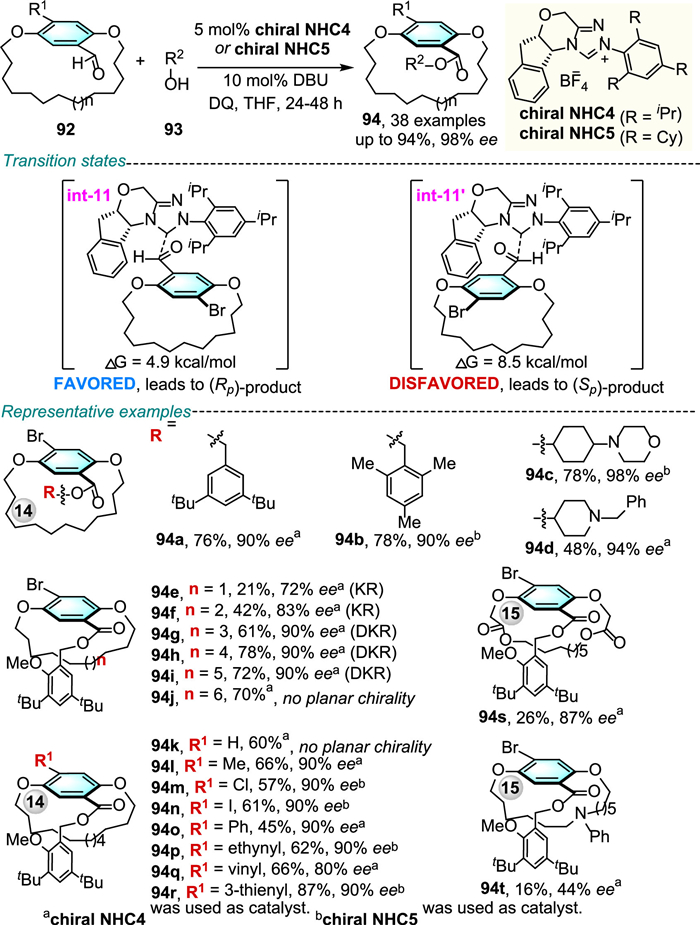
A wide range of planar chiral paracyclophanes 94 with different ansa chain lengths and aromatic substitutions were obtained in moderate to good yields and good to high enantiomeric excesses. The stereochemically enriched Breslow intermediate oxidized to acylazolium and then transformed into a bulky ester group that ensures the configurational stability of the final products. DFT calculations illuminate that the addition of chiral NHC4 to (Rp)−92 (int-11) is lower in energy barrier by 3.6 kcal/mol in comparison to that of (Sp)−92 (int-11′), thus, the R configurational isomer pathway is kinetically favored.
Nearly at the same time, Veselý, Dočekal and colleagues disclosed an elegant example of NHC-catalyzed enantioselective desymmetrization of prochiral diformyl[2.2]paracyclophanes to establish highly enantioenriched planar chiral [2.2]paracyclophanes (Scheme 31 and 32) [64].
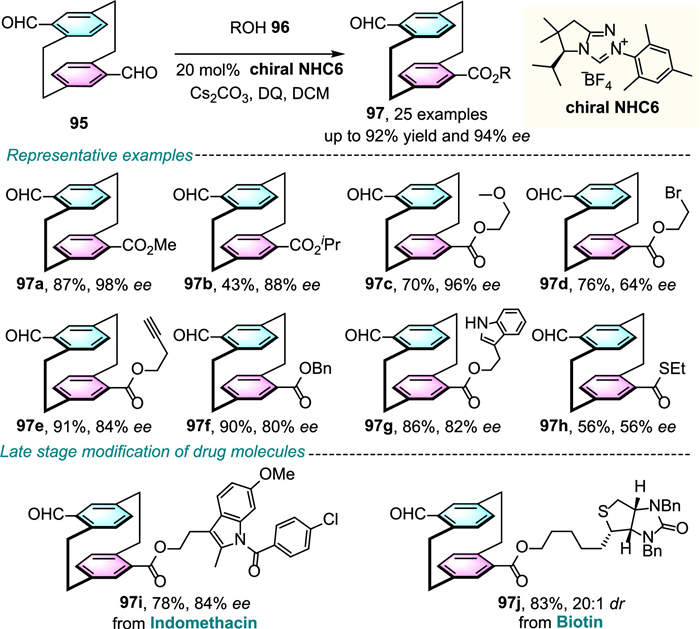

Under the catalytic system, a wide range of alcohols 96 with diverse substitution patterns were well tolerated and afforded the corresponding ester products 97 and 99 in good to high yields with high enantioselectivities. Interestingly, the corresponding mechanistic investigations in the origin of enantiocontrol suggested that the reaction took place through two distinct pathways by using different meso-dicarbaldehydes, albeit the reaction conditions were quite similar. The pseudo-para diformyl derivatives 95 underwent an enantiodivergent process that desymmetrization was followed by kinetic resolution, with a reversible formation of the key Breslow intermediate, whereas the perfect enantioselectivity generated from the pseudo-gem substrates 98 was the result of an irreversible enantioselective formation of Breslow intermediate.
Very recently, Chen, Xue and co-workers developed a chiral Lewis base catalyzed approach for the asymmetric electrophilic sulfenylation of various prochiral cyclophanes 100 (Scheme 33) [65].
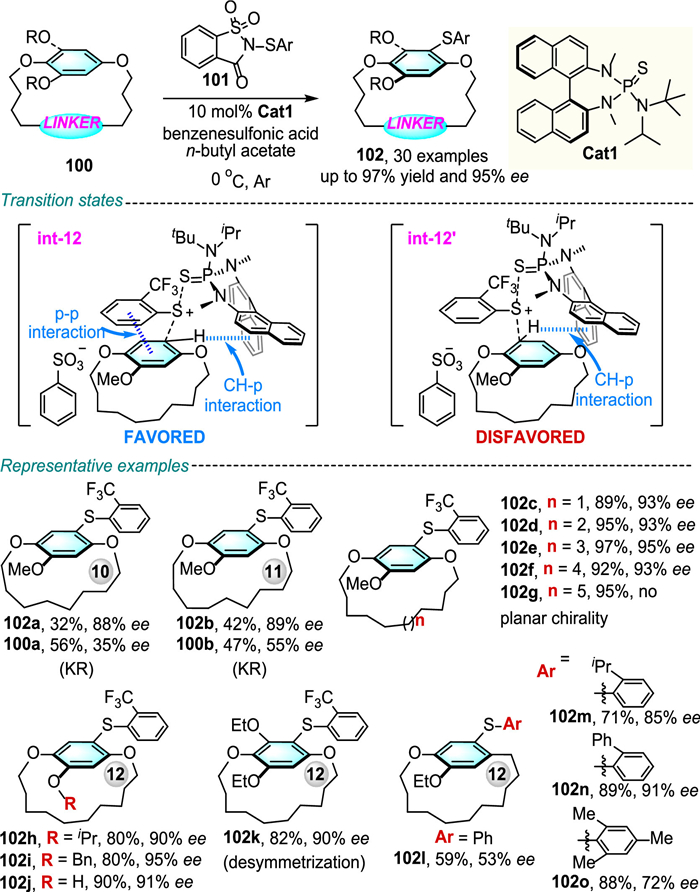
Upon treatment with ortho-trifluoromethyl-substituted sulfenylating reagent 101 and 10 mol% of catalyst Cat1, the reaction occurred smoothly to give the enantioenriched planar chiral sulfur-containing macrocycles 102 in moderate to excellent yields and enantioselectivities. It should be noted that the steric hindrance of the ortho substituent on the sulfenylating reagent had a dramatic effect on the enantioselectivity of this transformation. Mechanistically, transition state structure int-12 is a favored conformation due to the π-π interaction between the sulfenyl group of 101 and the phenyl ring of 100 together with a CH-π interaction between the phenyl ring of 100 and the naphthyl group of the catalyst Cat1. Further computational studies have also found that transition structure int-12 is more stable than int-12′ by 2.7 kcal/mol.
Over the last few years, catalytic enantioselective synthesis of planar chiral cyclophanes underwent a remarkable development and emerged as a fast evolving field within synthetic chemistry. Many efficient catalytic protocols steaming from transition metal promotion, organocatalysis, enzymatic chemistry and chiral capsule assembly strategy have been disclosed, enabling the construction of a series of value-added and previously difficult-to-access optically pure planar chiral cyclophanes, including [n]metacyclophanes and [2, 2]paracyclophanes. Nevertheless, practical access to potentially useful scaffolds remains elusive, as contemporary approaches are frequently limited by the scope of substrates. An overwhelming majority of conversions rely on the well-designed, highly reactive starting materials, thereby rendering the generation of a relatively narrow class of functionalized cyclophanes. Accordingly, the development of truly versatile and more efficient catalytic enantioselective methodologies would be developed in the future.
Moving forward, from the perspective of this review, innovative solutions that focus on the following suggested directions might significantly accelerate the exploration of planar chiral cyclophanes in both academia and industry: (1) developing new strategies and methods for the assembly of unconventional cyclophanes (e.g., [n]metacyclophanes and [n, n]metacyclophanes) to broaden the chemical space; (2) increasing the diversity and complexity of [2.2]paracyclophanes (beyond desymmetrization of diformyl[2.2]paracyclophanes); (3) implementing functionalized cyclophanes bearing multiple chiral elements (e.g., center chirality and axial chirality); (4) application of computational chemistry and statistical tools to reveal the underlying factors that influence conformational stability and rotational barriers of planar chiral cyclophanes (structure-chirality relationships), thus providing theoretical support for the rational design of new transformations; (5) leveraging the power of artificial intelligence chemistry, electrochemistry, or photochemistry to expedite discovery of novel reaction modes.
By summarizing and systematically discussing the advancements in this rapidly developing field, we hope that the present overview can help to inspire conceptually novel methodologies. In addition, this may produce hitherto unseen planar chiral cyclophanes with immense potential for applications in various fields, including chiral catalysis, medicinal chemistry, chiral material science, etc.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Kai Zhu: Writing – review & editing, Writing – original draft, Conceptualization. Lei Yang: Writing – original draft, Data curation. Yang Yang: Writing – original draft, Formal analysis, Data curation. Yanqi Wu: Writing – review & editing, Writing – original draft, Formal analysis, Data curation. Fengzhi Zhang: Writing – review & editing, Writing – original draft, Supervision, Conceptualization.
The authors gratefully acknowledge the financial support provided by Huanghuai University and Hangzhou Medical College.
Y. Cetinkaya, P. Falk, C.G. Mayhall, Clin. Microbiol. Rev. 13 (2000) 686–707. doi: 10.1128/CMR.13.4.686
H. Nakamura, E.E. Schultz, E.P. Balskus, Nat. Chem. Biol. 13 (2017) 916–921. doi: 10.1038/nchembio.2421
K.Y. Chen, H.Q. Wang, Y. Yuan, et al., Angew. Chem. Int. Ed. 62 (2023) e202307602.
K.E. Malterud, T. Anthonsen, J. Hjortås, Tetrahedron Lett. 17 (1976) 3069–3072.
T. Gulder, P.S. Baran, Nat. Prod. Rep. 29 (2012) 899–934. doi: 10.1039/c2np20034a
J. Li, J.X. Sun, H.Y. Yu, et al., Chin. Chem. Lett. 24 (2013) 521–523. http://www.ccspublishing.org.cn//article/id/100031514
P.J. Pye, K. Rossen, R.A. Reamer, et al., J. Am. Chem. Soc. 119 (1997) 6207–6208.
H. Liang, W. Guo, J. Li, et al., Angew. Chem. Int. Ed. 61 (2022) e202204926.
S.V. Kumar, P.J. Guiry, Angew. Chem. Int. Ed. 61 (2022) e202205516.
J.F. Schneider, R. Fröhlich, J. Paradies, Isr. J. Chem. 52 (2012) 76–91. doi: 10.1002/ijch.201100082
Y.W. Huo, P.P. Shen, W.Z. Duan, et al., Chin. Chem. Lett. 29 (2018) 1359–1362. doi: 10.1016/j.cclet.2017.12.005
M. Cakici, Z.G. Gu, M. Nieger, et al., Chem. Commun. 51 (2015) 4796–4798.
Y. Morisaki, M. Gon, T. Sasamori, et al., J. Am. Chem. Soc. 136 (2014) 3350–3353. doi: 10.1021/ja412197j
C.H. Chen, W.H. Zheng, Org. Lett. 23 (2021) 5554–5558. doi: 10.1021/acs.orglett.1c01924
Z.M. Fan, W.T. Sun, Y. Yang, et al., Chin. Chem. Lett. 34 (2023) 107729. doi: 10.1016/j.cclet.2022.08.009
R.S. Cahn, C.K. Ingold, V. Prelog, Experientia 12 (1956) 81–94.
E.L. Eliel, S. H, Wilen in Stereochemistry of Organic Compounds, Wiley, Hoboken, 1994.
R. López, C. Palomo, Angew. Chem. Int. Ed. 61 (2022) e202113504.
Z. Hassan, E. Spuling, D.M. Knoll, et al., Angew. Chem. Int. Ed. 59 (2020) 2156–2170. doi: 10.1002/anie.201904863
Z. Hassan, E. Spuling, D.M. Knoll, et al., Chem. Soc. Rev. 47 (2018) 6947–6963. doi: 10.1039/c7cs00803a
S. Felder, S. Wu, J. Brom, et al., Chirality 33 (2021) 506–527. doi: 10.1002/chir.23335
G. Yang, J. Wang, Angew. Chem. Int. Ed. 63 (2024) e202412805. doi: 10.1002/anie.202412805
K. Tanaka, Bull. Chem. Soc. Jpn. 91 (2018) 187–194. doi: 10.1246/bcsj.20170346
K. Tanaka, H. Sagae, K. Toyoda, et al., J. Am. Chem. Soc. 129 (2007) 1522–1523. doi: 10.1021/ja0679127
K. Tanaka, H. Sagae, K. Toyoda, et al., Tetrahedron 64 (2008) 831–846. doi: 10.1016/j.tet.2007.10.085
T. Shibata, T. Uchiyama, K. Endo, Org. Lett. 11 (2009) 3906–3908. doi: 10.1021/ol9014893
T. Araki, K. Noguchi, K. Tanaka, Angew. Chem. Int. Ed. 52 (2013) 5617–5621. doi: 10.1002/anie.201300696
Y. Aida, J. Nogami, H. Sugiyama, et al., Chem. Eur. J. 26 (2020) 12579–12588. doi: 10.1002/chem.202001450
Y. Aida, J. Nogami, Y. Nagashima, et al., Chem. Sci. 14 (2023) 3963–3972.
K. Tanaka, T. Hori, T. Osaka, et al., Org. Lett. 9 (2007) 4881–4884. doi: 10.1021/ol702242x
T. Hori, Y. Shibata, K. Tanaka, Tetrahedron: Asymmetry 21 (2010) 1303–1306.
K. Mori, K. Ohmori, K. Suzuki, Angew. Chem. Int. Ed. 48 (2009) 5638–5641. doi: 10.1002/anie.200901974
S. Jung, Y. Kitajima, Y. Ueda, et al., Synlett. 27 (2016) 1521–1526.
M.Q. Salih, C.M. Beaudry, Org. Lett. 15 (2013) 4540–4543. doi: 10.1021/ol402096k
S. Wei, L.Y. Chen, J. Li, ACS Catal. 13 (2023) 7450–7456. doi: 10.1021/acscatal.3c01147
G. Islas-Gonzalez, M. Bois-Choussy, J. Zhu, Org. Biomol. Chem. 1 (2003) 30–32.
Q. Ding, Q. Wang, H. He, et al., Org. Lett. 19 (2017) 1804–1807. doi: 10.1021/acs.orglett.7b00570
S.Z. Yu, G.S. Shen, F.Q. He, et al., Chem. Commun. 58 (2022) 7293–7296. doi: 10.1039/d2cc01690g
G. Yang, Y. He, T. Wang, et al., Angew. Chem. Int. Ed. 63 (2023) e202316739.
J. Wang, M. Wang, Y. Wen, et al., Org. Lett. 26 (2024) 1040–1045. doi: 10.1021/acs.orglett.3c04200
X. Lv, F. Su, H. Long, et al., Nat. Commun. 15 (2024) 958.
C. Gagnon, E. Godin, C. Minozzi, et al., Science 367 (2020) 917–921. doi: 10.1126/science.aaz7381
L.F. Tan, M. Sun, H.X. Wang, et al., Nat. Synth. 2 (2023) 1222–1231. doi: 10.1038/s44160-023-00360-0
K. Kanda, K. Endo, T. Shibata, Org. Lett. 12 (2010) 1980–1983. doi: 10.1021/ol100444u
K. Kanda, R. Hamanaka, K. Endo, et al., Tetrahedron 68 (2012) 1407–1416. doi: 10.1016/j.tet.2011.12.031
K. Kanda, T. Koike, K. Endo, et al., Chem. Commun. (2009) 1870–1872. doi: 10.1039/b818904h
M.L. Delcourt, S. Felder, E. Benedetti, et al., ACS Catal. 8 (2018) 6612–6616. doi: 10.1021/acscatal.8b01872
M.L. Delcourt, S. Felder, S. Turcaud, et al., J. Org. Chem. 84 (2019) 5369–5382. doi: 10.1021/acs.joc.9b00372
Z. Dong, J. Li, T. Yao, et al., Angew. Chem. Int. Ed. 62 (2023) e202315603.
D. Wang, Y.B. Shao, Y. Chen, et al., Angew. Chem. Int. Ed. 61 (2022) e202201064.
K. Rossen, P.J. Pye, A. Maliakal, et al., J. Org. Chem. 62 (1997) 6462–6463. doi: 10.1021/jo971300a
D.C. Braddock, I.D. MacGilp, B.G. Perry, J. Org. Chem. 67 (2002) 8679–8681. doi: 10.1021/jo020451x
K. Mori, H. Kishi, T. Akiyama, Synthesis 49 (2017) 365–370.
P. Dorizon, C. Martin, J.C. Daran, et al., Tetrahedron Asymmetry 12 (2001) 2625–2630.
M.L. Delcourt, S. Turcaud, E. Benedetti, et al., Adv. Synth. Catal. 358 (2016) 1213–1218. doi: 10.1002/adsc.201501153
M.L. Delcourt, S. Felder, S. Turcaud, et al., J. Org. Chem. 84 (2019) 5369–5382. doi: 10.1021/acs.joc.9b00372
C. Zippel, Z. Hassan, A.Q. Parsa, et al., Adv. Synth. Catal. 363 (2021) 2861–2865. doi: 10.1002/adsc.202001536
Y. Zhao, H. Wang, B. Wu, et al., Org. Chem. Front. 6 (2019) 3956–3960. doi: 10.1039/c9qo01011d
Y. Zhao, Y.X. Ding, B. Wu, et al., J. Org. Chem. 86 (2021) 10788–10798. doi: 10.1021/acs.joc.1c01011
Y. Zhao, X.Q. Wang, Y.J. Yu, et al., J. Org. Chem. 86 (2021) 1262–1272. doi: 10.1021/acs.joc.0c02509
S. Yu, H. Bao, D. Zhang, et al., Nat. Commun. 14 (2023) 5239.
J. Li, C. Zhao, ACS Catal. 13 (2023) 14155–14162. doi: 10.1021/acscatal.3c03718
J. Li, Z. Dong, Y. Chen, et al., Nat. Commun. 15 (2024) 2338.
V. Dočekal, F. Koucký, I. Císařová, et al., Nat. Commun. 15 (2024) 3090.
D. Zhu, T. Mu, Z.L. Li, et al., Angew. Chem. Int. Ed. 63 (2024) e202318625.

Scheme 2 Rh-catalyzed enantioselective synthesis of planar-chiral [7]-[10]metacyclophanes.

Scheme 5 Rh-catalyzed enantioselective synthesis of PAH-based planar chiral bent cyclophanes.

Scheme 7 Rh-catalyzed enantioselective synthesis of planar chiral dithiaparacyclophanes.

Scheme 8 Ru-catalyzed enantioselective synthesis of planar chiral [10]- and [12]paracyclophanes.

Scheme 10 Palladium-catalyzed enantioselective synthesis of planar chiral metacyclophanes.

Scheme 12 The first example of chiral PTC-catalyzed enantioselective cycloetherification.

Scheme 17 Construction of axial-planar indole macrocycles via kinetic resolution strategy.

Scheme 18 NHC-catalyzed stereoselective synthesis of [11-14]planar chiral macrocycles.

Scheme 19 NHC-catalyzed macrocyclization in the synthesis of planar chiral macrolactones.

Scheme 22 Catalytic asymmetric ortho-lithiation and dilithiation of achiral [n]paracyclophane.

Scheme 23 Enantioselective synthesis of [7.7]paracyclophanes via double Sonogashira coupling.

Scheme 24 Asymmetric synthesis of planar chiral [2.2]paracyclophane through catalytic desymmetrization.

Scheme 27 CPA-catalyzed enantioselective KR and desymmetrization of diamido[2.2]paracyclophane.

Scheme 30 NHC-catalyzed asymmetric aldehyde oxidation esterification of racemic cyclophanes through DKR.

Scheme 31 NHC-catalyzed enantioselective desymmetrization of pseudo-para diformyl[2.2]paracyclophanes.

Scheme 32 NHC-catalyzed enantioselective desymmetrization of pseudo-gem diformyl[2.2]paracyclophanes.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:

















 下载:
下载: