Figure 1.
Graphical representation of the discovery of the potent antibacterial agents targeting MraY. The figure was created using ChemDraw and Microsoft Office PowerPoint.

Despite the availability of effective antibacterial drugs, the increasing prevalence of antibiotic-resistant bacteria is primarily attributed to their excessive and inappropriate utilization. Antimicrobial resistance (AMR) represents one of the most concerning global health and development threats [1]. Thereby, the continuous escalation in AMR necessitates urgent advancements in novel antibacterial strategies. The MraY enzyme, which plays a pivotal role in synthesizing bacterial cell wall-composing polysaccharides, holds significant potential as a target for antibacterial agents [2]. However, its conformational dynamics have presented substantial challenges in developing MraY-targeting inhibitors.
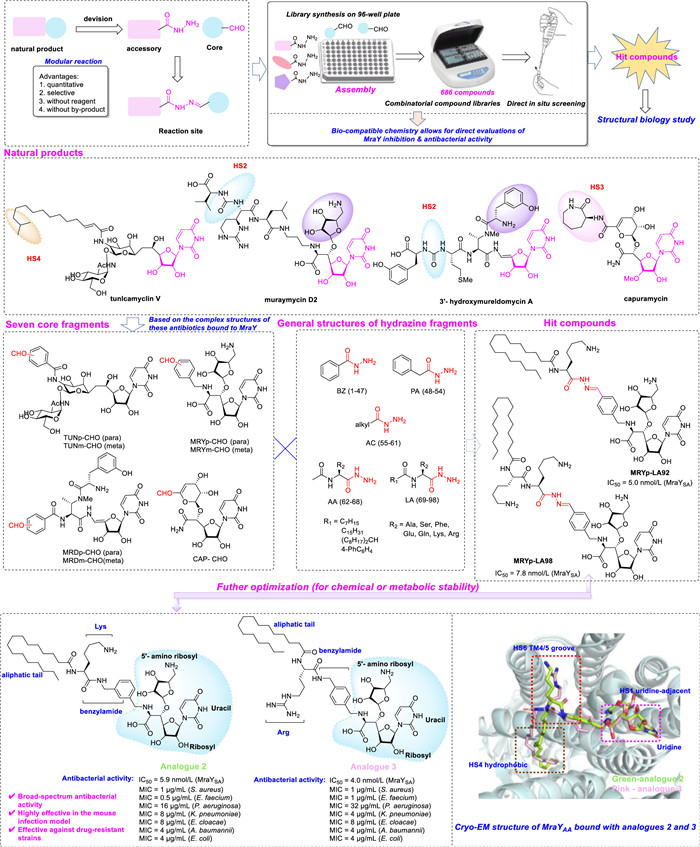
In a groundbreaking study published in Nature Communications, Yamamoto and Ichikawa et al. [3] deconstructed the structure of MraY inhibitory natural products into core and auxiliary fragments to facilitate the development of robust MraY inhibitors (Fig. 1). The core fragments are expected to play a crucial role in protein binding, while the auxiliary fragments serve to regulate affinity towards the target, selectivity against non-targets, and in vivo disposition properties. Subsequently, employing a highly selective nitrilimine bond formation reaction to establish a connection between the core and the auxiliary fragments, they constructed an extensive library comprising 686 compounds on a microplate platform, enabling nitrilimine bond formation in the absence of any additives through simple mixing of DMSO solutions containing the core aldehyde/ketone and auxiliary hydrazine. The compound library allows for direct synthesis and microplate-based evaluation of compounds, eliminating the need for conventional separation and purification steps. Next, two hit compounds, MRYp-LA92 and MRYp-LA98 were identified, demonstrating prominent inhibitory effects on MraY (half maximal inhibitory concentration (IC50) = 5.0 and 7.8 nmol/L, respectively). The minimum inhibitory concentration (MIC) of the compounds was determined using the microdilution broth method in a 96-well plate, with colistin and vancomycin served as positive controls. The results showed that MRYp-LA98, bearing a basic lysine residue, exhibited broad-spectrum antibacterial activity with the lowest overall MICs, ranging from 1 µg/mL to 16 µg/mL, against the entire ESKAPE pathogen panel, surpassing or comparable to colistin and vancomycin. Preliminary structure-activity relationships (SAR) analysis indicated that the presence of a long lipophilic chain is crucial for antibacterial activity.
Further SAR analysis revealed a strong correlation between the key chemical features of MraY inhibitory natural products and their biological activity. The study emphasizes the essential role of the uridine moiety, which serves as a fundamental structure binding to the active site of MraY. Additionally, the diversity of auxiliary fragments significantly impacts inhibitor affinity, selectivity, and pharmacokinetic properties. Specifically, compounds containing long-chain hydrophobic alkyl groups and positively charged amino acids (such as lysine or arginine) exhibit enhanced antibacterial activity due to their effective interaction with both the hydrophobic groove and polar head region of MraY SAR analysis also underscores how long-chain alkyl portions function as membrane anchors while positively charged amino acid residues enhance compound interaction with bacterial cell membranes. These findings establish a solid chemical and structural foundation for further optimization of MraY inhibitors and facilitate the design of novel antibacterial drug candidates with improved pharmacological properties.
Given the inherent susceptibility of hydrazones to hydrolysis, stable analogues incorporating amide or anilide groups based on MRYp-LA92 were designed and synthesized. These compounds consist of Lys or Arg residues and various linkers to enhance chemical or metabolic stability. Analogues 1 to 8 exhibited potent MraY inhibition (IC50 = 1.7–6.0 nmol/L). Subsequent assessment of antibacterial activity revealed excellent efficacy against S. aureus and E. faecium with MIC values between 0.5–1 and 0.25–1 µg/mL, respectively. Toxicity assessment demonstrated moderate cytotoxicity for all analogues (IC50 = 12–25 µmol/L, HepG2 cells), which was lower than that observed for known MraY inhibitors. It is noteworthy that these compounds displayed high efficacy against antimicrobial drug-resistant strains, such as methicillin-resistant S. aureus (MRSA) and vancomycin-resistant enterococci (VRE), as well as most clinical isolates. Suggesting a low likelihood of cross-resistance with existing drugs. Next, the researchers focused on analogue 2 and evaluated its potential as an antibacterial agent in vivo. The results demonstrated its efficacy as an ideal antibacterial agent due to its dual mechanism of action, inhibiting peptidoglycan biosynthesis while also exerting a membrane-disrupting effect.
Building on the above findings, the researchers determined the Cryo-EM structure of MraYAA complexed with a nanobody recognizing its periplasmic face (NB7-MraYAA) in the presence of two selected inhibitors (analogues 2 and 3). The findings unveiled the unique binding modes of compounds 2 and 3 with MraYAA, which is typically situated near the active center. Previously, the highly conserved binding pocket was divided into six hot spots (HSs). The inhibitors demonstrated an embedded uridine moiety within a highly conserved uridine pocket, surrounded by residues K70, G194, L195, D196, and F262, similar to interaction seen with other nucleoside natural product inhibitors, while their 5′-amino ribose moiety interacts with an adjacent site (HS1). Furthermore, the shape complementarity and electrostatic interactions between lysine or arginine side chains of inhibitors 2 and 3 and the TM4/5 groove (HS6) facilitated their binding to MraYAA. These pivotal interactions guided hydrophobic side chains into a hydrophobic groove (HS4), thereby enhancing inhibitor potency. Overall, these structural insights not only elucidate the interaction of inhibitors with the active site of MraY but also establish a foundation for designing novel MraY inhibitors.
Additionally, the researchers extended the utilization of the build-up library synthesis strategy to tubulin-binding natural products such as epothilone B, paclitaxel, and vinblastine. By strategically introducing diverse formyl groups at various positions of these natural products, they designed and synthesized six core aldehydes. These aldehydes were reacted with 98 auxiliary hydrazines to construct a comprehensive library comprising 588 hydrazone compounds. The compound library was then evaluated for its effects on microtubule protein polymerization/depolymerization activity and cell growth inhibition. The results revealed that certain analogues exhibited enhanced microtubule protein polymerization activity compared to the original natural products and demonstrated significant cytotoxicity against HCT-116 cells in vitro. In conclusion, this study successfully demonstrated the universality of the strategy for in situ evaluation of the build-up library, establishing a robust paradigm for the efficient discovery of promising drug candidates.
The innovative strategy highlighted here marks a significant leap forward in the field of natural product-based drug discovery. By employing a fragment-based approach to optimize MraY inhibitors and tubulin-binding natural products, their research opens new avenues for the development of next-generation antibacterial agents. The incorporation of core aldehydes and accessory hydrazines in constructing a diverse library of analogues exemplifies a modular and efficient chemical synthesis strategy that can be adapted to various bioactive natural products. Furthermore, compared to traditional medicinal chemistry, it significantly enhances the ability to rapidly generate and evaluate numerous compounds simultaneously, allowing for a deeper comprehension of SAR.
The research focuses on integrating structural biology with drug design, laying the groundwork for the future of medicinal chemistry. The cryo-EM structures of MraY-analogue complexes offer invaluable insights into the enzyme's interaction with inhibitors, guiding the rational design of more effective compounds. Detailed interactions revealed by the cryo-EM analyses illustrate how specific structural modifications influence binding affinity and mechanism of action, which is crucial for developing inhibitors with unique binding modes that can potentially bypass existing resistance mechanisms. Furthermore, this strategy's flexibility to accommodate different target proteins suggests it can be widely applied across various therapeutic areas, beyond just infectious diseases and cancer.
From our perspective, the research represents a significant advancement in the rational design of drugs derived from natural products. However, several challenges need to be addressed in this field. For instance, we must contend with the limited availability of modular reaction types and the high sensitivity requirements for direct screening techniques, as well as improve methods to assess the pharmacokinetics, pharmacodynamics, and toxicity of the newly synthesized analogues. Additionally, synthesizing natural products poses difficulties due to their intricate nature. Other obstacles include the issue of chiral isomerism in synthesizing fragment molecules, the diversity of molecules in modular reactions, and detection methods. While the strategy is broadly applicable, specific adaptations may be necessary for each new target or class of natural products, which calls for a flexible and iterative optimization process. These innovative strategies have improved efficiency in exploring novel drugs but also emphasize the importance of continuously updating and integrating traditional methods to better serve our needs [4]. In future endeavors, we anticipate integrating this strategy with protein degradation technologies to expedite the discovery of novel mechanism-based broad-spectrum antimicrobial drugs [5].
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Mianling Yang: Writing – review & editing, Writing – original draft. Meehyein Kim: Writing – review & editing. Peng Zhan: Writing – review & editing.
S. Barman, L.B. Kurnaz, R. Leighton, et al., Biomaterials 311 (2024) 122690.
T.D.H. Bugg, R.V. Kerr, J. Antibiot. 72 (2019) 865–876. doi: 10.1038/s41429-019-0227-3
K. Yamamoto, T. Sato, A. Hao, et al., Nat. Commun. 15 (2024) 5085. doi: 10.1038/s41467-024-49484-7
H. Sun, S. Li, Q. Liu, et al., Chin. Chem. Lett. (2024), doi: 10.1016/j.cclet.2024.109999.
K.N. Yan, Y.Q. Nie, J.Y. Wang, et al., Adv. Sci. 11 (2024) e2400594. doi: 10.1002/advs.202400594

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
 下载:
下载: