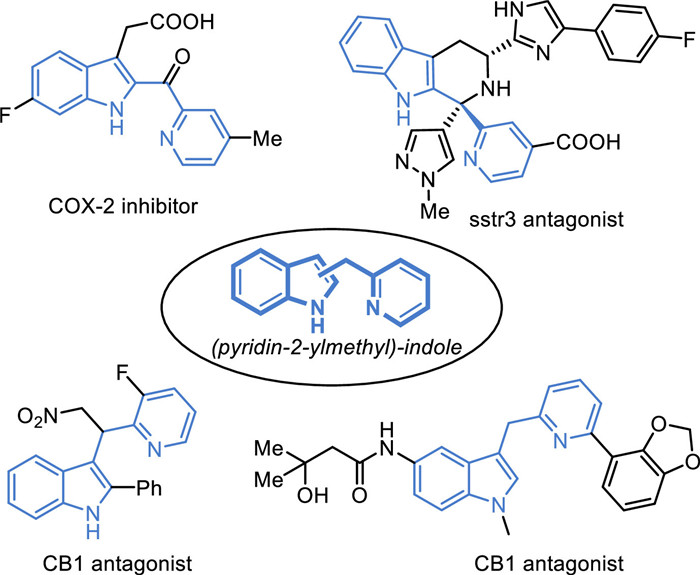
Figure 1.
Biologically active (pyridine-2-ylmethyl)-indole derivatives.

Nitrogen heterocycles are one of the most important structural fragments in pharmaceuticals. Recent data from the US FDA approved drugs indicates that over half of small molecule drugs contain nitrogen heterocycles [1]. Indole and pyridine are two classic "privileged structures" in drug molecules, which rank second and ninth, respectively, among FDA-approved heterocyclic drugs, highlighting their significant advantages in drug development. It is notable that (pyridin-2-ylmethyl)-indole, a structural unit formed by the combination of indole and pyridine, found in many pharmacologically active molecules with diverse and significant biological activities as COX-2 inhibitors, sstr3 agonists, and CB1 agonists (Fig. 1) [2-5]. Its synthetic method is typically limited to the pyridyl alkylation of indole through nucleophilic substitution by using picolinaldehyde under harsh reaction conditions, which poses a significant challenge for the functional group tolerance (Scheme 1A) [6]. The C−H activation provides a mild approach for the synthesis of heterocycles and their late-stage modifications under nearly neutral reaction conditions with good functional group tolerance and a broad range of substrate applicability [7-10]. Therefore, the directed C−H functionalization strategy is highly anticipated to offer a potential efficient method for the synthesis of highly valuable pharmaceutical (pyridin-2-ylmethyl)-indole derivatives.

The switchable C−H functionalization of indole at C2- and C3-positions with high regioselectivities has been always an attractive work [11, 12]. Although metal-catalyst-, ligand- or oxidate-dependent selective functionalization strategies have been developed to achieve selective arylation [13-21], alkenylation [22, 23], cyanation [24], and alkynylation [25] at the 2, 3-positions of indoles, the expanded C−H functionalization scope of indoles at C2- and C3-position is still a challenging field that continues to attract significant interests. Transition-metal catalyzed carbenoid coupling reactions have been well-established as powerful tools in C−C bond formation with diazo compounds and N-tosylhydrazones as precursors to carbenes [26]. 1, 2, 3-Triazoles are commonly used as directing groups or auxiliary directing groups in metal-catalyzed C−H activation reactions due to their strong coordination with the metal (Scheme 1B) [27-33]. Despite being able to participate in a variety of carbenoid functionalization as a carbene precursor, its application in C−H activation is not widely observed [34]. Recently, pyridotriazoles as precursors to carbenes have been successfully used in the directed ortho sp2 C−H activation process to access pyridyl methylated arenes by using pyridyl [35], sulfoximine [36], imidamides [37] or sulfonamide [38] as a directing group (Scheme 1C), however, switchable carbenoid functionalization at different sp2 C−H sites remains challenging. Due to the high side reactivity and low regioselectivity present in most current C−H activation reactions, developing effective strategy to achieve high regioselectivity in C−H functionalization is highly desired. Despite advances in the carbenoid functionalization of indoles with diazo compounds [39-42], the development of carbenoid functionalization of indoles with pyridotriazoles as carbene precursors is highly desired to expand the applicability of these processes. Considering the challenges of achieving selective functionalization at the C2- and C3-positions of indoles and the significant pharmaceutical value of pyridyl methylated indole derivatives, herein, we first report a switchable carbenoid coupling reactions of indoles and 1, 2, 3-triazoles to afford C2- and C3-carbenoid functionalized indoles by a strategy of synergistic dependence between Rh-catalysts and auxiliary groups. Strong coordinating group (2-pyridyl, 2-pym) directs the reaction to occur at the C2-position (Scheme 1D) [11], whereas non-coordinating group (benzyl, Bn) leads the reaction to occur at the more nucleophilic C3-position (Scheme 1E).
The ortho-directing C−H activation strategy is an important approach for the C2-functionalization of indoles. Due to the strong coordination with metal catalyst and good removal properties, the pyrimidyl (pym) is often selected as a suitable directing group to assist for C−H bond cleavage and C−metal bond formation [43, 44]. Initially, the C−H carbenoid functionalization of indole at C2-position was investigated by using N-pym indole (1a) as a model substrate with different solvents in the presence of 5 mol% [Cp*RhCl2]2 and 20 mol% AgOAc at 100 ℃ for 18 h (Table 1, entries 1–6). Compared to 1, 4-dioxane, toluene, DMF and DCE, MeCN or MeOH resulted in a good yield (88% or 89%, respectively). Additive screening showed that AgOAc was optimal, NaOAc, nBuNOAc or no additive gave a significantly decreased yield of the desired product 3a (Table 1, entries 7–9). Raising the temperature to 120 ℃ or lowering the temperature to 80 ℃ both resulted in a decreased yield, indicating that 100 ℃ was an appropriate reaction temperature (Table 1, entries 10 and 11). We then turned our attention to the C−H functionalization at the C3-position of indole. Inspired by the intermolecular reactions of electron-rich heterocycles with rhodium carbenoids [45], Rh2(OAc)4-catalyzed C3-selective cross-couplings of N-benzyl indole (4a) and pyridotriazole (2a) through carbenoid insertion was tested in different solvent at 110 ℃ for 18 h, and the results were summarized in Table 2. Toluene gave the desired C3-carbenoid functionalized product 5a in 62% yield, but other tested solvents led to significantly decreased yields (Table 2, entries 1–5). Further investigation on reaction temperature showed that lowering the temperature to 90 ℃ resulted in an incomplete conversion (Table 2, entry 6). It is worth noting that the yield of 5a could be increased to 71% when the reaction was carried out at 110 ℃ for 12 h and then 130 ℃ for 6 h (Table 2, entry 7). Other Rh-catalysts, including Rh(Ⅰ), and Rh(Ⅲ), did not give the satisfactory results, indicating the crucial importance of Rh2(OAc)4 for the C3-carbenoid functionalization (Table 2, entries 8–12). Reducing the Rh2(OAc)4 loading to 5% resulted in a slight decrease in yield of the desired C3-functionalized product 5a (Table 2, entry 13).
 DownLoad:
CSV
DownLoad:
CSV
 |
|||
| Entry | Additive | Solvent | Yield (%)b |
| 1 | AgOAc | 1, 4-Dioxane | 35 |
| 2 | AgOAc | Toluene | 66 |
| 3 | AgOAc | DMF | 45 |
| 4 | AgOAc | MeCN | 88 |
| 5 | AgOAc | DCE | 70 |
| 6 | AgOAc | MeOH | 89 |
| 7 | NaOAc | MeOH | 23 |
| 8 | nBuNOAc | MeOH | 47 |
| 9 | – | MeOH | 19 |
| 10c | AgOAc | MeOH | 69 |
| 11d | AgOAc | MeOH | 58 |
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (0.3 mmol, 1.5 equiv.), [RhCp*Cl2]2 (0.01 mmol, 5 mol%), additive (0.04 mmol, 20 mol%), solvent (2 mL), 100 ℃, 18 h. b Isolated yield. c 80 ℃. d 120 ℃. |
|||
DownLoad:
CSV
 |
|||
| Entry | Rh-catalyst | Solvent | Yield (%)b |
| 1 | Rh2(OAc)4 | 1, 4-Dioxane | 30 |
| 2 | Rh2(OAc)4 | DMF | 28 |
| 3 | Rh2(OAc)4 | MeOH | 15 |
| 4 | Rh2(OAc)4 | Toluene | 62 |
| 5 | Rh2(OAc)4 | DCE | 35 |
| 6c | Rh2(OAc)4 | Toluene | 46 |
| 7d | Rh2(OAc)4 | Toluene | 71 |
| 8d | [Rh(COE)Cl]2 | Toluene | 10 |
| 9d | [Rh(COD)Cl]2 | Toluene | 0 |
| 10d | Rh(PPh3)2(CO)Cl | Toluene | 0 |
| 11d | Rh(acac)3 | Toluene | 0 |
| 12d | [RhCp*Cl2]2 | Toluene | 14 |
| 13d, e | Rh2(OAc)4 | Toluene | 58 |
| COD = cyclooctadiene, COE = cyclooctene, acac = acetylacetonate. a Reaction conditions: 4a (0.2 mmol, 1.0 equiv.), 2a (0.3 mmol, 1.5 equiv.), Rh2(OAc)4 (0.02 mmol, 10 mol%), solvent (2 mL), 110 ℃, 18 h. b Isolated yield. c 90 ℃. d 110 ℃, 12 h then 130 ℃, 6 h. e 5 mol% Rh2(OAc)4. |
|||
With the optimal conditions in hand, the substrate scope of rhodium-catalyzed site-selective cross-couplings of indoles and pyridotriazoles at C2- or C3-position was investigated (Scheme 2). A series of indoles substituted at C4-, C5-, C6-, C7-position can be converted into switchable C2- or C3-products in moderate to excellent yields. A variety of electronically diverse indoles performed well under these two site-selective conditions to afford the desired C2 and C3-functionalized products, regardless of whether the substituent bears electron-donating or electron-withdrawing groups. It is worth noting that these two methods can be correspondingly used to obtain the C2- and C3-functionalized products with good functional group tolerance, respectively. Further investigation showed that the pyridyl and methyl can also effectively assist the switchable cross-coupling of indole and pyridotriazole at C2- and C3-position, respectively. In addition to structurally diverse pyridotriazoles, [1, 2, 3]triazolo[1, 5-a]pyrazine also could be used to afford the desired products in good yields.

The synthetic applicability of these site-selective cross-coupling methods was further evaluated. As shown in Scheme 3A, Method A and Method B are both capable of supporting the synthesis on a 1 mmol scale. The C−H carbenoid functionalized product 3a was smoothly converted to a decarboxylation product 6 under basic conditions. By treating with NaOEt in DMSO, the pyrimidyl group could be further removed to afford an indole derivative 7 (Scheme 3B). Additionally, these two methods could be effectively applied to the late-stage functionalization of indole drug derivatives. For example, compound 8, an inhibitor of 11βHSD1, could be easily transformed into the corresponding pyridyl- and benzyl-substituted indole compounds 9 and 10, which were further derived using this method to obtain indole derivatives with heterocyclic modifications at C2- and C3-positions, respectively (Scheme 3C).
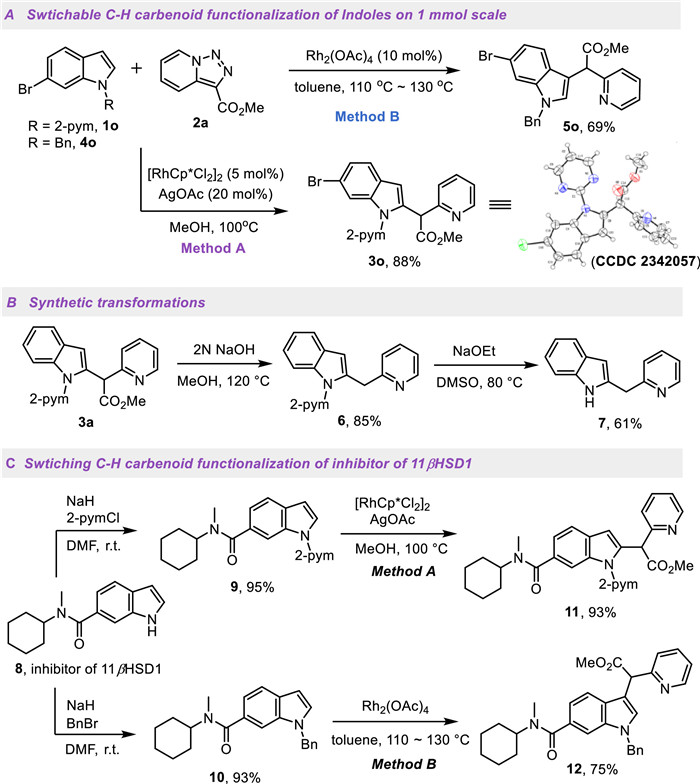
To explore the reaction mechanism, several preliminary studies were conducted (Scheme 4). H/D exchange experiment was carried out under standard conditions using N-pyrimidyl indole 1a in the absence of pyridotriazole 2a in [D4]-MeOD at 100 ℃; 98%, 65% and 67% deuteration were observed at the C2-, C3- and C7-position, indicating that the C−H bond cleavage step at C2-position is reversible (Scheme 4A). The intermolecular kinetic isotope effect (KIE) experiment on pyrimidyl directed C−H carbenoid functionalization gave a relatively small value (k/k = 1.1), indicating that the C−H bond cleavage was likely not the rate-determining step in C2-arylation process (Scheme 4B). As shown in Scheme 4C, treatment of N-pyridyl indole 1a with [Cp*RhCl2]2 and NaOAc gave the rhodacycle 13, which was found to effectively catalyze C2-carbenoid functionalization, giving the desired product 3a in 82% yield. However, a trace amount of the desired product 3a was detected by liquid chromatography-mass spectrometry (LC-MS) analysis when the rhodacycle 13 was used as reactant and treated with pyridotriazole in the presence of 20% AgOAc, which implied that the rhodacycle 13 might be an off-cycle intermediate [46-48]. An intermolecular competitive experiment was preformed using an equimolar mixture of 5-Me- and 5-CF3-substituted indoles, and the results indicated that the electron-rich indole was slightly kinetically favored (3c/3k = 1.7:1, Scheme 4D).
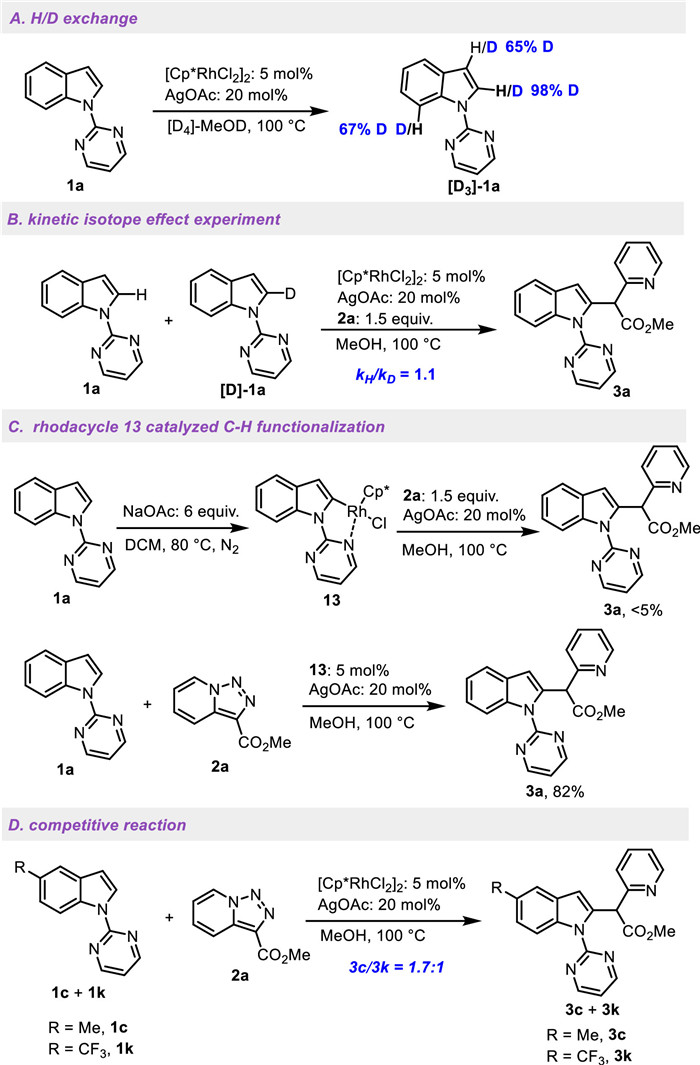
On a basis of the previous literatures and our preliminary experiments, two catalytic cycles are proposed for the site-selective cross-couplings of indoles and pyridotriazoles through carbene insertion in Scheme 5. As shown in catalytic cycle A [49-51], the C2-functionalization of N-pyrimidyl indole 1a is initiated by the ligand exchange of [RhCp*Cl2]2 in the presence of OAc− to give an active rhodium species, RhCp*(OAc)2, followed by the coordination of indole 1a and a reversible C−H bond cleavage to afford the rhodacycle A. Then the Rh-carbene intermediate C is formatted by the release of N2 from diazopyridine B, which is generated in situ from pyridotriazole 2a. The intermediate C undergoes migratory insertion to yield the rhodacycle D, followed by proto-demetalation to generate the desired C2-functionalized product 3a and RhCp*(OAc)2 to start a new catalytic cycle. As shown in catalytic cycle B [52, 53], the C3-functionalization of N-benzyl indole 4a is initiated by transformation of rhodium(Ⅱ) carbene E and its zwitterionic isomer F from diazopyridine B, followed by an intermolecular nucleophilic attack of indole 4a at high electrophilic C3-position to give the zwitterionic intermediate G (path a). The intramolecular aromatization results in intermediate H, which undergoes protodemetallation to afford the desired product 5a. Another possible path should also be considered (path b). The intermediate F undergoes cyclopropanation with indole 4a to give the intermediate I and RhⅡ for the next cycle. Subsequent ring opening generates the intermediate J, followed by the intramolecular aromatization to give the desired product 5a.
In summary, rhodium-catalyzed site-selective cross-couplings of indoles and pyridotriazoles through carbene insertion has been developed in this paper. Rhodium catalysts and auxiliary groups determine the high regioselectivity of this C−H carbenoid functionalization. This selective functionalization strategy can lead to a series of indole derivatives with pyridyl substitutions in switchable C2- and C3-position in moderate to excellent yields. More importantly, this method enables the late-stage modification of biological indole compounds due to its good functional group tolerance, which provides a concise and efficient method to synthesize drug molecules that possess a dual indole-pyridine pharmacophore.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Hua Tian: Writing – original draft, Investigation. Xin Yang: Investigation. Ge Shi: Investigation. Heng Xu: Writing – review & editing, Supervision. Yi Dong: Writing – review & editing, Supervision.
We gratefully acknowledge the CAMS Innovation Fund for Medical Sciences (CIFMS) (Nos. 2022-I2M-1–013, 2022-I2M-1–014, 2022-I2M-2–002).
Supplementary material associated with this article can be found, in the online version, at doi:
E. Vitaku, D.T. Smith, J.T. Njardarson, J. Med. Chem. 57 (2014) 10257–10274. doi: 10.1021/jm501100b
S. Hayashi, N. Ueno, A. Murase, Y. Nakagawa, J. Takada, Eur. J. Med. Chem. 50 (2012) 179–195. doi: 10.1016/j.ejmech.2012.01.053
X. Wu, K. Sudhakar, L.J. Alcock, Y.H. Lau, J. Med. Chem. 66 (2023) 11271–11281. doi: 10.1021/acs.jmedchem.3c00674
S. He, Z. Lai, Z. Ye, et al., ACS Med. Chem. Lett. 5 (2014) 748–753. doi: 10.1021/ml500028c
J.J. Letourneau, P. Jokiel, J. Olson, et al., Bioorg. Med. Chem. Lett. 20 (2010) 5449–5453. doi: 10.1016/j.bmcl.2018.10.047
R.J. Sundberg, R.L. Parton, J. Org. Chem. 41 (1976) 163–165. doi: 10.1021/jo00863a044
Y. Wu, C. Pi, Y. Wu, X. Cui, Chem. Soc. Rev. 50 (2021) 3677–3689. doi: 10.1039/d0cs00966k
C.M. Josephitis, H.M.H. Nguyen, A. McNally, Chem. Rev. 123 (2023) 7655–7691. doi: 10.1021/acs.chemrev.2c00881
J.A. Leitch, Y. Bhonoah, C.G. Frost, ACS Catal. 7 (2017) 5618–5627. doi: 10.1021/acscatal.7b01785
S.B. Ankade, A.B. Shabade, V. Soni, B. Punji, ACS Catal. 11 (2021) 3268–3292. doi: 10.1021/acscatal.0c05580
A.H. Sandtorv, Adv. Synth. Catal. 357 (2015) 2403–2435. doi: 10.1002/adsc.201500374
B. Prabagar, Y. Yang, Z. Shi, Chem. Soc. Rev. 50 (2021) 11249–11269. doi: 10.1039/d0cs00334d
A.N. Campbell, E.B. Meyer, S.S. Stahl, Chem. Commun. 47 (2011) 10257–10259. doi: 10.1039/c1cc13632a
Y.S. Yang, S. Lee, S.H. Son, et al., Org. Chem. Front. 9 (2022) 5906–5911. doi: 10.1039/d2qo01326f
R.J. Phipps, N.P. Grimster, M.J. Gaunt, J. Am. Chem. Soc. 130 (2008) 8172–8174. doi: 10.1021/ja801767s
D.R. Stuart, E. Villemure, K. Fagnou, J. Am. Chem. Soc. 129 (2007) 12072–12073. doi: 10.1021/ja0745862
D. Wang, C.A. Salazar, S.S. Stahl, Organometallics 40 (2021) 2198–2203. doi: 10.1021/acs.organomet.1c00139
B.S. Lane, M.A. Brown, D. Sames, J. Am. Chem. Soc. 127 (2005) 8050–8057. doi: 10.1021/ja043273t
R.J. Phipps, N.P. Grimster, M.J. Gaunt, J. Am. Chem. Soc. 130 (2008) 8172–8174. doi: 10.1021/ja801767s
L. Joucla, N. Batail, L. Djakovitch, Adv. Synth. Catal. 352 (2010) 2929–2936. doi: 10.1002/adsc.201000512
J. Zhou, P. Hu, M. Zhang, et al., Chem. Eur. J. 16 (2010) 5876–5881. doi: 10.1002/chem.201000529
N.P. Grimster, C. Gauntlett, C.R.A. Godfrey, M.J. Gaunt, Angew. Chem. Int. Ed. 44 (2005) 3125–3129. doi: 10.1002/anie.200500468
Y. Nakao, K.S. Kanyiva, S. Oda, T. Hiyama, J. Am. Chem. Soc. 128 (2006) 8146–8147. doi: 10.1021/ja0623459
S. Xu, X. Huang, X. Hong, B. Xu, Org. Lett. 14 (2012) 4614–4617. doi: 10.1021/ol302070t
G.N. Hermann, M.T. Unruh, S.H. Jung, M. Krings, C. Bolm, Angew. Chem. Int. Ed. 57 (2018) 10723–10727. doi: 10.1002/anie.201805778
Y. Xia, D. Qiu, J. Wang, Chem. Rev. 117 (2017) 13810–13889. doi: 10.1021/acs.chemrev.7b00382
Q. Gu, H.H. Al Mamari, K. Graczyk, L.Ackermann Diers, Angew. Chem. Int. Ed. 53 (2014) 3868–3871 doi: 10.1002/anie.201311024
X. Ye, Z. He, T. Ahmed, et al., Chem. Sci. 4 (2013) 3712–3716. doi: 10.1039/c3sc51211h
Y. Yang, M.B. Zhou, X.H. Ouyang, et al., Angew. Chem. Int. Ed. 54 (2015) 6595–6599. doi: 10.1002/anie.201501260
M. Bauer, W. Wang, M.M. Lorion, C. Dong, L. Ackermann, Angew. Chem. Int. Ed. 57 (2018) 203–207. doi: 10.1002/anie.201710136
H.H. Al Mamari, E. Diers, L. Ackermann, Chem. Eur. J. 20 (2014) 9739–9743. doi: 10.1002/chem.201403019
L. Ackermann, R. Vicente, Org. Lett. 11 (2009) 4922–4925. doi: 10.1021/ol9020354
L. Ackermann, R. Jeyachandran, H.K. Potukuchi, P. Novák, L. Büttner, Org. Lett. 12 (2010) 2056–2059. doi: 10.1021/ol1005517
M. Akter, K. Rupa, P. Anbarasan, Chem. Rev. 122 (2022) 13108–13205. doi: 10.1021/acs.chemrev.1c00991
J.H. Kim, T. Gensch, D. Zhao, et al., Angew. Chem. Int. Ed. 54 (2015) 10975–10979. doi: 10.1002/anie.201504757
W.H. Jeon, J.Y. Son, J.E. Kim, P.H. Lee, Org. Lett. 18 (2016) 3498–4501. doi: 10.1021/acs.orglett.6b01750
H.B. Xu, Y. Zhu, L. Dong, J. Org. Chem. 84 (2019) 16286–16292. doi: 10.1021/acs.joc.9b02468
Y. Dong, J. Chen, Y. Cui, L. Bao, H. Xu, Org. Lett. 22 (2020) 772–775. doi: 10.1021/acs.orglett.9b03904
X. Gao, B. Wu, Z. Yan, Y.G. Zhou, Org. Biomol. Chem. 14 (2016) 8237–8240.
J. Ghorai, M. Chaitanya, P. Anbarasan, Org. Biomol. Chem. 16 (2018) 7346–7350. doi: 10.1039/c8ob02111b
Y. Cai, S.F. Zhu, G.P. Wang, Q.L. Zhou, Adv. Synth. Catal. 353 (2018) 2939–2944.
X. Gao, B. Wu, W.X. Huang, M.W. Chen, Y.G. Zhou, Angew. Chem. Int. Ed. 54 (2015) 11956–11960. doi: 10.1002/anie.201504483
P. Kumar, P.J. Nagtilak, M. Kapur, New J. Chem. 45 (2021) 13692–13746. doi: 10.1039/d1nj01696b
L.H. Wang, D. Xiong, L.H. Jie, C.D. Yu, X.L. Cui, Chin. Chem. Lett. 29 (2018) 907–910.
H.M.L. Davies, S.J. Hedley, Chem. Soc. Rev. 36 (2007) 1109–1119. doi: 10.1039/b607983k
J. Xia, Z. Huang, X. Zhou, et al., Org. Lett. 20 (2018) 740–743. doi: 10.1021/acs.orglett.7b03881
S.S. Zhang, Y.C. Zheng, Z.W. Zhang, et al., Org. Lett. 23 (2021) 5719–5723. doi: 10.1021/acs.orglett.1c01832
H. Hwang, J. Kim, J. Jeong, S. Chang, J. Am. Chem. Soc. 136 (2014) 10770–10776. doi: 10.1021/ja5053768
J. Xia, L. Kong, X. Zhou, G. Zheng, X. Li, Org. Lett. 19 (2017) 5972–5975. doi: 10.1021/acs.orglett.7b02983
H. Xu, Y. Gu, S. Zhang, et al., Angew. Chem. Int. Ed. 59 (2020) 13273–13280. doi: 10.1002/anie.202003595
C. Ma, C. Li, J. Bai, et al., ACS Catal. 12 (2022) 10141–10146. doi: 10.1021/acscatal.2c01433
J. Huang, Y. Yang, Z. Chen, Adv. Synth. Catal. 358 (2016) 201–206. doi: 10.1002/adsc.201500792
A. DeAngelis, V.W. Shurtleff, O. Dmitrenko, J.M. Fox, J. Am. Chem. Soc. 133 (2011) 1650–1653. doi: 10.1021/ja1093309

Scheme 2 Substrate scope. Reaction conditions. Method A: 1 (0.2 mmol), 2 (0.3 mmol), [RhCp*Cl2]2 (5 mol%), AgOAc (20 mol%), MeOH (2 mL), 100 ℃, 18 h. Method B: 4 (0.2 mmol), 2 (0.3 mmol), Rh2(OAc)4 (10 mol%), toluene (2 mL), 110 ℃, 12 h then 130 ℃, 6 h. Isolated yield.

Scheme 5 Proposed catalytic cycles for site-selective cross-couplings of indoles and pyridotriazoles.
Table 1. Optimal conditions for C2-functionalization.a
|
|||
| Entry | Additive | Solvent | Yield (%)b |
| 1 | AgOAc | 1, 4-Dioxane | 35 |
| 2 | AgOAc | Toluene | 66 |
| 3 | AgOAc | DMF | 45 |
| 4 | AgOAc | MeCN | 88 |
| 5 | AgOAc | DCE | 70 |
| 6 | AgOAc | MeOH | 89 |
| 7 | NaOAc | MeOH | 23 |
| 8 | nBuNOAc | MeOH | 47 |
| 9 | – | MeOH | 19 |
| 10c | AgOAc | MeOH | 69 |
| 11d | AgOAc | MeOH | 58 |
| a Reaction conditions: 1a (0.2 mmol, 1.0 equiv.), 2a (0.3 mmol, 1.5 equiv.), [RhCp*Cl2]2 (0.01 mmol, 5 mol%), additive (0.04 mmol, 20 mol%), solvent (2 mL), 100 ℃, 18 h. b Isolated yield. c 80 ℃. d 120 ℃. |
|||
 下载: 导出CSV
下载: 导出CSV
Table 2. Optimal conditions for C3-functionalization.a
|
|||
| Entry | Rh-catalyst | Solvent | Yield (%)b |
| 1 | Rh2(OAc)4 | 1, 4-Dioxane | 30 |
| 2 | Rh2(OAc)4 | DMF | 28 |
| 3 | Rh2(OAc)4 | MeOH | 15 |
| 4 | Rh2(OAc)4 | Toluene | 62 |
| 5 | Rh2(OAc)4 | DCE | 35 |
| 6c | Rh2(OAc)4 | Toluene | 46 |
| 7d | Rh2(OAc)4 | Toluene | 71 |
| 8d | [Rh(COE)Cl]2 | Toluene | 10 |
| 9d | [Rh(COD)Cl]2 | Toluene | 0 |
| 10d | Rh(PPh3)2(CO)Cl | Toluene | 0 |
| 11d | Rh(acac)3 | Toluene | 0 |
| 12d | [RhCp*Cl2]2 | Toluene | 14 |
| 13d, e | Rh2(OAc)4 | Toluene | 58 |
| COD = cyclooctadiene, COE = cyclooctene, acac = acetylacetonate. a Reaction conditions: 4a (0.2 mmol, 1.0 equiv.), 2a (0.3 mmol, 1.5 equiv.), Rh2(OAc)4 (0.02 mmol, 10 mol%), solvent (2 mL), 110 ℃, 18 h. b Isolated yield. c 90 ℃. d 110 ℃, 12 h then 130 ℃, 6 h. e 5 mol% Rh2(OAc)4. |
|||
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们




 下载:
下载: