Scheme 1.
Catalytic asymmetric IEDDA reactions of 2-pyrones with aryl enol ethers.
Catalytic asymmetric inverse-electron-demand Diels–Alder reaction of 2-pyrones with aryl enol ethers
Fangqing Zhang , Yu Wang , Zhenda Tan , Yangbin Liu , Lijuan Song , Xiaoming Feng

Chiral aryl cyclohex-3-en ether scaffold is widely present in biologically active natural products and drug molecules (Scheme 1a) [1-5], such as xylariacyclone A, xculeatusquinone C, magmenthane G and galantamine. Among these, magmenthane G showed great neuroprotective effects against glutamic acid- and oxygen glucose deprivation (OGD)-induced SK-N-SH cell injury [3]. Moreover, galantamine is an acetylcholinesterase inhibitor for the treatment of Alzheimer’s disease [4,5]. Considering the significance and utility of these structures, a general and efficient method to construct the fascinating chiral aryl cyclohex-3-en ether scaffold is highly desirable.
Catalytic asymmetric all-carbon-based Diels-Alder reaction is one of the most fundamental and efficient protocols for creating optically active six-membered carbocycles, which are key structural motifs in bioactive molecules and functional materials [6-10]. The inverse-electron-demand Diels–Alder (IEDDA) reactions featuring an electron-enriched dienophile and an electron-deficient diene provide an alternative and promising strategy to construct such chiral six-membered carbocycles [11-14]. However, compared to the well-established asymmetric normal-electron-demand Diels–Alder (NEDDA) reactions, the type of asymmetric IEDDA reactions is still underdeveloped thus far (Scheme 1b) [15-21]. Electron-deficient 2-pyrone derivatives are privileged diene components for IEDDA reactions to afford bridged bicyclic lactone structures as versatile intermediates, which have a wide range of applications in complex natural synthesis via a diverse array of transformations, including retro-Diels–Alder reaction, ring opening or aromatization [22-29]. As such, the chiral aryl cyclohex-3-en ether scaffold can be readily accessed via the ring-opening of a bridged bicyclic lactone, which is readily assembled from an electron-deficient diene 2-pyrone and an aryl enol ether via an enantioselective IEDDA reaction (Scheme 1c).
Since the pioneering work by Posner [30-32] and Markó [33-35], catalytic asymmetric IEDDA reactions involving 2-pyrone derivatives have attracted increasing attention from synthetic chemists [36-53]. Among various electron-enriched dienophiles, the readily available enol ethers have been an important partner for the asymmetric IEDDA reactions of 2-pyrones. Although recent impressive works have been contributed from the groups of Cai [38,43], Torre [48,49] and Guo [50], these reactions were mainly limited to alkyl enol ethers regardless of acyclic or cyclic dienophiles. Comparingly, the enantioselective IEDDA reaction of 2-pyrones with aryl enol ethers remains elusive. Until now, only one example was reported where a phenyl enol ether underwent an IEDDA reaction with a 2-pyrone derivative to afford the corresponding bridged bicyclic lactone in 11% yield and 75% ee catalyzed by Yb(OTf)3 and BINOL [34]. Moreover, the use of 1,2-disubstituted aryl enol ethers as dienophiles has never been explored in the catalytic asymmetric IEDDA reactions (Scheme 1d). The possible challenges are due to: (1) Aryl enol ethers possess a p-π conjugated effect between the O-atom and the aryl group, which would reduce the electron density of the enol moiety. (2) The steric hindrance of aryl enol ethers is larger than that of alkyl enol ethers, which is also unfavorable to the activity of IEDDA reaction. (3) Additional challenges of precise multi-stereoinduction (diastereoselectivity and enantioselectivity) and potentially diverse mechanisms (pericyclic or stepwise) also limit the development of aryl enol ethers in asymmetric IEDDA reactions [9,13,28].
Based on our previous works on asymmetric IEDDA reaction of 2-pyrones [46,47], herein we demonstrate a chiral N, N’-dioxide/Lewis acid complex [54-60] catalyzed all-carbon-based IEDDA reaction using 3-carboalkoxyl-2-pyrones and aryl enol ethers as the reaction partners (Scheme 1e). This broadly applicable method provides an excellent diastereo- and enantioselective approach to access the chiral bridged lactone structures in high yields (up to 96%) and stereoselectivities (up to 20:1 dr, 97:3 er) under mild conditions. Furthermore, the aryl cyclohex-3-en ether scaffold is facilely prepared by transesterification, highlighting the potential of this reaction in total synthesis.
As summarized in Table 1, our studies commenced by selecting 3-carbomethoxy-2-pyrone A1 and phenyl vinyl ether B1 as model substrates to optimize the reaction conditions (for detailed optimization see Tables S1–S5 in Supporting information). At the beginning, a preliminary investigation of Lewis acids was carried out in DCE at 35 ℃ in the presence of N, N′-dioxide ligand L (entries 1–4). To our delight, when Eu(OTf)3 was employed as the central metal, the catalytic asymmetric IEDDA reaction occurred smoothly, generating the desired C1 in 76% yield and 81:19 er. Then, various chiral N, N’-dioxide ligands with different backbones of chiral amino acids and substituents on the aromatic amide group were evaluated. However, none of other ligands performed a better result (entries 5–8). The subsequent screening of solvents suggested that dichloromethane could increase both yield and enantioselectivity (95%, 88:12 er, entry 11). When the reaction was conducted at 10 ℃ and 4 Å MS was added as additive, C1 could be obtained in 76% yield with 91:9 er (entry 12). Intriguingly, the use of Sm(OTf)3 as Lewis acid could slightly increase the enantioselectivity in the current reaction condition (92:8 er, entry 13). Furthermore, enhanced results of 83% yield and 95:5 er for the cycloaddition product C2 were obtained by using 3-carboethoxy-2-pyrone as the diene and 4-(vinyloxy)−1,1′-biphenyl as the dienophile (entry 14).
 DownLoad:
CSV
DownLoad:
CSV
 |
|||||
| Entry | Ligand | Lewis acid | Solvent | C1 yield (%) b | C1 er c |
| 1 | L3-PrMe2 | Mg(OTf)2 | DCE | 27 | 59:41 |
| 2 | L3-PrMe2 | Yb(OTf)3 | DCE | 50 | 57:43 |
| 3 | L3-PrMe2 | Sm(OTf)3 | DCE | 57 | 76:24 |
| 4 | L3-PrMe2 | Eu(OTf)3 | DCE | 76 | 81:19 |
| 5 | L3-PiMe2 | Eu(OTf)3 | DCE | 24 | 55:45 |
| 6 | L3-RaMe2 | Eu(OTf)3 | DCE | 77 | 79:21 |
| 7 | L3-PrPr2 | Eu(OTf)3 | DCE | 42 | 72:28 |
| 8 | L3-PrMe(4-tBu) | Eu(OTf)3 | DCE | 70 | 80:20 |
| 9 | L3-PrMe2 | Eu(OTf)3 | MeCN | 34 | 77:23 |
| 10 | L3-PrMe2 | Eu(OTf)3 | CHCl3 | 70 | 74:26 |
| 11 | L3-PrMe2 | Eu(OTf)3 | DCM | 95 | 88:12 |
| 12 d | L3-PrMe2 | Eu(OTf)3 | DCM | 76 | 91:9 |
| 13 d | L3-PrMe2 | Sm(OTf)3 | DCM | 73 | 92:8 |
| 14 d, e | L3-PrMe2 | Sm(OTf)3 | DCM | 83 | 95:5 |
| a General reaction conditions: A (0.1 mmol, 1.0 equiv.), B (0.15 mmol, 1.5 equiv.), ligand (10 mol%), Lewis acid (10 mol%) and DCE (1.6 mL) at 35 ℃ for 24 h. b NMR yield detected by using p-xylene as an internal standard. c The er value was determined by HPLC analysis on a chiral stationary phase. d The reaction was conducted at 10 ℃ for 96 h, and 4 Å MS (50 mg) was added. e Product was C2. |
|||||
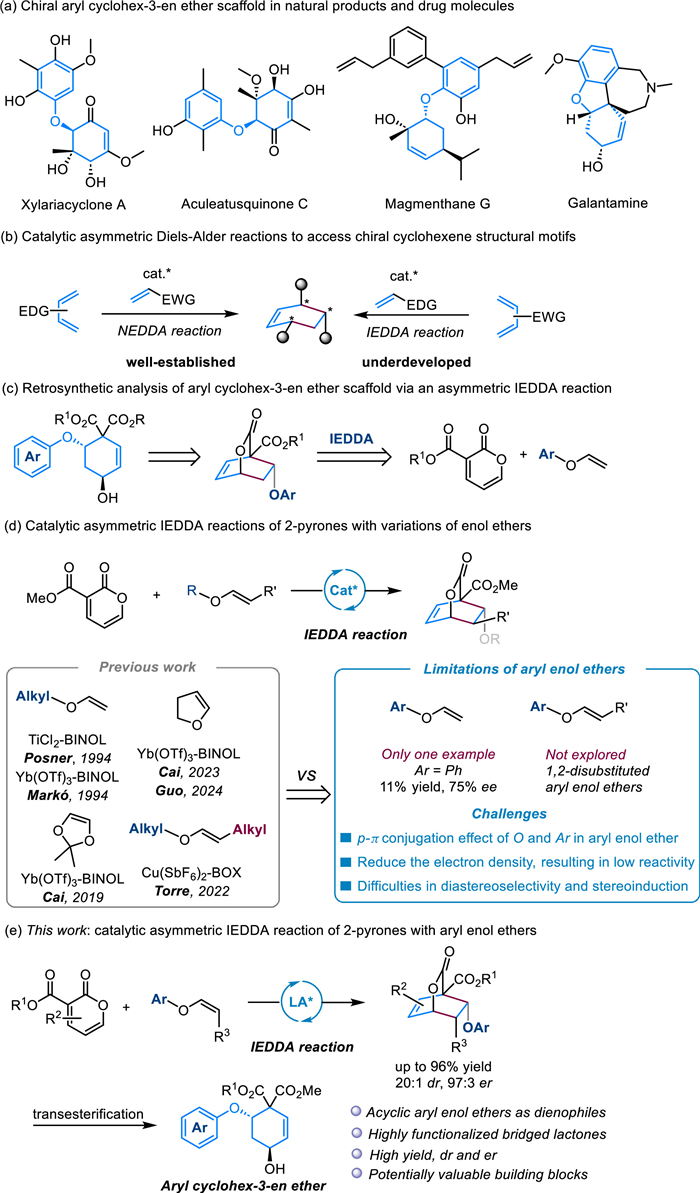
With the optimized reaction conditions in hand, the substrate scope of this catalytic asymmetric IEDDA reaction was explored. Firstly, we chose 3-carboethoxy-2-pyrone as the electron-deficient diene to evaluate the scope of non-substituted aryl vinyl ethers (Scheme 2). A variety of electron-donating alkyl (Me, iPr and tBu) and alkoxyl (OMe, OPh and OTBS) substituents on different positions of the phenyl moiety were found to be well tolerated, thus delivering the corresponding products C4–C11 in high yields and enantioselectivities (76%–91% yield, > 20:1 dr, 94:6–95:5 er). Substrates bearing diverse sensitive functional groups, such as -SMe, -Bpin, -TMS, -NMeBoc, morpholine and -CH2CO2Me, were also compatible with this protocol, providing C12–C17 in good yields but with slightly decreased er values in some cases. Notably, 4-Bpin-substituted aryl enol ether took part in the reaction smoothly to give product C13 in excellent results (80% yield, 97:3 er), which potentially could be employed for further down-stream transformations. In addition, multiple substituents on the phenyl ring including the complex groups, such as vanitrope-derived aryl enol ether (C20) were well accommodated, and the desired products C18–C21 were afforded with good results. Remarkably, when the challenging electron-deficient halogens (4-F, 4-Cl, 4-Br, and 4-I) were incorporated into the aryl enol ethers, the catalytic IEDDA reaction also proceeded smoothly at an increased temperature (20 ℃), giving C22–C25 in moderate yields and enantioselectivities. Subsequently, fused-/heteroaryl-substituted bridged lactone products, such as 1,2,3,4-tetrahydronaphthalene (C26), 1,4-benzodioxan (C27), naphthalene (C28), benzofuran (C29) and indole (C30) were obtained in 67%–96% yield with 92:8–95:5 er. What’s more, estrone-derived enol ether also underwent the diastereoselective IEDDA reaction efficiently, delivering an exclusive diastereoisomer C31 (> 20:1 dr). 2-Pyrones bearing various ester groups such as methyl, isopropyl, allyl and benzyl were well tolerated, affording C32-C35 in good yields and enantioselectivities (65%–85% yield, 92:8–94:6 er). Meanwhile, the absolute configuration of product C32 was determined by X-ray crystallography analysis. A methyl group at the C4 position of 2-pyrone resulted a decrease of reactivity and enantioselectivity, giving C36 in 46% yield with 90:10 er. However, C6 substituted 2-pyrone was unreactive in the current system, probably due to the significant steric effect.
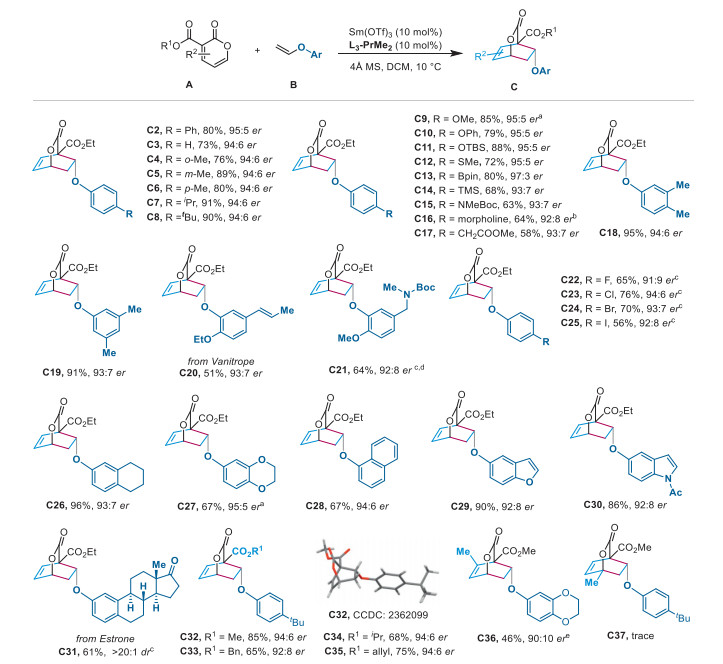
Next, we turned our attention to the enantioselective IEDDA reaction of 2-pyrones with acyclic disubstituted aryl enol ethers. Potential challenges associated with such dienophiles lie in the increased steric hindrance resulting in lower reactivity for IEDDA cycloaddition, and the diastereospecificity for cis or trans product from Z or E substrates. After slightly optimizing the reaction conditions, the combination of Eu(OTf)3 and L could efficiently catalyze the asymmetric IEDDA reaction of 2-pyrones with Z-1,2-disubstituted aryl enol ethers. As shown in Scheme 3, substrates with a Z-2-methly at the enol ether were smoothly converted to the densely functionalized bridged bicyclic lactones C38–C42 in good yields with excellent diastereo- and enantioselectivities (51%–91% yield, > 20:1 dr, 93:7–97:3 er). When bulky tert–butyldimethylsilyl-protected aryl enol ethers were investigated, the Z-disubstituted dienophile could afford the desired product C43 in good yield with moderate er, while the E-dienophile was not reactive enough to undergo the cycloaddition (C44). Intriguingly, we were pleased to find that the more challenging 1,1-disubstituted aryl enol ether was reactive as well, providing the desired cycloaddition product C45 with sterically encumbered contiguous quaternary stereocenters. In addition, the enantioselective IEDDA reaction of alkyl enol ethers such as 2,3-dihydrofuran (C46), cyclohexyl enol ether (C47) and benzyl enol ether (C48) with 2-pyrone gave the corresponding products in good yields and moderate enantioselectivities.
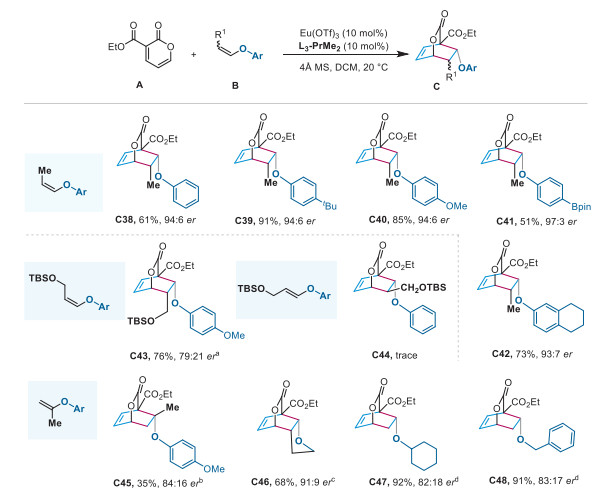
To gain more insight into the reaction mechanism and the origins of stereoselectivity, we have conducted quantum chemical calculations through density functional theory (DFT) using 3-methoxycarbonyl-2-pyrone (1) and aryl enol ether (2) as the model substrates (Fig. 1A). The computational analysis shows that the chiral Lewis acid [Sm]* coordinates with 2-pyrone 1 to form an octahedral structure [Sm]*_1 with four oxygen atoms of L and two carbonyl oxygen atoms of 1. Initially, the complex [Sm]*_1 associates with the enol ether 2 via weak non-covalent interactions to generate a stabilized reactant complex [Sm]*_1_2 (–0.8 kcal/mol).
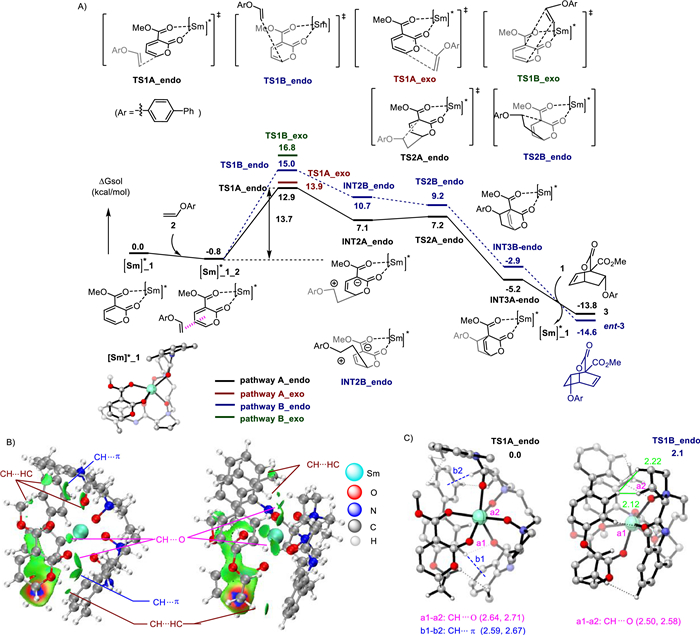
Next, we have explored both concerted and stepwise mechanisms for the attack of the aryl enol ether in an endo or exo fashion. The calculations strongly support that the endo pathway is a stepwise mechanism, while the exo pathway is a concerted mechanism. The endo attack of 2 from different sides of pyrone results in the formation of the INT2A_endo and INT2B_endo with an energy barrier of 13.7 kcal/mol (TS1A_endo) and 15.8 kcal/mol (TS1B_endo), respectively. The following C—C bond formation via a barrierless transition state TS2 leads to the product complexes. Along the reaction path, transition state TS1 is the rate-determining and enantioselectivity-determining step. The most favorable transition state TS1A_endo leading to the major product 3 has a lower energy barrier than that of TS1B_endo, which is consistent with our experimental observations. We also located that the concerted exo transition states TS1A_exo and TS1B_exo are energetically less favored than TS1A_endo at least by 1.0 kcal/mol, thus correlating well with the high diastereoselectivity for the cycloaddition products. We also performed the Hirshfeld partition of molecular density (IGMH) to investigate the non-covalent interactions between the [Sm]*_1 fragment and the aryl enol ether 2 in both TS1A_endo and TS1B_endo. As shown in Fig. 1B, strong CH···O, CH···π and CH···HC interactions between the catalyst and substrates are formed in these two endo-transition states. The greater stability of TS1A_endo is partly accounted for by the CH···π interactions between the chiral ligand and enol ether 2 (the distances of the CH···π interaction are 2.59 Å and 2.67 Å, Fig. 1C). TS1A_endo also exhibits stronger CH···HC interactions in comparison to TS1B_endo, as indicated by the number of CH···HC interactions. Furthermore, steric repulsion between the ligand and the phenyl group on enol ether 2 are evident through the H···H short contacts (H···H = 2.12 Å and 2.22 Å in TS1B_endo). These findings suggest that the enantioselectivity is determined by the steric repulsion and the attractive dispersion interactions such as CH···π and CH···HC interaction.
Next, distortion-interaction analysis was used to investigate the origin of the enantioselectivity. The computed activation energies (ΔE⧧) were dissected into two terms: distortion energy ΔEdist (including ΔEdist(TS1A_endo_1) and ΔEdist(TS1A_endo_2) and interaction energy ΔEint (Fig. S5A in Supporting information). The results indicates that the lower energy barrier for TS1A_endo can be attributed to the stronger interactions between the [Sm]*_1 and the substrate 2, in which the interaction energy (ΔEint) is 2.6 kcal/mol higher than that in TS1B_endo (ΔEint = –12.9 kcal/mol in TS1A_endo vs. ΔEint = –10.3 kcal/mol in TS1B_endo). The energy decomposition analysis (EDA) was then performed to identify the primary factor that governs the interaction energy (Fig. S5B in Supporting information). The interaction energies are divided into five terms, namely electrostatic (ΔEels), Pauli repulsion (ΔErep), exchange (ΔEx), orbital (ΔEorb), and Coulomb correlation (ΔEc). As a result, electrostatic energy (ΔΔEels = –5.2 kcal/mol) dominants the interaction energy, indicating that the electrostatic interaction has a significant effect on stabilizing TS1A_endo.
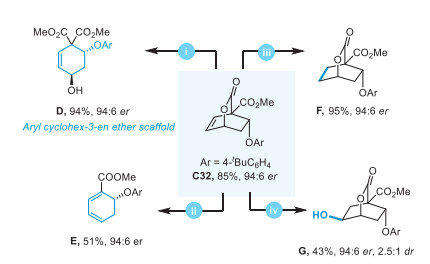
To demonstrate the potential utility of this methodology, diverse post-functionalizations were conducted by using enantiomerically pure product C32 (Scheme 4). Firstly, bridged bicyclic lactone structure could be readily ring-opened by transesterification with methanol, thus furnishing the aryl cyclohex-3-en ether scaffold D in excellent yield, which is widely existed in natural products. Then, extrusion of CO2 of the bicyclic lactone product via a retro-Diels–Alder reaction generated the cyclohexadiene ether E as a versatile intermediate for the synthesis of polysubstituted cyclohexane/cyclohexene motifs. Hydrogenation of the internal olefin of C32 smoothly afforded the saturated bicyclic compound F in almost quantitative yield. Additionally, a sequence hydroboration-oxidation (BH3 and then NaBO3) provided straightforward access to chiral cyclohexanol scaffold in 43% yield. These results declared that chiral bridged bicyclic lactones with an aryl ether moiety can be readily transferred into various optically pure and useful frameworks.
In conclusion, we have developed an asymmetric all-carbon-based IEDDA reaction of electron-deficient 2-pyrones with electronically unfavorable aryl enol ethers catalyzed by the chiral N,N’-dioxide/Lewis acid complex. This IEDDA reaction is applicable to a wide range of non- and 1,2-disubstituted acyclic aryl enol ethers and 2-pyrones, delivering diverse chiral bridged bicyclic lactones in high yields with excellent stereoselectivities (up to 96% yield, > 20:1 dr, 97:3 er). Particularly, the bridged bicyclic lactone core could be easily converted to a chiral aryl cyclohex-3-en ether scaffold, which existed in numerous natural products and pharmaceuticals. In addition, DFT calculations revealed a stepwise and endo mechanism, explaining the high enantioselectivity of the reaction controlled by the cooperative effect of the steric factors and the dispersion interactions.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Fangqing Zhang: Writing – original draft, Methodology, Investigation. Yu Wang: Software, Investigation. Zhenda Tan: Validation. Yangbin Liu: Writing – review & editing, Supervision, Project administration, Funding acquisition, Conceptualization. Lijuan Song: Writing – review & editing, Supervision, Software, Funding acquisition. Xiaoming Feng: Writing – review & editing, Supervision, Project administration, Funding acquisition, Conceptualization.
We appreciate the National Natural Science Foundation of China (Nos. 22001177, 22203023), Guangdong Pearl River Talent Program (no. 2021QN020268), the Natural Science Foundation of Guangdong Province (Nos. 2024A1515012381, 2022A1515011859), Shenzhen Bay Laboratory Startup Fund (No. S201100003), Major Program of Shenzhen Bay Laboratory (No. S211101001-4) and Shenzhen Bay Qihang Fellow Program (No. QH23001) for generous financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
V. Rukachaisirikul, S. Buadam, S. Phongpaichit, J. Sakayaroj, Tetrahedron 69 (2013) 10711–10717.
L. Chen, Q. Zhang, W.W. Zhang, et al., Heterocycles 87 (2013) 861–868.
C. Li, C.J. Li, J. Ma, et al., Bioorg. Chem. 88 (2019) 102948.
J.J. Sramek, E.J. Frackiewicz, N.R. Cutler, Expert Opin. Invest. 9 (2000) 2393–2402. doi: 10.1517/13543784.9.10.2393
N. Yamamoto, H. Fujii, S. Imaide, et al., J. Org. Chem. 76 (2011) 2257–2260. doi: 10.1021/jo1022487
E.J. Corey, Angew. Chem. Int. Ed. 41 (2002) 1650–1667.
K.I. Takao, R. Munakata, K.I. Tadano, Chem. Rev. 105 (2005) 4779–4807. doi: 10.1021/cr040632u
J.A. Funel, S. Abele, Angew. Chem. Int. Ed. 52 (2013) 3822–3863. doi: 10.1002/anie.201201636
K.N. Houk, F. Liu, Z. Yang, J.I. Seeman, Angew. Chem. Int. Ed. 60 (2021) 12660–12681. doi: 10.1002/anie.202001654
B. Briou, B. Améduri, B. Boutevin, Chem. Soc. Rev. 50 (2021) 11055–11097. doi: 10.1039/d0cs01382j
X. Jiang, R. Wang, Chem. Rev. 113 (2013) 5515–5546. doi: 10.1021/cr300436a
A.C. Knall, C. Slugovc, Chem. Soc. Rev. 42 (2013) 5131–5142. doi: 10.1039/c3cs60049a
Z.M. Png, H. Zeng, Q. Ye, J. Xu, Chem. Asian J. 12 (2017) 2142–2159. doi: 10.1002/asia.201700442
B.L. Oliveira, Z. Guo, G.J.L. Bernardes, Chem. Soc. Rev. 46 (2017) 4895–4950.
I.E. Markó, S.L. Warriner, B. Augustyns, Org. Lett. 2 (2000) 3123–3125.
P. Li, H. Yamamoto, J. Am. Chem. Soc. 131 (2009) 16628–16629. doi: 10.1021/ja908127f
J.L. Li, T.R. Kang, S.L. Zhou, et al., Angew. Chem. Int. Ed. 49 (2010) 6418–6420. doi: 10.1002/anie.201002912
J.L. Li, S.L. Zhou, P.Q. Chen, et al., Chem. Sci. 3 (2012) 1879–1882. doi: 10.1039/c2sc20096a
P. Burch, M. Binaghi, M. Scherer, et al., Chem. Eur. J. 19 (2013) 2589–2591. doi: 10.1002/chem.201203746
B.Q. Gu, W.L. Yang, S.X. Wu, Y.B. Wang, W.P. Deng, Org. Chem. Front. 5 (2018) 3430–3434. doi: 10.1039/c8qo01042k
S.X. Wu, B.Q. Gu, H. Xu, et al., Adv. Synth. Catal. 361 (2019) 4302–4313. doi: 10.1002/adsc.201900664
K. Afarinkia, V. Vinader, T.D. Nelson, G.H. Posner, Tetrahedron 48 (1992) 9111–9171.
M. Feng, X. Jiang, Chem. Commun. 50 (2014) 9690–9692.
N. Wang, S. Du, D. Li, X. Jiang, Org. Lett. 19 (2017) 3167–3170. doi: 10.1021/acs.orglett.7b01292
N. Wang, J. Liu, C. Wang, L. Bai, X. Jiang, Org. Lett. 20 (2018) 292–295. doi: 10.1021/acs.orglett.7b03694
Q. Cai, Chin. J. Chem. 37 (2019) 946–976. doi: 10.1002/cjoc.201900048
X.G. Si, Z.M. Zhang, Q. Cai, Synlett 32 (2021) 947–954.
G. Huang, C. Kouklovsky, A. de la Torre, Chem. Eur. J. 27 (2021) 4760–4788. doi: 10.1002/chem.202003980
B. Huang, D. Xing, H. Jiang, L. Huang, J. Org. Chem. 89 (2024) 7280–7285. doi: 10.1021/acs.joc.4c00288
G.H. Posner, F. Eydoux, J.K. Lee, D.S. Bull, Tetrahedron Lett. 35 (1994) 7541–7544.
G.H. Posner, J.C. Carry, J. Kyoo Lee, D.S. Bull, H. Dai, Tetrahedron Lett. 35 (1994) 1321–1324.
G.H. Posner, H. Dai, D.S. Bull, et al., J. Org. Chem. 61 (1996) 671–676.
I.E. Markó, G.R. Evans, Tetrahedron Lett. 35 (1994) 2771–2774.
I.E. Markó, G.R. Evans, J.P. Declercq, Tetrahedron 50 (1994) 4557–4574.
I.E. Markó, G.R. Evans, P. Seres, I. Chellé, Z. Janousek, Pure Appl. Chem. 68 (1996) 113–122. doi: 10.1351/pac199668010113
Y. Zhou, Z. Zhou, W. Du, Y. Chen, Acta Chim. Sinica 76 (2018) 382–386. doi: 10.6023/a18040131
C.J.F. Cole, L. Fuentes, S.A. Snyder, Chem. Sci. 11 (2020) 2175–2180. doi: 10.1039/c9sc05738b
X.W. Liang, Y. Zhao, X.G. Si, et al., Angew. Chem. Int. Ed. 58 (2019) 14562–14567. doi: 10.1002/anie.201908284
X.G. Si, Z.M. Zhang, C.G. Zheng, Z.T. Li, Q. Cai, Angew. Chem. Int. Ed. 59 (2020) 18412–18417. doi: 10.1002/anie.202006841
M.M. Xu, L. Yang, K. Tan, et al., Nat. Catal. 4 (2021) 892–900. doi: 10.1038/s41929-021-00687-x
M.M. Xu, X.Y. You, Y.Z. Zhang, et al., J. Am. Chem. Soc. 143 (2021) 8993–9001. doi: 10.1021/jacs.1c04759
Y. Lu, M.M. Xu, Z.M. Zhang, J. Zhang, Q. Cai, Angew. Chem. Int. Ed. 60 (2021) 26610–26615. doi: 10.1002/anie.202112223
J.X. He, X.G. Si, Q.T. Lu, Q.W. Zhang, Q. Cai, Chin. J. Chem. 41 (2023) 21–26. doi: 10.1002/cjoc.202200441
X.Y. You, Q. Cai, Synlett 34 (2023) 948–952.
X.G. Si, S.X. Feng, Z.Y. Wang, et al., Angew. Chem. Int. Ed. 62 (2023) e202303876.
F. Zhang, B.T. Ren, Y. Zhou, Y. Liu, X. Feng, Chem. Sci. 13 (2022) 5562–5567. doi: 10.1039/d2sc01458k
F. Zhang, B.T. Ren, Y. Liu, X. Feng, Org. Chem. Front. 9 (2022) 3956–3960. doi: 10.1039/d2qo00493c
G. Huang, R. Guillot, C. Kouklovsky, B. Maryasin, A. de la Torre, Angew. Chem. Int. Ed. 61 (2022) e202208185.
G. Huang, C. Kouklovsky, A. de la Torre, J. Am. Chem. Soc. 144 (2022) 17803–17807. doi: 10.1021/jacs.2c08760
H.B. Yu, Y.G. Chen, Y. Tian, M.S. Xie, H.M. Guo, ACS Catal. 14 (2024) 8930–8938. doi: 10.1021/acscatal.4c02072
I.E. Markó, I. Chellé-Regnaut, B. Leroy, S.L. Warriner, Tetrahedron Lett. 38 (1997) 4269–4272.
M. Saktura, P. Grzelak, J. Dybowska, Ł. Albrecht, Org. Lett. 22 (2020) 1813–1817. doi: 10.1021/acs.orglett.0c00138
M.M. Xu, P.P. Xie, J.X. He, et al., J. Am. Chem. Soc. 146 (2024) 6936–6946. doi: 10.1021/jacs.3c14409
W. Cao, X. Liu, X. Feng, Chin. Sci. Bull. 65 (2020) 2941–2951. doi: 10.1360/tb-2020-0158
S. Dong, X. Liu, X. Feng, Acc. Chem. Res. 55 (2022) 415–428. doi: 10.1021/acs.accounts.1c00664
D.F. Chen, L.Z. Gong, Org. Chem. Front. 10 (2023) 3676–3683. doi: 10.1039/d3qo00566f
Y. Liu, X. Liu, X. Feng, Chem. Sci. 13 (2022) 12290–12308. doi: 10.1039/d2sc03806d
W. Yang, Z. Yang, L. Chen, et al., Chin. Chem. Lett. 34 (2023) 107791–107795.
T. Zhan, L. Zhou, Y. Zhou, et al., Chem. Sci. 15 (2024) 4797–4803. doi: 10.1039/d4sc00078a
Y. Mo, L. Ning, Z. Luo, et al., ACS Catal. 14 (2024) 6687–6695. doi: 10.1021/acscatal.4c01264

Scheme 2 Substrate scope of non-substituted aryl enol ethers. General reaction conditions: A (0.1 mmol, 1.0 equiv.), B (0.15 mmol, 1.5 equiv.), Sm(OTf)3 (10 mol%), L3-PrMe2 (10 mol%), DCM (1.6 mL), 4 Å MS (50 mg), 10 ℃, 96 h. Isolated yield. The er value was determined by HPLC analysis on a chiral stationary phase. a At 0 ℃. b At –10 ℃. c At 20 ℃ for 96 h. d Sm(OTf)3 (15 mol%) and L3-PrMe2(4-tBu) (15 mol%). e Eu(OTf)3 (15 mol%) and L3-PrEt2 (15 mol%).

Scheme 3 Substrate scope regarding to disubstituted aryl enol ethers. General reaction conditions: A (0.1 mmol, 1.0 equiv.), B (0.15 mmol, 1.5 equiv.), Eu(OTf)3 (10 mol%), L3-PrMe2 (10 mol%), DCM (1.6 mL), 4 Å MS (50 mg), 20 ℃, 96 h. Isolated yield. The er value was determined by HPLC analysis on a chiral stationary phase. a Eu(OTf)3 (15 mol%) and L3-PrMe3 (15 mol%). b Sm(OTf)3 (15 mol%) and L3-PrMe2(4-tBu) (15 mol%). c Sm(OTf)3 (10 mol%) and −10 ℃ for 96 h. d Sm(OTf)3 (10 mol%) and 10 ℃ for 24 h.

Figure 1 Quantum chemical calculations. (A) Computed reaction profiles for the asymmetric IEDDA reaction of the 2-pyrone 1 and aryl enol ether 2 at the PBE0-D3(BJ)/6–311+G(d)/Sm(MWB51) level. (B) The IGMH of non-covalent interactions between the [Sm]*_1 fragment and aryl enol ether 2 fragment in both TS1A_endo and TS1B_endo. (C) Optimized structures of TS1A_endo and TS1B_endo (the distances are in Å).

Scheme 4 Post functionalization of the cycloaddition product. (ⅰ) KOH (1.5 equiv.), MeOH, 0 ℃ to r.t., 0.5 h. (ⅱ) PhCl, 90 ℃, 20 h. (ⅲ) [Ir(cod)(PCy3)(Py)]PF6 (15 mol%), H2 (1.0 atm), THF, r.t., 24 h. (ⅳ) BH3·Me2S (6.0 equiv.), THF, r.t., 24 h; then NaBO3·4H2O (5.0 equiv.), H2O, 0 ℃ to r.t., 4.0 h.
Table 1. Optimization of the reaction conditions.a
|
|||||
| Entry | Ligand | Lewis acid | Solvent | C1 yield (%) b | C1 er c |
| 1 | L3-PrMe2 | Mg(OTf)2 | DCE | 27 | 59:41 |
| 2 | L3-PrMe2 | Yb(OTf)3 | DCE | 50 | 57:43 |
| 3 | L3-PrMe2 | Sm(OTf)3 | DCE | 57 | 76:24 |
| 4 | L3-PrMe2 | Eu(OTf)3 | DCE | 76 | 81:19 |
| 5 | L3-PiMe2 | Eu(OTf)3 | DCE | 24 | 55:45 |
| 6 | L3-RaMe2 | Eu(OTf)3 | DCE | 77 | 79:21 |
| 7 | L3-PrPr2 | Eu(OTf)3 | DCE | 42 | 72:28 |
| 8 | L3-PrMe(4-tBu) | Eu(OTf)3 | DCE | 70 | 80:20 |
| 9 | L3-PrMe2 | Eu(OTf)3 | MeCN | 34 | 77:23 |
| 10 | L3-PrMe2 | Eu(OTf)3 | CHCl3 | 70 | 74:26 |
| 11 | L3-PrMe2 | Eu(OTf)3 | DCM | 95 | 88:12 |
| 12 d | L3-PrMe2 | Eu(OTf)3 | DCM | 76 | 91:9 |
| 13 d | L3-PrMe2 | Sm(OTf)3 | DCM | 73 | 92:8 |
| 14 d, e | L3-PrMe2 | Sm(OTf)3 | DCM | 83 | 95:5 |
| a General reaction conditions: A (0.1 mmol, 1.0 equiv.), B (0.15 mmol, 1.5 equiv.), ligand (10 mol%), Lewis acid (10 mol%) and DCE (1.6 mL) at 35 ℃ for 24 h. b NMR yield detected by using p-xylene as an internal standard. c The er value was determined by HPLC analysis on a chiral stationary phase. d The reaction was conducted at 10 ℃ for 96 h, and 4 Å MS (50 mg) was added. e Product was C2. |
|||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们

 下载:
下载: