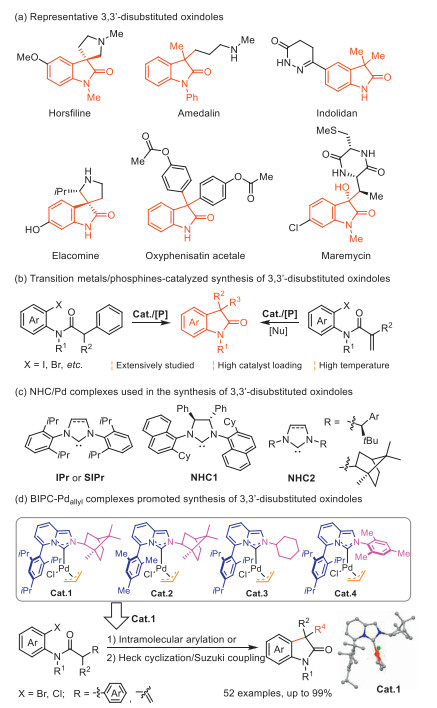
Scheme 1.
Representative examples of (a) useful 3,3′-disubstituted oxindoles and their catalytic synthesis involving (b) phosphines, (c) NHCs and (d) BIPC-Pdallyl.
Bornylimidazo[1,5–a]pyridin-3-ylidene allylic Pd catalyst with optimal electronic and steric properties for synthesis of 3,3′-disubstituted oxindoles
Kun Wang , Tianxue Gong , Yaohuang Huang , Boyang Han , Hanxiao Yang , Pavlo O. Dral , Weiwei Fang

3,3′-Disubstituted oxindoles, as the privileged heterocyclic framework, are widely present in natural products and bioactive pharmaceuticals (Scheme 1a) [1-7]. In the past decades, a great effort has been devoted to their catalytic synthesis, and among them, transition metal-catalyzed α-arylation and Heck cyclization/sequential coupling reactions have constituted one of the most practical methods [8-16]. Since the seminal contributions from Grigg [17,18], diverse sophisticated phosphorous ligands were designed to promote these reactions (Scheme 1b) [10-12]. Though good enantioselectivity was achieved, some limitations still remain, which remarkedly hampered their potential practical applicability. One of them is the high loading of Pd center (5–10 mmol%) and phosphines (10–20 mmol%), which seriously increased the economic cost. Additionally, most of the highly active phosphines are air-sensitive. Another limitation is that high temperature (mostly > 100 ℃) was always required. Consequently, the development of alternative approaches to access 3,3′-disubstituted oxindoles by using new ligands at milder reaction conditions is of great importance.
In this sense, N-heterocyclic carbenes (NHCs) represent a type of environmentally friendly robust ligands [19] and have exhibited much promise in transition metal catalysis due to their strong σ-donor and weak π-acceptor ability [20-23]. Most importantly, besides the NHC skeleton [24,25], the catalytic activity could be elegantly tuned by varying throw-away ligands [26,27]. However, in sharp contrast to phosphines, the use of NHC-Pd complexes in promoting the catalytic conversion towards desired 3,3′-disubstituted oxindoles has been widely neglected, and only a few conventional skeletons of NHCs, such as 1,3-disubstituted imidazolylidenes (ImCs), were studied for the α-arylations [13,28]. Though these methods featured a lower reaction temperature (25–50 ℃), a high catalyst loading (5 mol%) was still required. To the best of our knowledge, no examples of NHC-Pd catalyzed Heck cyclization/Suzuki coupling towards 3,3′-disubstituted oxindoles have been reported to date.
In this work, we designed a novel NHC-Pd catalyst enabling milder reaction conditions and showcased their efficacy for both intramolecular α-arylations and Heck cyclization/Suzuki couplings for useful 3,3′-disubstituted oxindoles. We based the design on the NHC electronic structure considerations, and aimed at a ligand featuring both strong σ-donor and π-acceptor abilities that would favor the Heck cyclization/sequential coupling process considering the following two points: 1) The strong σ-donating ability of NHC would increase the electron density of Pd improving the catalytic activity toward the oxidative addition; 2) The strong π-accepting property would favor the coordination of the olefin to Pd followed by the migratory insertion. Therefore, such a catalyst could furnish a promising protocol realizing the Heck cyclization/Suzuki coupling at mild reaction conditions.
The recipe for the NHC featuring both strong π-accepting and σ-donating characters was first suggested by Lassaletta and Glorius [29,30] in their imidazo[1,5-a]pyridine-derived carbenes (IPCs) [31,32]. This unique property endowed the IPC-M complexes with superior catalytic activity in diverse types of reactions [31]. Especially, the substituents at the C5- and N2-positions of IPCs strongly affected the catalytic performance in the denitrative cross-couplings [33-38]. It was attributed to the elongation of catalyst lifetime via an in situ generated η1-π interaction [39] between the Pd with a carbon atom of C5-oligoTripp (Tripp = 2,4,6-triisopropylphenyl), and the acceleration of reductive elimination by tuning the steric bulk of IPCs [38]. Considering all the above-mentioned points, IPCs might meet well the prerequisites to design an NHC-Pd complex to catalyze intramolecular Heck cyclization/Suzuki couplings towards 3,3′-disubstituted oxindoles. Therefore, following our interests in the design of diverse NHC-Pd catalysts [40-44], herein, we introduced a rigid bicycloskeleton of the bornyl unit at the N2-position to modify the steric property of IPCs, affording a rationally designed IPC allylic Pd catalyst (BIPC-Pdallyl, Cat.1, Scheme 1d). Due to a synergistic combination of electronic and steric properties, Cat.1 enabled a general catalytic protocol achieving 3,3′-disubstituted oxindoles with a low catalyst loading at mild reaction conditions (Scheme 1d). Moreover, useful transformations to several bioactive heterocycle-fused indoline alkaloids were also successfully proved.
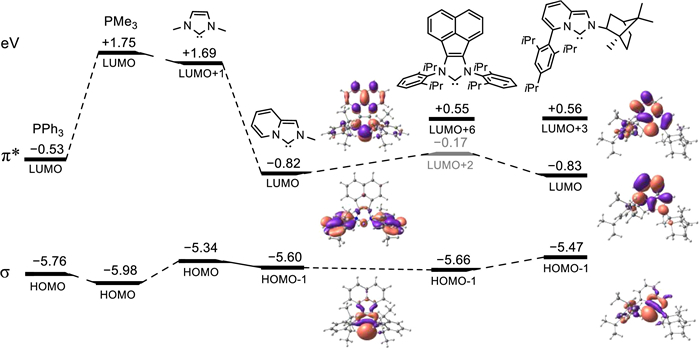
We commenced our investigation by designing the new BIPC-Pdallyl catalysts. The first one Cat.1 was introduced with a bornyl moiety at the N2-position as the rigid bicycloskeleton and was attached with the bulky Tripp group at the C5-position (Scheme 1d). The IPC ligand in Cat.1 possesses the desired property of strong σ-donating and π-accepting character as its relevant occupied orbital (HOMO−1) is higher in energy while its unoccupied orbital LUMO is much lower in energy than the corresponding orbitals of other NHCs [31] including acenaphthoimidazol-2-ylidene (AnIPr, Cat.5, in Table 1) and imidazolylidene, as well as common phosphines of PPh3 and PMe3 (Fig. 1, see the details of DFT calculations in Supporting information). For a comparative investigation of different steric and electronic effects, we also prepared a series of other IPC-Pd catalysts (Cat.2–4, Scheme 1d). Cat.2 replaces the Tripp moiety with a less bulky, rigid flat unit of Mes (Mes = 2,4,6-trimethylphenyl). To study the effect of rigid bicycloring of the bornyl group, IPCs with cyclohexyl or Mes units as the control ligands were also synthesized (CyIPCs in Cat.3 and MesIPCs in Cat.4). All these IPC-Pdallyl catalysts Cat.1–Cat.4 were obtained in moderate yields and synthetic routes were shown in Scheme S1 (Supporting information).
 DownLoad:
CSV
DownLoad:
CSV
 |
||||
| Entry | Cat. | Solvent | Base | Yield (%) |
| 1 | Cat.1 | DMA | tBuONa | 99/54b |
| 2 | Cat.1 | DMA | tBuOK | 90 |
| 3 | Cat.1 | CPME | tBuONa | 78 |
| 4 | Cat.1 | DMSO | tBuONa | 81 |
| 5 | Cat.1 | DMF | tBuONa | 65 |
| 6 | Cat.2 | DMA | tBuONa | 56 |
| 7 | Cat.3 | DMA | tBuONa | 66 |
| 8 | Cat.4 | DMA | tBuONa | 68 |
| 9 | Cat.5 | DMA | tBuONa | 47 |
| 10 | Cat.6 | DMA | tBuONa | 51 |
| 11 | Cat.7 | DMA | tBuONa | 42 |
| 12 | Cat.8 | DMA | tBuONa | 48 |
| a Standard conditions: 1a (0.4 mmol), Cat.1 (2 mol%), base (0.48 mmol), solvent (6 mL), 50 ℃, 12 h. Isolated yield. b Cat. (0.5 mol%). |
||||
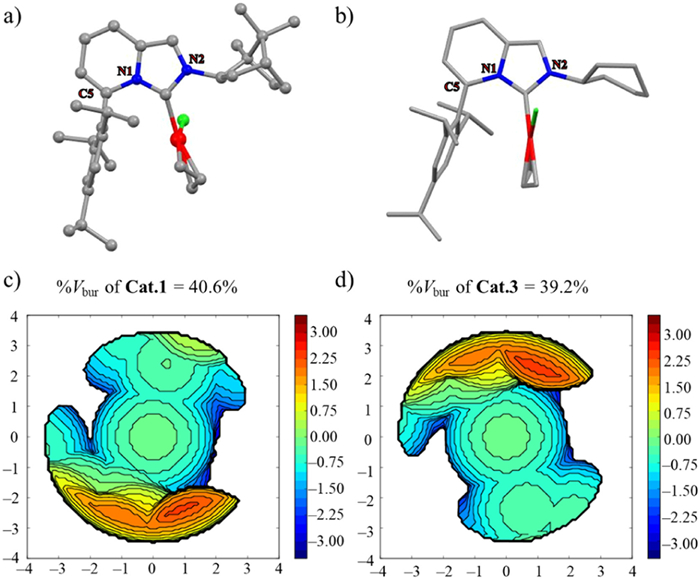
The structure of Cat.1 was studied by the X-ray diffraction and a L-shape conformation is depicted in Fig. 2, which shows two coordination modes between the Pd and allylic unit. The phenyl ring of Tripp is almost perpendicular to the IPC plane, while the bornyl ring of Cat.1 arranges a bit closer to the Pd center due to the rigid bicycloskeleton in comparison with the flexible Cy of Cat.3 in its simulated geometry optimized with DFT (Supporting information). Therefore, the space around the Pd center of Cat.1 is quite congested, exhibiting a larger percent buried volume (%Vbur = 40.6%) [45-48] in contrast to Cat.3 (%Vbur = 39.2%). For comparison, we also calculated the %Vbur of AnIPr-derived Pd-catalysts Cat.5–Cat.8 (their structures were shown in Table 1), which showed smaller %Vbur (34.4%–35.6%, Fig. S4 in Supporting information). Importantly, in an example of Cat.5, we can see that it is weaker σ-donor and π-acceptor in comparison with Cat.1 (Fig. 1). The distances of Pd-CIPC in Cat.1 and Cat.3 range from 2.041 Å to 2.053 Å, which are similar to those of Cat.5 (2.048 Å, Table S2 in Supporting information). However, their distances of Pd-Callyl are slightly shorter (Table S2), which indicates the strong back-donation from the d-orbitals of Pd to the allyl at the trans position [49]. In a word, though IPC acted as a π-accepting ligand, Cat.1 still could present the strong electron shift between Pd and allyl, suggesting that the strong σ-donor property of IPC contributes to the higher electron density of Pd in Cat.1. All these factors lead to the higher activity of Cat.1 compared to Cat.5, as we will see later.
With the catalysts in hands, we performed an intramolecular α-arylation of N-(2-bromophenyl)-N-methyl-2-phenylpropanamide 1a as the initial model reaction to evaluate the catalytic efficiency (Table 1). At room temperature, the combination of 2 mol% Cat.1 with tBuONa as a base led to 2a in 25% yield in N,N-dimethylacetamide (DMA). The reaction temperature disclosed a crucial effect on the reaction efficiency (Table S3 in Supporting information), and an almost quantitative yield was obtained at 50 ℃ (99%, entry 1). Screening other bases such as tBuOK, and organic solvents including cyclopentyl methyl ether (CPME), dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF) all afforded inferior yields (≤90%, Tables S3–S5 in Supporting information) with the best combination of tBuONa and DMA. A 54% yield was still observed at only 0.5 mol% catalyst loading, indicating the high activity of Cat.1. Unfortunately, no enantioselectivity was observed in all cases (Tables S3-S6 in Supporting information), which might be attributed to the faraway arrangement between the chiral center of bornyl moiety and Pd center based on the single crystal structure of Cat.1.
Subsequently, other NHC-Pdallyl catalysts (Cat.2–5) were tested to study the structure–activity relationship. The replacement of Tripp by Mes at the C5-position resulted in a decrease of 43% yield (Cat.2, 56%), and Cat.3 bearing the flexible Cy at the N2-position also exhibited a lower activity (66%) in comparison with Cat.1. Additionally, Cat.4 with the combination of Tripp and Mes units, the best catalyst in the denitrative Suzuki coupling reactions [33,35], still presented a lower efficiency (68%). It might be attributed to the deeper buried position of Pd in the rigid V-shaped pocket leading to a slightly bulkier steric environment (%Vbur = 42.1%, Fig. S5 in Supporting information) [38]. The substantial difference in the reactivity of Cat.1–4 suggested the important role of substituents connected to the IPC skeleton, which endowed Cat.1 with the matched steric property rather than the possible relevant effect on the electronic property of IPC. This point was further supported by the similar results of σ-donating and π-accepting (π*) molecular orbital energies of control IPC ligands, among which, one only possesses a N2-bornyl moiety (IPCN-bornyl, CH), and the other bears a C5-Tripp and N2-Me (IPCN−Me, C-Tripp). The results are shown in Fig. S3 (Supporting information).
The comparison of Cat.1 with Cat. 5 (99% vs. 47%) further indicated its higher catalytic ability. Notably, the best catalyst Cat.6 used in our previously developed intramolecular α-arylation (98%, at 110 ℃) [42], as well as other rationally designed AnIPr-derived palladacycles all gave lower yields (Cat.6–8, 42%–51%). Importantly, all IPC-Pdallyl catalysts (Cat.1–4, 56%–99%) exhibited higher catalytic performances than those of AnIPr-Pd complexes (Cat.5–Cat.8, 42%–51%), indicating the key role of electronic property of IPC ligand in dictating the excellent activity of Cat.1. To put it briefly, Cat.1 bearing Tripp and bornyl units was proved to be the best catalyst at lower reaction temperature due to the matched electronic and steric properties.
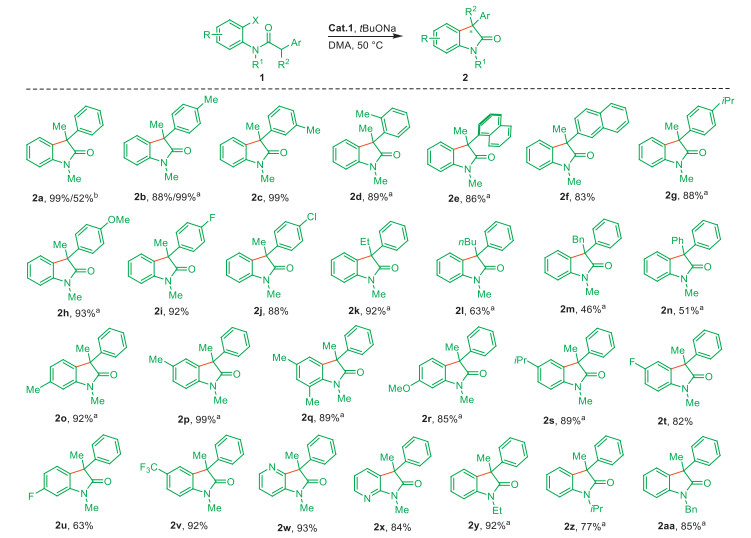
Subsequently, we investigated the scope of N-(2-bromophenyl)-N-substituted-2-phenylpropanamides. As depicted in Scheme 2, amides with diverse functional groups were rapidly converted into desired 3,3′-disubstituted oxindoles in good to excellent yields (63%–99%). The relative position of substituents in the phenyl moiety at α-carbon (α-Ph) of the carbonyl group greatly affected the efficiency at 50 ℃: meta-substrate showed a better yield than ortho- and para-isomers (2c vs. 2b and 2d, 99% vs. 88% and 20%, respectively). Increasing the reaction temperature to 90 ℃ could also result in a good yield of ortho-product (2d, 89%). The steric effect was also observed in the case of naphthylpropanamides (2e and 2f, 86% and 83%, respectively). A higher activity was obtained for α-Ph with electron-withdrawing groups (F, Cl) than those with electron-donating groups (iPr, OMe), all leading to good yields (2g–2j, 88%–93%). Furthermore, the variation of R2 substituents such as Me, Et, nBu and Bn as well as aromatic one of Ph were tested. All of them were compatible in the intramolecular α-arylation (2k–2n, 46%–92%), though the steric bulky affected the coupling efficiency (92% and 99% for 2a and 2k vs. 2l–2n, 46%–63%). Unfortunately, alkyl R2 with heteroatoms such as O and N were not tested due the failed synthesis of relevant substrates. The aniline moiety showed a slight positional effect (2o–2q, 89%–99%), while a similar electronic effect was found that the efficient conversion of anilines bearing electron-donating groups into products required a higher reaction temperature than those with electron-withdrawing groups (2o-2s, 85%−99% at 100 ℃ vs. 2r-2v, 63%−92% at 50 ℃). Heterocyclic rings of pyridine (2w and 2x, 93% and 84%) and other N-substitutions such as Et, iPr and Bn (2y–2aa) were well tolerated (77%–93%). Additionally, product 2a could also be formed from the inactive chloro–analog of 1b in a moderate yield (52%), further indicating the high activity of Cat.1.
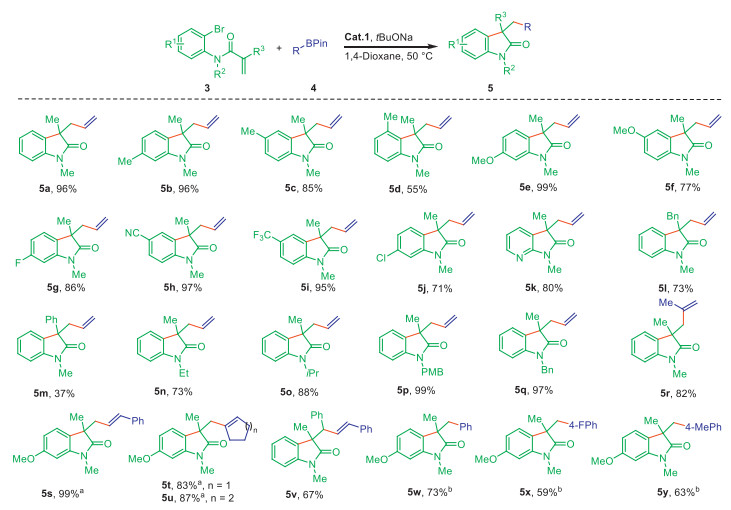
Considering the strong π-accepting property of IPC favoring the coordination of Pd with unsaturated bonds, we further explored the catalytic activity of Cat.1 in the intramolecular Heck cyclization/Suzuki coupling of N-substituted-N-(2-bromophenyl)acrylamides with vinyl boron reagents (Scheme 3). It would readily afford valuable 3,3-substituted oxindoles featuring an allylic unit, as a key intermediate for the synthesis of heterocycle-fused indoline alkaloids (Scheme 4) [50]. N-Methyl-N-(2-bromophenyl)methacrylamide 3a and vinylboronic acid pinacol ester 4a were examined as the model substrates, and an 88% yield was obtained for 3-allyl-1,3-dimethylindolin-2-one 5a. Further screening other solvents and prolonging the reaction time to 24 h, Cat.1 could efficiently promote this reaction in 96% yield using 1,4-dioxane at 50 ℃ (Table S7 in Supporting information). As expected, a variety of N-(2-bromophenyl)acrylamides were well tolerated, leading to diverse 3-allyl oxindoles in good to excellent yields. For example, the acrylamide possessing a 2–bromo-5-methylphenyl (defined as para-position) produced 5b in almost quantitative yield (96%), while only a moderate yield was found for the ortho-isomer (5d, 55%), indicating a nonnegligible steric effect. There were no big catalytic differences between substrates with electron-donating (Me, OMe), and electron-withdrawing groups (F, CN, CF3, Cl), all presenting good yields (5b–5j, 71%–97%). Especially, the presence of CN and Cl provides the opportunity for useful post-transformations [51-55]. The substrate with a pyridyl skeleton also readily participated in the domino reaction, leading to 5k in 80% yield. Besides Me, the Bn group at 3-position of oxindole and alkyls (such as Et and iPr) at the N-atom were also compatible (5l–5o, 73%–88%), even the Ph group worked albeit in a low yield (5m, 37%). Despite the unsuccessful attempt with N-free 3-allyl-3-dimethylindolin-2-one, it could be easily obtained from the deprotection of 5p or 5q possessing PMB or Bn groups (99% and 97%). Besides, other alkenyl boron reagents bearing a terminal double bond unit, such as 1-phenyl vinyl boronic acid and 1-methyl vinylboronic acid pinacol cyclic ester were also explored, which indicated the later one as a suitable coupling partner, giving desired product in good yield (5r, 82%). Whereas, only a poor yield (11%) was obtained for the 1-phenyl vinyl boronic acid (Scheme S1). Apart from the terminal alkenyl boron reagents, other internal vinyl boronic acids were also involved, which delivered different products featuring allylic units in good yields (5s–5u, 83%–99%). Additionally, N-methyl-N-(2-bromophenyl)-α-methyl-β-phenylacrylamide was also tolerated, leading to product 5v in 67% yield. However, no conversion of N-(2-bromophenyl)-N-methyl-3-phenylacrylamide was observed under the same reaction condition (Scheme S1). As expected, arylboronic acids bearing electron-withdrawing and donating groups could also efficiently couple with N-methyl-N-(2–bromo-4-methoxylphenyl)acrylamide, affording desired products in acceptable yields (5w-5y, 59%−70%).
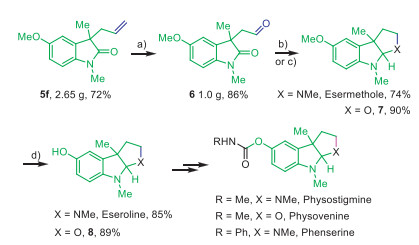
The applicability of this catalytic protocol was examined in the concise synthesis of heterocycle-fused indoline alkaloids, as depicted in Scheme 4. First, it could easily be scaled up to 16 mmol leading to 2.65 g of 5f in a 72% yield. Second, the formation of 3-formyloxindoles (6, 1.0 g, 86%) from 5f was achieved in good yield using the combined catalytic system of K2OsO4·2H2O, N-methylmorpholine-n-oxide (NMO) and NaIO4, which served as a key precursor for the synthesis of bioactive natural products (±)-desoxynoreseroline, (±)-esermethole and their derivatives such as (±)-physostigmine and (±)-phenserine. The racemic esermethole was successfully obtained in a 74% yield by using a reductive amination and condensation cascade one-pot process. The deprotection of the OMe group at esermethole by using BBr3 led to the generation of (±)-eseroline in 85% yield. Additionally, the O-congener (7) of (±)-esermethole was obtained in excellent yield (90%), and the subsequent deprotection led to 8 in 89% yield, which was the key intermediate of (±)-physovenine.
In summary, we developed a catalytic protocol for the synthesis of valuable 3,3′-disubstituted oxindoles via the intramolecular α-arylation and Heck cyclization/Suzuki coupling under mild reaction conditions by applying a new bulky BIPC-Pdallyl catalyst Cat.1. Critical to the success of Cat.1 is a combination of the unique σ-donor and π-acceptor of IPC and matched steric effect from rigid bornyl and Tripp skeletons. It enabled an outstandingly broad substrate scope bearing diverse functional groups, electronic properties, and steric bulkiness, affording desired products in good to excellent yields. Particularly, different oxindoles featuring the allyl unit were also obtained, proving their great applicability in the concise total synthesis of heterocycle-fused indoline alkaloids.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Kun Wang: Writing – original draft, Validation, Project administration, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Tianxue Gong: Investigation, Formal analysis, Data curation. Yaohuang Huang: Formal analysis, Investigation, Data curation, Visualization. Boyang Han: Data curation. Hanxiao Yang: Data curation. Pavlo O. Dral: Supervision, Writing – review & editing. Weiwei Fang: Writing – review & editing, Supervision, Funding acquisition, Conceptualization, Project administration.
Financial supports from the National Natural Science Foundation of China (No. 22101133), Natural Science Foundation of Jiangsu Province (No. BK20200768), Nanjing Forestry University, the National Natural Science Foundation of China (the Outstanding Youth Scholars (Overseas, 2021) project) and the Lab project of the State Key Laboratory of Physical Chemistry of Solid Surfaces are greatly acknowledged.
Supplementary material associated with this article can be found, in the online version, at doi:
C. Sheng, G. Dong, Z. Miao, et al., Chem. Soc. Rev. 44 (2015) 8238–8259.
M. Kaur, M. Singh, N. Chadha, et al., Eur. J. Med. Chem. 123 (2016) 858–894.
B. Han, X.H. He, Y.Q. Liu, et al., Chem. Soc. Rev. 50 (2021) 1522–1586. doi: 10.1039/d0cs00196a
Y.M. Khetmalis, M. Shivani, S. Murugesan, et al., Biomed. Pharmacother. 141 (2021) 111842.
F. Shi, H. Zhang, Chin. J. Org. Chem. 42 (2022) 3351–3372.
H.H. Zhang, F. Shi, Acc. Chem. Res. 55 (2022) 2562–2580. doi: 10.1021/acs.accounts.2c00465
Y.C. Zhang, F. Jiang, F. Shi, Acc. Chem. Res. 53 (2019) 425–446. doi: 10.3390/ma12030425
F. Zhou, Y.L. Liu, J. Zhou, Adv. Synth. Catal. 352 (2010) 1381–1407. doi: 10.1002/adsc.201000161
R. Dalpozzo, Org. Chem. Front. 4 (2017) 2063–2078.
J. Li, S. Yang, W. Wu, et al., Chem. Asian J. 14 (2019) 4114–4128. doi: 10.1002/asia.201901202
Y. Ping, Y. Li, J. Zhu, et al., Angew. Chem. Int. Ed. 58 (2019) 1562–1573. doi: 10.1002/anie.201806088
A.D. Marchese, E.M. Larin, B. Mirabi, et al., Acc. Chem. Res. 53 (2020) 1605–1619. doi: 10.1021/acs.accounts.0c00297
S. Ostrowska, T. Scattolin, S.P. Nolan, Chem. Commun. 57 (2021) 4354–4375. doi: 10.1039/d1cc00913c
A.N. Reznikov, M.A. Ashatkina, Y.N. Klimochkin, Org. Biomol. Chem. 19 (2021) 5673–5701. doi: 10.1039/d1ob00496d
H. Mei, K. Aradi, L. Kiss, et al., Chin. Chem. Lett. 34 (2023) 108657.
J. He, H. Mei, J. Escorihuela, J. Han, Chin. J. Chem. 42 (2024) 1691–1698. doi: 10.1002/cjoc.202400127
R. Grigg, V. Santhakumar, V. Sridharan, Tetrahedron Lett. 34 (1993) 3163–3164.
R. Grigg, P. Fretwell, C. Meerholtz, et al., Tetrahedron 50 (1994) 359–370.
M.N. Hopkinson, C. Richter, M. Schedler, et al., Nature 510 (2014) 485–496. doi: 10.1038/nature13384
W. Wang, L. Cui, P. Sun, et al., Chem. Rev. 118 (2018) 9843–9929. doi: 10.1021/acs.chemrev.8b00057
Q. Zhao, G. Meng, S.P. Nolan, et al., Chem. Rev. 120 (2020) 1981–2048. doi: 10.1021/acs.chemrev.9b00634
F. Nahra, C.S.J. Cazin, Chem. Soc. Rev. 50 (2021) 3094–3142. doi: 10.1039/c8cs00836a
P.Y. Choy, K.B. Gan, F.Y. Kwong, Chin. J. Chem. 41 (2023) 1099–1118. doi: 10.1002/cjoc.202200703
P.L. Arnold, S. Pearson, Coord. Chem. Rev. 251 (2007) 596–609.
C. Chen, F.S. Liu, M. Szostak, Chem. Eur. J. 27 (2021) 4478–4499. doi: 10.1002/chem.202003923
C. Valente, S. Calimsiz, K.H. Hoi, et al., Angew. Chem. Int. Ed. 51 (2012) 3314–3332. doi: 10.1002/anie.201106131
K. Wang, R. Fan, X. Wei, et al., Green. Synth. Catal. 3 (2022) 327–338.
T. Delcaillau, H.L. Schmitt, P. Boehm, et al., ACS Catal. 12 (2022) 6081–6091. doi: 10.1021/acscatal.2c01178
M. Alcarazo, S.J. Roseblade, A.R. Cowley, et al., J. Am. Chem. Soc. 127 (2005) 3290–3291. doi: 10.1021/ja0423769
C. Burstein, C.W. Lehmann, F. Glorius, Tetrahedron 61 (2005) 6207–6217.
F. Shibahara, Y. Shibata, T. Murai, Chem. Lett. 50 (2021) 1892–1900. doi: 10.1246/cl.210461
Y. Koto, F. Shibahara, T. Murai, Org. Biomol. Chem. 15 (2017) 1810–1820.
K. Chen, W. Chen, X. Yi, et al., Chem. Commun. 55 (2019) 9287–9290. doi: 10.1039/c9cc04634h
W. Chen, K. Chen, W. Chen, et al., ACS Catal. 9 (2019) 8110–8115. doi: 10.1021/acscatal.9b02760
M. Kashihara, R.L. Zhong, K. Semba, et al., Chem. Commun. 55 (2019) 9291–9294. doi: 10.1039/c9cc05055h
W. Chen, W. Chen, M. Liu, et al., Org. Lett. 24 (2022) 6983–6987. doi: 10.1021/acs.orglett.2c02796
Y. Ma, A.A. Hussein, Z. Wang, J. Org. Chem. 87 (2022) 531–539. doi: 10.1021/acs.joc.1c02536
T. Zhou, P. Gao, E. Bisz, et al., Catal. Sci. Technol. 12 (2022) 6581–6589. doi: 10.1039/d2cy01136k
M. Miura, Angew. Chem. Int. Ed. 43 (2004) 2201–2203.
K. Wang, H. Yang, F. Bauer, et al., Chem. Eur. J. 29 (2023) e202300719.
X. Wei, K. Wang, W. Fang, Org. Biomol. Chem. 21 (2023) 3858–3862. doi: 10.1039/d3ob00354j
H. Yang, K. Wang, W. Fang, Asian J. Org. Chem. 12 (2023) e202300120.
R. Fan, H. Wen, Z. Chen, et al., Org. Lett. 26 (2024) 22–28. doi: 10.1021/acs.orglett.3c03438
R. Fan, M. Kuai, D. Lin, et al., Org. Lett. 24 (2022) 8688–8693. doi: 10.1021/acs.orglett.2c03580
A. Poater, B. Cosenza, A. Correa, et al., Eur. J. Inorg. Chem. 2009 (2009) 1759–1766. doi: 10.1002/ejic.200801160
L. Falivene, Z. Cao, A. Petta, et al., Nat. Chem. 11 (2019) 872–879. doi: 10.1038/s41557-019-0319-5
A. Poater, F. Ragone, S. Giudice, et al., Organometallics 27 (2008) 2679–2681. doi: 10.1021/om8001119
H. Clavier, S.P. Nolan, Chem. Commun. 46 (2010) 841–861. doi: 10.1039/b922984a
T. Tu, Y.G. Zhou, X.L. Hou, et al., Organometallics 22 (2003) 1255–1265.
N.H. Greig, X.F. Pei, T.T. Soncrant, et al., Med. Res. Rev. 15 (1995) 3–31. doi: 10.1002/med.2610150103
P. Ruiz-Castillo, S.L. Buchwald, Chem. Rev. 116 (2016) 12564–12649. doi: 10.1021/acs.chemrev.6b00512
Y. Lin, J. Yu, X. Zhang, et al., Chin. Chem. Lett. 33 (2022) 186–196.
P.Y. Choy, K.B. Gan, F.Y. Kwong, Chin. J. Chem. 41 (2023) 1099–1118. doi: 10.1002/cjoc.202200703
R.M.B. Carrilho, M.J.F. Calvete, G. Mikle, et al., Chin. J. Chem. 42 (2024) 199–221. doi: 10.1002/cjoc.202300384
Y. Nakao, Chem. Rev. 121 (2021) 327–344. doi: 10.1021/acs.chemrev.0c00301

Scheme 1 Representative examples of (a) useful 3,3′-disubstituted oxindoles and their catalytic synthesis involving (b) phosphines, (c) NHCs and (d) BIPC-Pdallyl.

Figure 1 The relevant σ-donating and π-accepting (π*) molecular orbital energies of ligands, and molecular orbitals of the IPC in Cat.1 and AnIPr in Cat.5 at B3LYP/6-31G(d, p).

Figure 2 (a) Crystal structure of Cat.1 (the ellipsoid contour at 50% probability levels), (b) the simulated structure of Cat.3, %Vbur and steric maps of (c) Cat.1 and (d) Cat.3.

Scheme 2 Substrate scope for the intramolecular α-arylation. Standard conditions: 1 (X = Br, 0.4 mmol), Cat.1 (2 mol%), tBuONa (0.48 mmol), DMA (6 mL), 50 ℃, 12 h. Isolated yield. a 90 ℃. b X = Cl. Bn = benzyl.

Scheme 3 Substrate scope for the intramolecular Heck cyclization/Suzuki coupling. Standard conditions: 3 (0.4 mmol), 4 (0.8 mmol), Cat.1 (2 mol%), tBuONa (0.48 mmol), 1,4-dioxane (6 mL), 50 ℃, 24 h. Isolated yield. a 70 ℃. b 100 ℃. PMB = p-methoxybenzyl.

Scheme 4 (a) K2OsO4·2H2O, NMO, 2,6-lutidine, NaIO4, dioxane/H2O (3/1), 25 ℃; (b) MeNH2·HCl, Et3N, MgSO4, LiAlH4, THF, reflux; (c) LiAlH4, THF, 0 ℃; (d) BBr3, DCM.
Table 1. Screening reaction conditions.a
|
||||
| Entry | Cat. | Solvent | Base | Yield (%) |
| 1 | Cat.1 | DMA | tBuONa | 99/54b |
| 2 | Cat.1 | DMA | tBuOK | 90 |
| 3 | Cat.1 | CPME | tBuONa | 78 |
| 4 | Cat.1 | DMSO | tBuONa | 81 |
| 5 | Cat.1 | DMF | tBuONa | 65 |
| 6 | Cat.2 | DMA | tBuONa | 56 |
| 7 | Cat.3 | DMA | tBuONa | 66 |
| 8 | Cat.4 | DMA | tBuONa | 68 |
| 9 | Cat.5 | DMA | tBuONa | 47 |
| 10 | Cat.6 | DMA | tBuONa | 51 |
| 11 | Cat.7 | DMA | tBuONa | 42 |
| 12 | Cat.8 | DMA | tBuONa | 48 |
| a Standard conditions: 1a (0.4 mmol), Cat.1 (2 mol%), base (0.48 mmol), solvent (6 mL), 50 ℃, 12 h. Isolated yield. b Cat. (0.5 mol%). |
||||
 下载: 导出CSV
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 下载:
下载: