State Key Laboratory of Coordination Chemistry, Jiangsu Key Laboratory of Advanced Organic Materials, School of Chemistry and Chemical Engineering, Nanjing University, Nanjing 210023, China
☆ This manuscript is dedicated to Prof. Fengling Qing's 60th birthday. * Corresponding authors. E-mail addresses: wgzhang@nju.edu.cn (W. Zhang)
Received Date:
14 July 2024 Accepted Date:
13 September 2024 Revised Date:
30 August 2024 Available Online:
15 July 2025
Abstract:
Transition-metal-catalyzed tandem cross-coupling reactions can rapidly construct complex molecules, but they often suffer from site- and regio- selectivity issues. Here, we designed a novel nickel-catalyzed three-component cross-electrophile coupling (cXEC) platform that enables access to valuable gem-difluoroalkenes. This multicomponent reaction proceeds through a chemoselective alkenylation of aryl halides, followed by alkylation of α-(trifluoromethyl)styrenes, providing a streamlined pathway towards this kind of building blocks.
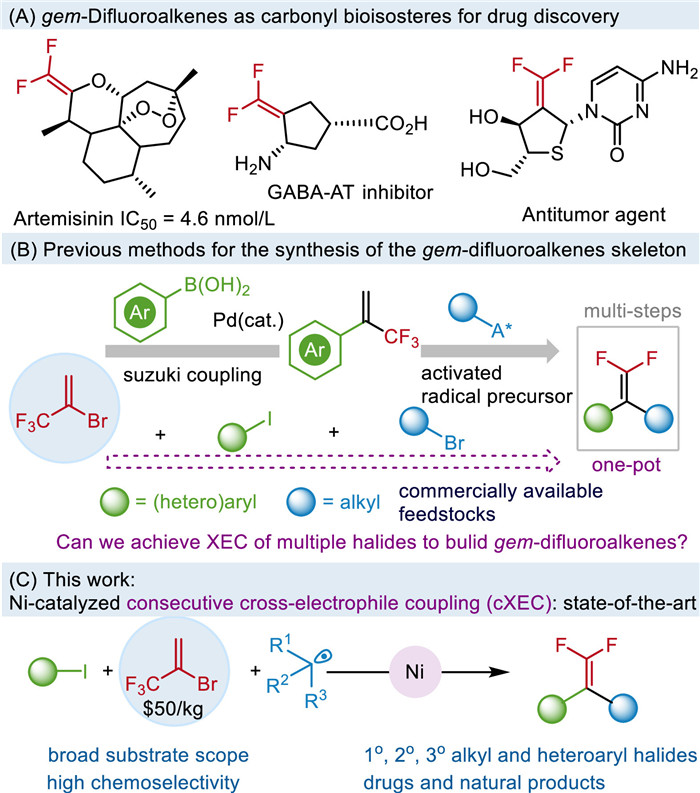
As the bioisosteres of carbonyl compounds, gem-difluoroalkenes are found in many medicinal scaffolds and they are a privileged motif in drug discovery [1]. Introducing gem-difluoroalkenes into pharmaceuticals can significantly enhance the bioactivity of drug molecules [2-5]. However, a modular three-component platform for the synthesis of gem-difluoroalkenes remains elusive. Conventional methods to access gem-difluoroalkenes usually started with carbonyl [6-12] or diazo compounds [13-15], or by SN2'-type addition-elimination with organometallic reagents [16-18], which might be problematic due to the unstable intermediates.
Recently, defluoroalkylation of α-(trifluoromethyl)styrenes with alkyl radical precursors has been reported [19-32]. However, the need to pre-synthesize the α-(trifluoromethyl)styrenes makes the late-stage functionalization (LSF) challenging [33]. α-(Trifluoromethyl)styrenes are commonly synthesized through Suzuki-Miyaura coupling reaction of 2-bromo-3, 3, 3-trifluoropropene (BTP) with arylborons [34]. Although there is a considerable number of commercially available arylborons, their availability is limited in comparison to commercially accessible (het)aryl halides. Furthermore, the protocol for the construction of heteroaryl-substituted gem-difluoroalkenes has been infrequently reported [35-37], primarily attributed to the challenges associated with accessing heteroaryl-substituted trifluoroalkenes and the complexities involved in multi-step synthesis process. Therefore, it is crucial to develop efficient catalytic systems for constructing this crucial building block.
The nickel-catalyzed cross-electrophile coupling reactions (XEC) typically employ aryl and alkyl halides as electrophiles, thereby circumventing the need for highly reactive organometallic reagents and enhancing functional group tolerance [38-41]. We assumed whether a nickel-catalyzed electrophilic cross-coupling reaction could enable the access to gem-difluoroalkenes. However, three-component XECs are generally limited to the difunctionalization of olefins [42-48], expanding this strategy to other substrates remains challenge due to the potential chemoselectivity and byproducts formation. Intrigued by this, we sought to develop a Ni-catalyzed consecutive cross-electrophile coupling (cXEC) relying on specific catalytic mode of various halide reagents. Initially, two electrophilic halides would selectively couple to form a stable intermediate, which could be stitched with another coupling partner to afford highly functionalized skeletons. Particularly, a single catalyst continuously governs both catalytic cycles, which may result in potential intersections of the reaction and a diversity of coupling products (Fig. 1C).
Figure 1
Figure 1.
(A) gem-Difluoroalkenes as carbonyl bioisosteres for drug discovery. (B) Previous methods for the synthesis of the gem-difluoroalkenes skeleton. (C) This work: Ni-catalyzed consecutive cross-electrophile coupling (cXEC).
Our investigations began with the reaction of ethyl 4-iodobenzoate (1), 2-bromo-3, 3, 3-trifluoropropylene (BTP) (2) and 2-bromo-2-methylpropane (3) (Table 1). Using NiBr2·glyme as the catalyst, L1 as the ligand, Mn as the reductant, and NaI as an additive in DMA, the corresponding product (4) was delivered with a 76% yield. When the ligand was switched to L2, with increased steric hindrance, the yield decreased to 57%. Bidentate ligands, like bipyridine (L3), proved to be ineffective (entry 3 and Supporting information for details). Surprisingly, terpyridine resulted in only trace amounts of the product (entry 4). The exact reason is unknown but could be due to electronic effects. Other nickel sources could also be used as catalysts, though with slightly lower yields (entries 5–7). NaI is crucial to the reaction, MgBr2 proved no effect on the reaction (entries 8 and 9), while TBAI had a similar effect to NaI (entry 10). Solvents was also evaluated in the reaction, including DMF and DMSO (entries 11 and 12). Zn can be used as a substitute for Mn, but lowing the amount of BTP yielded a lower yield (entries 13 and 14). Finally, by changing the catalyst to NiBr2 and adding TBAI (0.2 equiv.), a higher yield could be afforded (89%).
a Reaction condition: Aryl iodide (0.2 mmol), BTP (0.4 mmol), alkyl bromide (0.4 mmol), NiBr2 (10 mol%), L1 (12 mol%), TBAI (0.2 equiv.), Mn (3 equiv.), in DMA (1 mL) under argon for 8 h. TBAI = Tetrabutylammonium iodide. b Yield was determined by GC using dodecane as the internal standard. c Isolated yield.
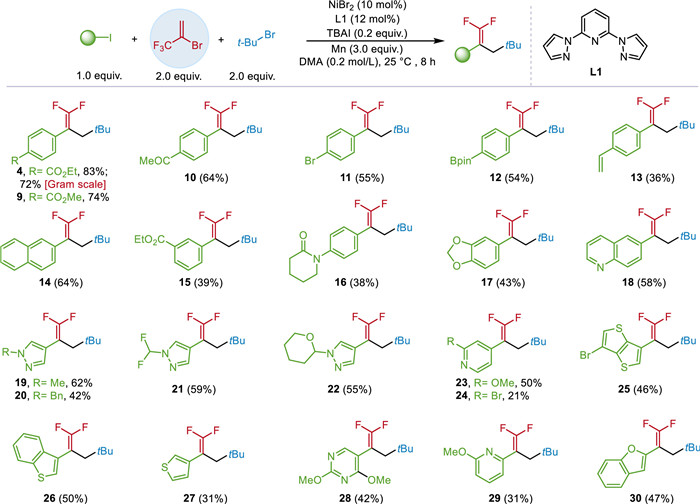
With the optimized conditions in hand, we first explored the substrate scope of aryl and heteroaryl iodides using BTP (2) and 2-bromo-2-methylpropane (3) as the model substrates (Scheme 1). The protocol reported here was found to be applicable to a broad set of aryl iodides, leading to the successful formation of target products in good yields and excellent chemoselectivity. Functional groups including esters (4, 9, 15), amides (16), carbonyls (10), boronates (12), and olefins (13) could be tolerated in the reaction. Naphthalene iodide (14) and benzodioxole iodide (17) yielded the gem-difluoroalkenes in 64% and 43% yield, without solubility issues. Heteroaryl iodides also could yield the corresponding products. Pyrazoles containing methyl, benzyl, difluoromethyl and tetrahydro-2H-pyran substituents afforded products 19–22 in moderate to good yields. Electron-deficient heterocycles, including pyridines (23, 24, 29) and pyrimidines (28) delivered the target products in 21%–50% yield. Electron-rich heterocycles including dithiophene (25), benzothiophene (26), thiophenes (27) and benzofurans (30) can also furnish the target products (31%–50% yields). Remarkably, this Ni-catalyzed system exhibits good site-selectivity with 1-bromo-4-iodobenzene affording the desired product (11) in 55% yield with the bromide remained unreactive.
Scheme 1
Scheme 1.
Substrate scope of aryl and heteroaryl iodides. Condition A: (hetero)aryl iodides (0.2 mmol), BTP (0.4 mmol), alkyl bromides (0.4 mmol), NiBr2 (10 mol%), L1 (12 mol%), TBAI (0.2 equiv.) and Mn (3 equiv.) in DMA (1 mL) under argon for 8 h.
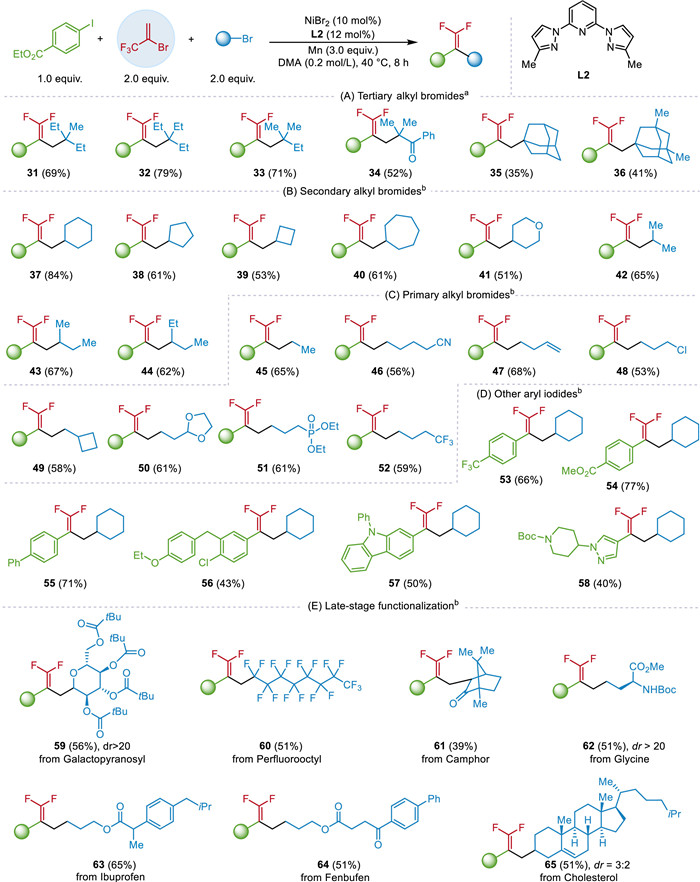
The scope of the alkyl bromides was also tested (Scheme 2). A diverse array of tertiary alkyl bromides could be converted to the coupling products in 35%–79% yields (Scheme 2A, 31–36). Primary and secondary alkyl bromides necessitating minor adjustments to the reaction conditions (Schemes 2B-D), including raising the temperature from 25 ℃ to 40 ℃, and changing the ligand to L2. Under the modified conditions, cyclic (37–41) and acyclic (42–44) alkyl bromides could be both participated in the reaction (51%–84%). Functional groups, including cyanoyl (46), alkenyl (47), ketal (50), phosphonate (51), and trifluoromethyl (52), were tolerated. In addition, a range of aryl iodides, such as substituted phenyl iodides (53–55), and carbazole (57) or pyrazole iodides (58), were also compatible in this protocol. Notably, halogens can be remained during the reaction (48, 56). Finally, we explored this protocol in the late-stage functionalization of natural and bioactive molecules. Galactopyranosyl (59), perfluorooctyl (60), camphor (61), glycine (62), ibuprofen (63), fenbufen (64), and cholesterol (65), exhibited good tolerance, providing the corresponding products in 39%–65% yields.
Scheme 2
Scheme 2.
Substrate scope of alkyl bromides and late-stage modification of complex molecules. a Condition A: (hetero)aryl iodides (0.2 mmol), BTP (0.4 mmol), alkyl bromides (0.4 mmol), NiBr2 (10 mol%), L1 (12 mol%), TBAI (0.2 equiv.), Mn (3 equiv.), in DMA (1 mL) 25 ℃, under argon for 8 h. b Condition B: (hetero)aryl iodides (0.2 mmol), BTP (0.4 mmol), alkyl bromides (0.4 mmol), NiBr2 (10 mol%), L2 (12 mol%), Mn (3 equiv.), in DMA (1 mL), 40 ℃, under argon for 8 h.
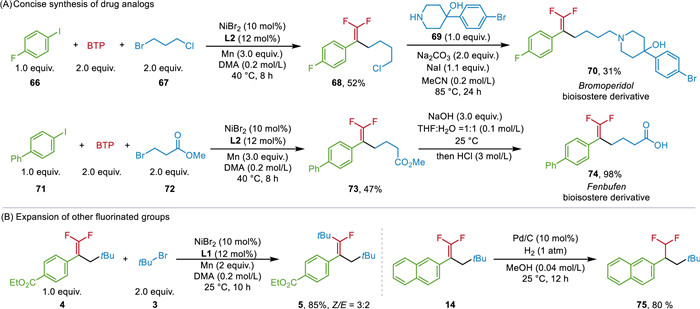
In order to further demonstrate the practical utility of this method, we focused our attention on using our method for the synthesis of bioisostere derivative of drug molecules (Scheme 3A). Bromperidol, an antipsychotic drug known for its specific efficacy in treating hallucinations and delusions [49], was chosen as a target compound. The tandem cross-electrophile coupling using commercially available aryl iodine 66 and alkyl bromide 67 with BTP, followed by nucleophilic substitution, enabled the Bromoperidol bioisostere derivative (70). Moreover, this protocol could also be employed to construct bioisostere derivative of the anti-inflammatory drug Fenbufen (74). Furthermore, the obtained difluoroalkenes were further elaborated (Scheme 3B). Adding excess of alkyl halides under the standard conditions, the defluoroalkylated product 5 was isolated in 85% yield through a C-F bond activation process [50-53]. The hydrogenation of the gem-difluoroalkene 14 can produce CF2H-subtituted naphthalene 75 in 80% yield [54, 55].
In order to investigate the mechanism of this consecutive cross-electrophile coupling, a series of experiments had been implemented. Aryl iodide 1 and BTP were able to efficiently yield 76 under standard conditions, and then the reaction between 76 and alkyl bromide 3 can smoothly generate the target product in 84% yield (Scheme 4A1). This result validated that the reaction is a sequential process. The primary kinetic profile also showed the aryl iodide 1 reacted with BTP first, to afford the compound 76, which could be added by alkyl bromide to deliver the final product (Scheme 4A2). Besides, the nickel-aryl complex (77) could react with BTP, afforded the trifluoromethyl alkene (76) in 33% yield, providing evidence for the reaction starting form Ni(0). In the absence of Mn, stoichiometric amounts of Ni(COD)2 were stirred at room temperature with 76 and alkyl bromide 3 for 8 h afforded product 4 in 78% yield (Scheme 4A3). Addition of 2 equiv. of TEMPO, the reaction was suppressed and the TEMPO adduct 83 was detected by HRMS (see Supporting information for detail). Additionally, radical clock experiments were carried out by adding cyclopropyl-substituted styrene 78 in the reaction, offering the ring-opening product 79 in 25% yield. These results demonstrated the Ni(0) reacted with alkyl bromides is a radical process instead of oxidative addition (Scheme 4A4).
Based on these results and previous reports [56, 57], a possible mechanism is proposed in Scheme 4B. The consecutive electrophilic cross-coupling reaction involves two catalytic cycles. Initially, the Ni(Ⅱ) catalyst undergoes reduction by Mn, generating the Ni(0) species A. Oxidative addition of ArI to Ni(0) resulted in the formation of the Ni(Ⅱ) species B, which could be reduced by Mn, leading to the Ni(Ⅰ) species C. The Ar-Ni(Ⅰ) intermediate C was further reacted with BTP, delivering the Ni(Ⅲ) intermediate D. After reductive elimination, liberating the intermediate I and regenerating the Ni(Ⅰ) species E. Concurrently, in the second catalytic cycle, the Ni(0) species A undergoes SET process with alkyl bromides, producing the Ni(Ⅰ) species F and the alkyl radical. The afforded alkyl radicals could be added to intermediate I, forming a new radical, which could be trapped by the Ni(Ⅰ) species to form G. The intermediate G undergoes β-fluorine elimination, resulting in the generation of the desired products. Finally, the initial Ni(0) is regenerated through the reduction of Ni(Ⅱ) species H by Mn. According to the previous reports [58, 59], the mechanism process of Ni(Ⅰ) intermediate as the initial active catalytic species cannot be ruled out.
In summary, we have disclosed a sequential and modular nickel-catalyzed cross-electrophile coupling approach that enables the synthesis of functionalized gem-difluoroalkenes with high efficiency and chemoselectivity. This protocol employs readily available chemical feedstock with single nickel catalyst to operate in two separate catalytic cycles simultaneously within a one-pot setup. The late-stage functionalization of complex bioactive molecules illustrates the potential application in drug discovery.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Jiyang Liu: Writing – review & editing, Writing – original draft, Methodology, Investigation, Formal analysis, Data curation, Conceptualization. Xiangzhang Tao: Methodology, Conceptualization. Zhenlei Zou: Data curation. Jia Xu: Software, Data curation. Hui Shu: Data curation. Yi Pan: Writing – review & editing. Weigang Zhang: Writing – review & editing, Funding acquisition. Shengyang Ni: Writing – review & editing, Funding acquisition. Yi Wang: Writing – review & editing, Funding acquisition, Conceptualization.
Acknowledgments
We gratefully acknowledge the financial support from the National Natural Science Foundation of China (Nos. 22071101 and 22271147), China Postdoctoral Science Foundation (Nos. 2021T140309 and 2021M691511) and Natural Science Foundation of Jiangsu Province (No. BK20230771).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.110461.
[1]
C. Leriche, X. He, C.W.T. Chang, H.W. Liu, J. Am. Chem. Soc. 125 (2003) 6348–6349. doi: 10.1021/ja021487+
[2]
N.A. Meanwell, J. Med. Chem. 54 (2011) 2529–2591. doi: 10.1021/jm1013693
[3]
S. Messaoudi, B. Tréguier, A. Hamze, et al., J. Med. Chem. 52 (2009) 4538–4542. doi: 10.1021/jm900321u
[4]
G. Magueur, B. Crousse, M. Ourévitch, D. Bonnet-Delpon, J.P. Bégué, J. Fluorine Chem. 127 (2006) 637–642.
[5]
C. Yao, S. Wang, J. Norton, M. Hammond, J. Am. Chem. Soc. 142 (2020) 4793–4799. doi: 10.1021/jacs.9b13757
[6]
I. Nowak, M.J. Robins, Org. Lett. 7 (2005) 721–724. doi: 10.1021/ol047416s
[7]
O. Schutz, A. Masarwa, I. Zilbermann, et al., Eur. J. Org. Chem. 2014 (2014) 932–940. doi: 10.1002/ejic.201301115
[8]
B. Gao, Y. Zhao, J. Hu, J. Hu, Org. Chem. Front. 2 (2015) 163–168. doi: 10.1039/C4QO00291A
[9]
Y. Zhao, W. Huang, L. Zhu, J. Hu, Org. Lett. 12 (2010) 1444–1447. doi: 10.1021/ol100090r
[10]
B. Gao, Y. Zhao, M. Hu, C. Ni, J. Hu, Chem. Eur. J. 20 (2014) 7803–7810. doi: 10.1002/chem.201402183
[11]
J. Zheng, J. Cai, J.H. Lin, Y. Guo, J.C. Xiao, Chem. Commun. 49 (2013) 7513–7515. doi: 10.1039/c3cc44271c
[12]
K. Aikawa, W. Toya, Y. Nakamura, K. Mikami, Org. Lett. 17 (2015) 4996–4999. doi: 10.1021/acs.orglett.5b02439
[13]
M. Hu, Z.B. He, B. Gao, et al., J. Am. Chem. Soc. 135 (2013) 17302–17305. doi: 10.1021/ja409941r
[14]
M. Hu, C. Ni, L. Li, Y. Han, J. Hu, J. Am. Chem. Soc. 137 (2015) 14496–14501. doi: 10.1021/jacs.5b09888
[15]
Z. Zhang, W.Z. Yu, C.G. Wu, et al., Angew. Chem. Int. Ed. 55 (2016) 273–277. doi: 10.1002/anie.201509711
[16]
K. Fuchibe, M. Takahashi, J. Ichikawa, Angew. Chem. Int. Ed. 51 (2012) 12059–12062. doi: 10.1002/anie.201206946
[17]
J. Ichikawa, Y. Ishibashi, H. Fukui, Tetrahedron Lett. 44 (2003) 707–710.
[18]
W. Dai, Y. Lin, Y. Wan, S. Cao, Org. Chem. Front. 5 (2018) 55–58. doi: 10.1039/C7QO00716G
[19]
Y.J. Jang, D. Rose, B. Mirabi, M. Lautens, Angew. Chem. Int. Ed. 57 (2018) 16147–16151. doi: 10.1002/anie.201808509
[20]
C. Zhang, Z. Lin, Y. Zhu, C. Wang, J. Am. Chem. Soc. 143 (2021) 11602–11610. doi: 10.1021/jacs.1c04531
Q. Lin, T. Diao, J. Am. Chem. Soc. 141 (2019) 17937–17948. doi: 10.1021/jacs.9b10026
[59]
W. Shu, A. García-Domínguez, M.T. Quirós, et al., J. Am. Chem. Soc. 141 (2019) 13812–13821. doi: 10.1021/jacs.9b02973
Figure 1
(A) gem-Difluoroalkenes as carbonyl bioisosteres for drug discovery. (B) Previous methods for the synthesis of the gem-difluoroalkenes skeleton. (C) This work: Ni-catalyzed consecutive cross-electrophile coupling (cXEC).
a Reaction condition: Aryl iodide (0.2 mmol), BTP (0.4 mmol), alkyl bromide (0.4 mmol), NiBr2 (10 mol%), L1 (12 mol%), TBAI (0.2 equiv.), Mn (3 equiv.), in DMA (1 mL) under argon for 8 h. TBAI = Tetrabutylammonium iodide. b Yield was determined by GC using dodecane as the internal standard. c Isolated yield.

 DownLoad:
DownLoad:



 下载:
下载:






 下载:
下载:
