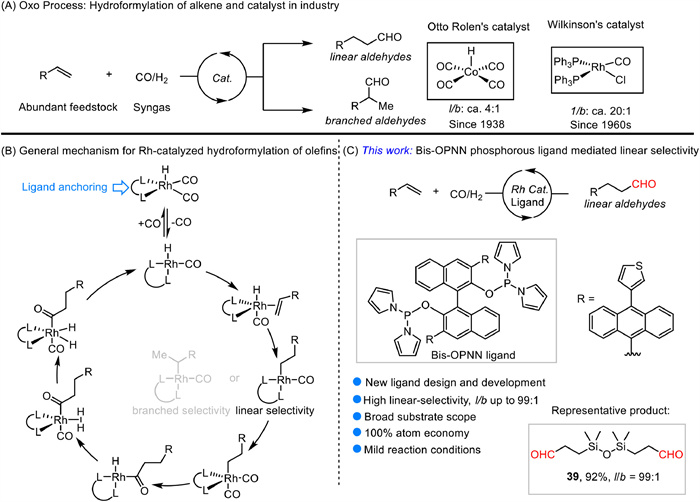
Scheme 1.
(A) Oxo process: Hydroformylation of olefins in industry. (B) General mechanism. (C) This work: Bis-OPNN phosphorus ligand controlled Rh-catalyzed linear-selective hydroformylation of alkenes.
A modified Bis-OPNN phosphorus ligand for Rh-catalyzed linear-selective hydroformylation of alkenes
Luyun Zhang , Ding Liu , Huri Piao , Zhenhua Jia , Fen-Er Chen

Hydroformylation, or the "oxo reaction", is a critical transformation in industrial process and also represents a cornerstone of transition-metal-catalyzed organic synthesis [1–4]. The ability to introduce a carbon-carbon bond in a single step, while simultaneously increasing the molecular complexity of the substrate, makes hydroformylation an indispensable tool in the production of fine chemicals, pharmaceuticals, agrochemicals, and a myriad of other value-added products [5–11]. Since its discovery by Otto Roelen in 1938, this reaction has become a vital process for the conversion of alkenes into aldehydes by the addition of a formyl group (−CHO) using syngas, a mixture of carbon monoxide (CO) and hydrogen (H₂) [12]. However, the hydroformylation process is often complicated by side reactions such as olefin isomerization and hydrogenation, leading to a mixture of products and necessitating further refinement to improve selectivity and yield. Therefore, controlling the regio-selectivity of the reaction for desired aldehydes over unexpected products still remains a significant challenge.
Since Wilkinson et al. introduced the rhodium catalyst into the field of hydroformylation, demonstrating superior activity and selectivity compared to cobalt catalysts (Scheme 1A) [1,13]. The development of ligands to effectively regulate the coordination environment of the rhodium catalyst has become paramount for steering the reaction towards the desired product [14,15]. In recent years, significant advancements have been made in the design of ligands for hydroformylation, with a particular focus on bidentate phosphorus ligands, such as Xantphos [16–18], Biphephos ligands [19], which feature high steric hindrance due to the incorporation of phosphite ester bonds.
For instance, Eastman Kodak company and Kohlpaintner et al. designed a novel bidentate phosphorus ligand and employed in Rh-catalyzed hydroformylation reactions [20–22]. These ligands demonstrated exceptionally high linear selectivity in hydroformylation of propylene (l/b = 30) [20]. Casey et al. discovered that the bond angle between the phosphorus atoms in the P-Rh-P complex played a crucial role in determining the selectivity of the linear hydroformylation reaction [23–25]. Building on this insight, Kamer et al. explored ligands with greater bond angle between P-Rh-P which further advanced the field [26,27]. However, these studies primarily focused on selected aliphatic olefins. For styrene, Matt et al. utilized a hemispherical diphosphanes ligand with a large bite angle, resulting in an l/b ratio of 3.3 [28]. Zhang et al. developed a series of tetraphosphorus ligand based on the biphenyl backbone to control the regio-selective hydroformylation of styrene, allyl, and vinyl derivatives, but achieving lower selectivity for allylic substrates with 31% isomerization by-product [29–31].
As shown in Scheme 1B, the probable mechanism of hydroformylation with bidentate phosphorus ligands initiated from ligand anchoring the rhodium catalyst, facilitating the formation of Rh-H intermediate (I). Following the alkene coordination, hydrometallation to produce an alkyl metal species, coordination to carbon monoxide followed by insertion, oxidative addition of hydrogen, and finally reductive elimination, the desired aldehyde was generated [32–34]. To continue our interest in the design and development of ligand for hydroformylation [35–39], we herein report the application of a Bis-OPNN phosphorus ligand in Rh-catalyzed hydroformylation of various alkenes with highly linear selectivity under mild conditions, using BINOL as the skeleton with bulky substitutes at its 3, 3' positions.
We started our study with styrene 1a as the model substrate to screen various ligands (Table 1). Inspired by previous works, we prepared a series of bidentate phosphorus ligands and tested their abilities to control the Rh-catalyzed hydroformylation with linear selectivity. To our delight, L1 yielded the linear product benzenepropanal 2 in 72% yield with 76:23 linear to branched ratio (l/b). The moderate regioselectivity indicated that the high π-acidity of the ligand probably plays a critical role in favoring linear product formation, suggesting that the pyrrole moiety enhances regioselectivity. Next, we modified the ligand's structure by replacing the biphenyl group with a naphthalene moiety (See details in Supporting information) [40]. This alteration slightly improved both the yield and regioselectivity, likely due to the increased rigidity of the naphthalene backbone (L3). However, when we changed the naphthalene skeleton with spiroindene, the results were not satisfactory (L2). Next, we selected L3 as the promising ligand and modified the ortho positions with diverse substitutes. Substituting the ortho position with a phenyl group (L4) produced the comparable performance to L3. However, when 4-methyl benzene group was installed on the naphthalene, both the yield and regioselectivity decreased slightly (L5). In contrast, substituting trifluoromethyl at the ortho position of the benzene ring increased the yield, although the selectivity remained similar (L6). These findings suggest that the electronic properties of substituents at the ortho position of the aromatic substitutes have minimal impact on regioselectivity. Subsequently, we designed and synthesized ligands L7 and L8, which possess significant steric hindrance. Surprisingly, these ligands led to a notable improvement in both yield and regioselectivity. However, when the pyrrole group was replaced with the indole moiety, the reaction exhibited lower efficiency (L9). Moreover, the phosphinite ligand L10 exhibited poor selectivity as well. Notably, commercially available ligands such as Xantphos (L11) and DPPB (L12), provided poor regioselectivities.
 DownLoad:
CSV
DownLoad:
CSV
 |
|||
| Entry | Change from the "standard conditions" | Yield of 2a (%)b | l/b ratio |
| 1 | None | 90 (86) | 94:6 |
| 2 | [Rh(cod)Cl]2 instead of Rh(acac)(CO)2 | 74 | 94:6 |
| 3 | L8 (0.2 mol%) | 16 | 94:6 |
| 4 | L8 (0.4 mol%) | 93 (90) | 96:4 |
| 5 | L8 (0.8 mol%) | 69 | 94:6 |
| 6 | 60 ℃ | 47 | 88:12 |
| 7 | 120 ℃ | 83 | 87:13 |
| 8 | CO/H2 (3:1) = 1.0 MPa | 92 | 96:4 |
| 9 | CO/H2 (1:3) = 1.0 MPa | 90 | 95:5 |
| a Reactions were performed with 1a (0.5 mmol), Rh(acac)(CO) 2 (0.2 mol%) in toluene (2.0 mL). Yields and l/b ratios were determined by GC–MS analysis using hexadecane as an internal standard. b Isolated yield. | |||
With the promising results, we selected L8 as the optimal ligand and further optimized the reaction conditions (Table 2 and Tables S1–S5 in Supporting information). Upon screening various Rh complexes, [Rh(acac)(CO)2] emerged as the most effective catalyst precursor (entries 1 and 2, Table S1 in Supporting information). The best results were obtained with a 1:2 ratio of rhodium to ligand (entry 3), with deviations in this ratio leading to lower yields (entries 3 and 5, Table S2 in Supporting information). Optimal conditions were found to be a reaction temperature of 80 ℃ and a syngas pressure of 1 MPa in a 1:1 CO/H2 ratio (entries 6–9, Tables S3 and S4 in Supporting information). Other reaction parameters were also systematically screened, and the results are detailed in Table S5 (Supporting information).
In general, this Bis-OPNN phosphorus ligand induced Rh-catalyzed hydroformylation demonstrated broad functional group tolerance, enabling the efficient conversion of various olefins into the linear aldehydes with high regioselectivity (Scheme 2). Under optimial conditions, styrene and its derivatives with various substituents on the benzene ring underwent the desired hydroformylation, producing the corresponding aldehydes with high linear-to-branched (l/b) ratios (2–24). Notably, a methyl group substitued at the ortho postion, rather than at para position (3) or meta position (4), retarded the hydroformylation process to give 5 in 73% yield with high regioselectivity. However, the methoxyl group substituted at different positions on the benzene ring did not show a significant substitution effect (6–8). Then, we systematically explored the scope of halogenated styrenes with fluoride, chloride and bromide at various postions, affording the desired aldehydes in 80%−88% yields with good linear selectivities (9–17). Moreover, the stertic tert‑butyl group at para position was also applicable in this reaction to generate 18 in 88% yield with 97:3 l/b ratio. In addition, the electron-withdrawing groups, such as cyano, ester, and trifluoromethyl, were well tolerated to give 19–21 in 79%−88% yields with good selectivity. To be noted, the free carboxylic acid was also suitable with standard conditions to yield 22 with excellent regioselectivity up to 99:1. The di-substituted styrenes with methoxyl and methyl groups, were next examined to produce 23 and 24 respectively. Poly-aromatic alkenes, including 2-vinylnaphthalene and 4-vinyl-1, 1′-biphenyl, were accommodated to afford 25 and 26 successfully.
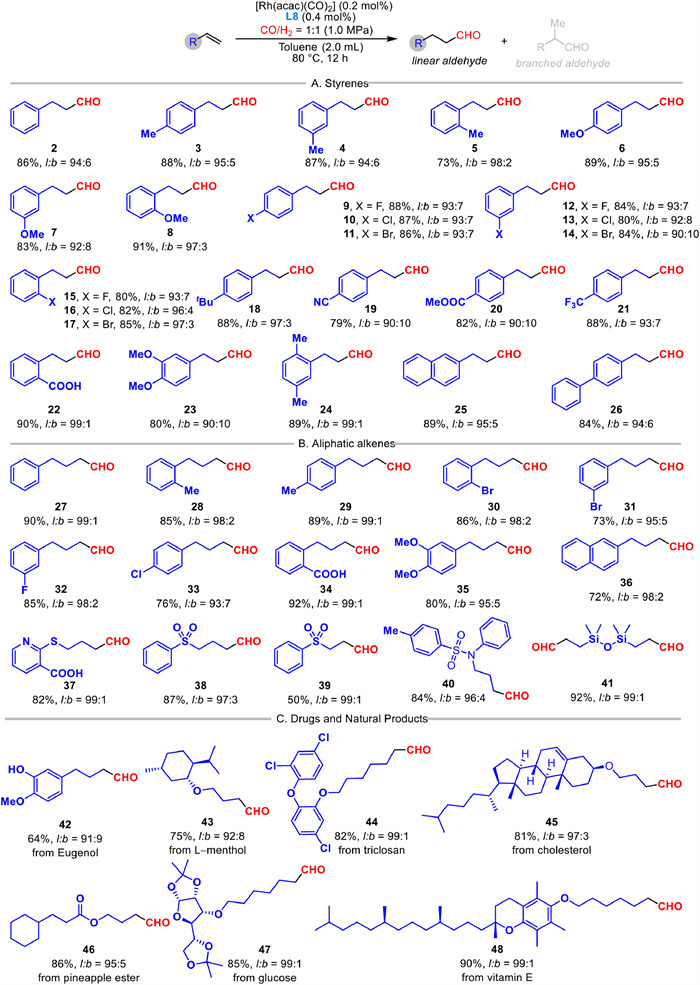
As shown in Scheme 2B, we next explore the scope of aliphatic alkenes to afford the corresponding linear aldehydes under standard conditions with excellent regioselectivites. We found that a series of phenylbutanals were obtained from allylbenzene and its derivatives in 73%−92% yields with good to excellent linear selectivities (27–35). Moreover, both electron-donating and electron-withdrawing substitutes at diverse positions of benzene ring or naphthalene ring were well accommodated with this efficient hydroformylation process. Furthermore, this reaction tolerated diverse functional groups, such as hetero atoms, pyridine, free carboxylic acids, sulfonamides, sulfides, and silyl ethers, producing the desired linear aldehydes in good to excellent yields and regioselectivities (37–41). To illustrate the potential application, we applied the Bis-OPNN phosphorus ligand in Rh-catalyzed hydroformylation to modify natural products and small-molecule drugs, offering a direct approach to generate a diverse range of target molecules. For instance, eugenol was directly converted into its corresponding aldehyde in 64% yield with 91:9 regioselectivity (42). Other aliphatic alkenes with natural product skeletons, inculding l-menthol, triclosan, and cholesterol, were subjected to the optimal comditions to afford the aldehydes 43–45 respectively. The pineapple ester, protected glucose and vitamin E derivative tethering with the terminal double bonds, were efficiently modified via our hydroformylation protocol to generate the linear aldehydes in excellent regioselectivities (46–48).
Subsequently, our protocol was successfully applied for the synthesis of pharmaceutical compounds through a two-step sequential route (Scheme 3A). For instance, cinacalcet, for the treatment of hyperparathyroidism in patients with chronic kidney disease, was prepared via the reductive amination of its aldehyde precursor in 89% yield, which was generated through our hydroformylation process in 85% yield (49). Following the similar strategy, piperazine-core drugs, such as buspirone (50), brexpiprazole (51), and aripiprazole (52) were also synthesized efficiently, which demonstrated the effectiveness of this strategy in converting readily available olefins into amine containing pharmaceuticals through a linear synthesis route.
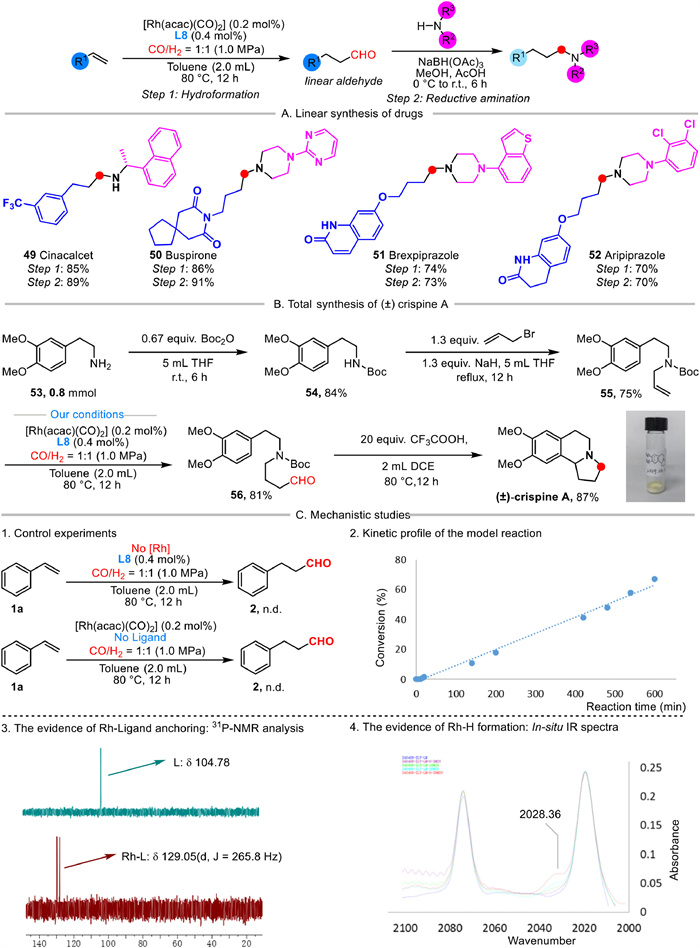
Furthermore, our hydroformylation protocol was applied in the concise total synthesis of (±)-crispine A, a biologically active pyrroloisoquinoline natural product (Scheme 3B). The synthetic route commenced with commercially available starting material 2-(3, 4-dimethoxy)−2-phenylethan-1-amine 53, involving BOC protected 54 and subsequent allylation to yield N-Boc protected allylamine 55 in 84% and 85% yields respectively. Then, we utilized 55 as the sustrate for hydroformylation to produce the aldehyde 56 in 81% yield. Finally, (±)-crispine A was achieved through N-Boc deprotection and simultaneously intramolecular Pictet–Spengler cyclization, delivering with 87% yield.
To gain more mechanistic insights of this ligand controlled Rh-catalyzed linear selective hydroformylation, the preliminary studies were conducted (Scheme 3C). In the absence of either the rhodium catalyst or the ligand, the desired aldehyde product 2 was not detected, which indicated that rhodium catalyst and this Bis-OPNN phosphorus ligand are both essential for the reaction. Moreover, the kinetic studies suggested that the reaction likely proceeded in the first-order kinetics. Furthermore, the coordination between rhodium and the two phosphorus atoms of the ligand was also confirmed by 31P-NMR analysis, highlighting the ligand's crucial role in controlling regioselectivity. In addition, the presence of the active Rh-H species was detected using in-situ high-pressure infrared spectroscopy, providing additional support for the Rh-catalyzed hydroformylation pathway.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Luyun Zhang: Investigation. Ding Liu: Investigation. Huri Piao: Supervision. Zhenhua Jia: Writing – original draft. Fen-Er Chen: Supervision, Conceptualization.
We gratefully acknowledge the financial support from the National Key Research and Development Program of China (No. 2021YFF0600704).
Supplementary material associated with this article can be found, in the online version, at doi:
O. Roelen, German patent DE 849548, 1938/1952; U.S. Patent 2327066.
H. Adkins, G. Krsek, J. Am. Chem. Soc. 71 (1949) 3051. doi: 10.1021/ja01177a032
X.F. Wu, X. Fang, L. Wu, et al., Acc. Chem. Res. 47 (2014) 1041–1053. doi: 10.1021/ar400222k
P. Kalck, M. Urrutigoïty, Chem. Rev. 118 (2018) 3833–3861. doi: 10.1021/acs.chemrev.7b00667
F. Ungváry, Coordin. Chem. Rev. 248 (2004) 867–880.
F. Ungváry, Coordin. Chem. Rev. 249 (2005) 2946–2961.
F. Ungváry, Coordin. Chem. Rev. 251 (2007) 2087–2102.
E.V. Gusevskaya, J. Jiménez-Pinto, A. Börner, ChemCatChem 6 (2014) 382–411. doi: 10.1002/cctc.201300474
R. Franke, D. Selent, A. Börner, Chem. Rev. 112 (2012) 5675–5732. doi: 10.1021/cr3001803
R.M.B. Carrilho, M.J.F. Calvete, G. Mikle, L. Kollár, M.M. Pereira, Chin. J. Chem. 42 (2024) 199–221. doi: 10.1002/cjoc.202300384
C. Botteghi, R. Ganzerla, M. Lenarda, G. Moretti, J. Mol. Catal. 40 (1987) 129–182.
Y. Ning, T. Ohwada, F.E. Chen, Green Synth. Catal. 2 (2021) 247–266.
D. Evans, A. Osborn, J. Chem. Soc. 12 (1968) 3133–3142.
C. Li, Z. Li, K. Tan, G. Liu, Eur. J. Org. Chem. 26 (2023) e202300398.
R. Bellini, S.H. Chikkali, G. Berthon-Gelloz, N. H J, Reek Angew. Chem. Int. Ed. 50 (2011) 7342–7345. doi: 10.1002/anie.201101653
L.A. Van Der Veen, M.D.K. Boele, C. Bo, et al., J. Am. Chem. Soc. 120 (1998) 11616–11626.
M. Kranenburg, Y.E.M. Van Der Burgt, J. Fraanje, et al., Organometallics 14 (1995) 3081–3089. doi: 10.1021/om00006a057
J.J. Carbó, C. Bo F. Maseras, P.W.N.M. Van Leeuwen, J. Am. Chem. Soc. 123 (2001) 7630–7637.
G.D. Cuny, S.L. Buchwald, J. Am. Chem. Soc. 115 (1993) 2066–2068. doi: 10.1021/ja00058a079
T.J. Devon, G.W. Phillips, T.A. Puckette, J.L. Stavinoha, J.J. Vanderbilt, (to Eastman Kodak Company), U.S. Patent 4694109, 1987.
W.A. Herrmann, C.W. Kohlpaintner, E. Herdtweck, P. Kiprof, Inorg. Chem. 30 (1991) 4271–4275. doi: 10.1021/ic00022a032
W.A. Herrmann, R. Schmid, C.W. Kohlpaintner, T. Priermeier, Organometallics 14 (1995) 1961–1968. doi: 10.1021/om00004a057
C.P. Casey, G.T. Whiteker, C.F. Campana, D.R. Powell, Inorg. Chem. 29 (1990) 3376–3381. doi: 10.1021/ic00343a023
C.P. Casey, G.T. Whiteker, M.G. Melville, et al., J. Am. Chem. Soc. 114 (1992) 5535–5543. doi: 10.1021/ja00040a008
C.P. Casey, E.L. Paulsen, D.R. Powell, et al., J. Am. Chem. Soc. 119 (1997) 11817–11825.
P.C.J. Kamer, P.W.N.M. Van Leeuwen, J.N.H. Reek, Acc. Chem. Res. 34 (2001) 895–904.
A. Van Rooy, P.C.J. Kamer, A.L. Spek, et al., Organometallics 15 (1996) 835–847.
D. Sémeril, C. Jeunesse, D. Matt, L. Toupet, Angew. Chem. Int. Ed. 45 (2006) 5810–5814. doi: 10.1002/anie.200601978
S. Yu, Y. Chie, X. Zhang, et al., Org. Lett. 11 (2009) 241–244. doi: 10.1021/ol802479y
C. Cai, S. Yu, B. Cao, X. Zhang, Chem. Eur. J. 18 (2012) 9992–9998. doi: 10.1002/chem.201201396
R.T. Zhang, X. Yana, X.M. Zhang, Green Synth. Catal. 3 (2022) 40–45.
R.F. Heck, D.S. Breslow, J. Am. Chem. Soc. 83 (1961) 4023–4027. doi: 10.1021/ja01480a017
D. Evans, G. Yagupsky, G. Wilkinson, J. Chem. Soc. A (1968) 2660–2665.
B. Breit, W. Seiche, Synthesis 1 (2001) 1–36.
P. Gao, G. Liang, F.E. Chen, et al., Nat. Commun. 12 (2021) 4698.
T. Ru, G. Liang, L. Zhang, Y. Ning, F.E. Chen, ChemCatChem 13 (2021) 5073–5077. doi: 10.1002/cctc.202101352
P. Gao, M. Ke, T. Ru, G. Liang, F.E. Chen, Chin. Chem. Lett. 33 (2022) 830–834.
L. Zhang, Y. Ning, B. Ye, T. Ru, F.E. Chen, Green Chem. 24 (2022) 4420–4424. doi: 10.1039/d2gc00802e
T. Ru, Y. Zhang, F.E. Chen, et al., Molecules 29 (2024) 2039. doi: 10.3390/molecules29092039
S. Liu, Y. Lu, J. Chen, et al., Green Synth. Catal. 4 (2023) 71–75.

Scheme 1 (A) Oxo process: Hydroformylation of olefins in industry. (B) General mechanism. (C) This work: Bis-OPNN phosphorus ligand controlled Rh-catalyzed linear-selective hydroformylation of alkenes.

Scheme 2 Scope of substrate. (A) Scope of styrenes. (B) Scope of aliphatic alkenes. (C) Drugs and natural products. Reactions were performed with alkenes (0.5 mmol), Rh(acac)(CO)2 (0.2 mol%) in toluene (2.0 mL). Regioselectivity were determined with GC–MS and 1H NMR analysis using the crude reaction mixture. Isolated yield.

Scheme 3 Practical application and preliminary mechanistic studies. (A) Linear syntheis of drugs. (B) Total synthesis of (±)-crispine A. (C) Mechanistic studies.
Table 1. Ligand screening and selected optimization of reaction conditions.a
|
|||
| Entry | Change from the "standard conditions" | Yield of 2a (%)b | l/b ratio |
| 1 | None | 90 (86) | 94:6 |
| 2 | [Rh(cod)Cl]2 instead of Rh(acac)(CO)2 | 74 | 94:6 |
| 3 | L8 (0.2 mol%) | 16 | 94:6 |
| 4 | L8 (0.4 mol%) | 93 (90) | 96:4 |
| 5 | L8 (0.8 mol%) | 69 | 94:6 |
| 6 | 60 ℃ | 47 | 88:12 |
| 7 | 120 ℃ | 83 | 87:13 |
| 8 | CO/H2 (3:1) = 1.0 MPa | 92 | 96:4 |
| 9 | CO/H2 (1:3) = 1.0 MPa | 90 | 95:5 |
| a Reactions were performed with 1a (0.5 mmol), Rh(acac)(CO) 2 (0.2 mol%) in toluene (2.0 mL). Yields and l/b ratios were determined by GC–MS analysis using hexadecane as an internal standard. b Isolated yield. | |||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: