Coating colloidal particles with a well-defined polymer layer by surface-initiated photoinduced polymerization-induced self-assembly and the subsequent seeded polymerization
Citation:
Bing Niu, Honggao Huang, Liwei Luo, Li Zhang, Jianbo Tan. Coating colloidal particles with a well-defined polymer layer by surface-initiated photoinduced polymerization-induced self-assembly and the subsequent seeded polymerization[J]. Chinese Chemical Letters,
2025, 36(2): 110431.
doi:
10.1016/j.cclet.2024.110431
Coating colloidal particles with a well-defined polymer layer by surface-initiated photoinduced polymerization-induced self-assembly and the subsequent seeded polymerization
English
Coating colloidal particles with a well-defined polymer layer by surface-initiated photoinduced polymerization-induced self-assembly and the subsequent seeded polymerization
Department of Polymeric Materials and Engineering, School of Materials and Energy, Guangdong University of Technology, Guangzhou 510006, China
b.
Guangdong Provincial Key Laboratory of Functional Soft Condensed Matter, Guangzhou 510006, China
* Corresponding author. E-mail address: tanjianbo@gdut.edu.cn (J. Tan). 1 These authors contributed equally to this work.
Received Date:
12 July 2024 Accepted Date:
06 September 2024 Revised Date:
05 September 2024 Available Online:
15 February 2025
Abstract:
Core-shell colloidal particles with a polymer layer have broad applications in different areas. Herein, we developed a two-step method combining aqueous surface-initiated photoinduced polymerization-induced self-assembly and photoinduced seeded reversible addition-fragmentation chain transfer (RAFT) polymerization to prepare a diverse set of core-shell colloidal particles with a well-defined polymer layer. Chemical compositions, structures, and thicknesses of polymer layers could be conveniently regulated by using different types of monomers and feed [monomer]/[chain transfer agent] ratios during seeded RAFT polymerization.
Over the past three decades or so, surface-initiated grafting via reversible deactivation radical polymerization (RDRP) techniques has been utilized for coating well-defined polymer layers to solid substrates [1–3]. Surface modification of colloidal particles via surface-initiated RDRP to prepare surface-functional colloidal particles is an attractive strategy for tuning their properties that find potential applications in different areas such as photonic crystals, biomedical analysis, molecular imprinting [4–8]. During surface-initiated RDRP, polymer chains grow from the attached initiators or chain transfer agents (CTAs) on the colloidal particles, leading to the formation of polymer layers with tunable chemical compositions. However, it is challenging to fabricate polymer layers with well-defined nanostructures via the traditional surface-initiated RDRP. In addition, aggregation of colloidal particles would occur during surface-initiated RDRP under some specific conditions [4]. Moreover, the formed core-shell colloidal particles cannot disperse in water when the formed polymer is hydrophobic, which is not beneficial for biomedical applications.
Self-assembly of block copolymers in solution is an attractive method to prepare well-defined block copolymer assemblies with various morphologies [9,10]. Recently, the solution self-assembly of block copolymers has been employed to modify the surface property of colloidal particles [11–13]. For example, Jia et al. [11]. reported the crystallization-driven self-assembly of block copolymers and silica particles with attached seeds, allowing the fabrication of colloidal mesoscale hybrid assemblies with cylindrical micelles on the surface. However, this approach can only be performed at diluted concentrations (typically < 1% w/w), which is not beneficial for the large-scale production. As an alternative, polymerization-induced self-assembly (PISA) has become a versatile method for the preparation of well-defined block copolymer assemblies at high solids (> 10% w/w) [14–32]. Very recently, the combination of surface-initiated RDRP and PISA has been explored to prepare colloidal mesoscale hybrid assemblies with various surface morphologies at high concentrations [33–35]. Particularly, under some specific conditions, colloidal particles with a polymer layer on the surface can be prepared via the fusion of polymer micelles at the surface [33,34]. However, our preliminary experiments demonstrated that concentric core–shell particles were formed when further increased the polymer layer thickness. Furthermore, the control of surface structures of the polymer layer has not yet been explored.
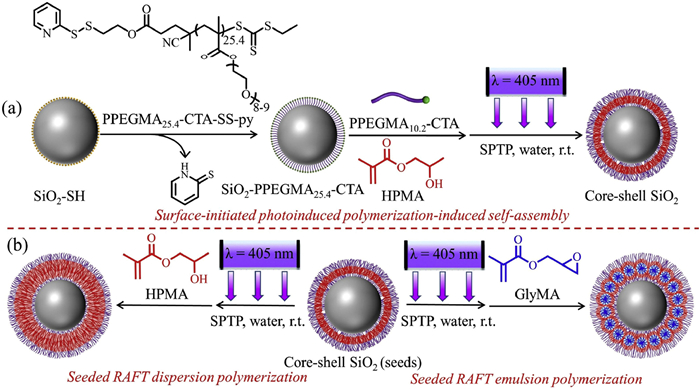
In this study, we reported the rational preparation of core-shell particles with a well-defined polymer layer by surface-initiated photoinduced PISA (photo-PISA) and the subsequent photoinduced seeded reversible addition-fragmentation chain transfer (RAFT) polymerization in water at room temperature (Scheme 1). Thiol-functionalized silica (SiO2-SH) particles with an average diameter of 280 nm were used as the model colloidal particles and modified with a poly(poly(ethylene glycol) methyl ether methacrylate) macromolecular chain transfer agent (PPEGMA25.4—CTA-SS-py) via disulfide exchange (Scheme 1a).
Scheme 1
Scheme 1.
(a) Schematic illustration for the synthesis of core-shell SiO2 particles by surface-initiated photo-PISA of HPMA using SiO2-PPEGMA25.4—CTA and PPEGMA10.2—CTA. (b) Schematic illustration for the synthesis of core-shell SiO2 particles by aqueous seeded RAFT dispersion polymerization of HPMA (left) or aqueous seeded RAFT emulsion polymerization of GlyMA (right).
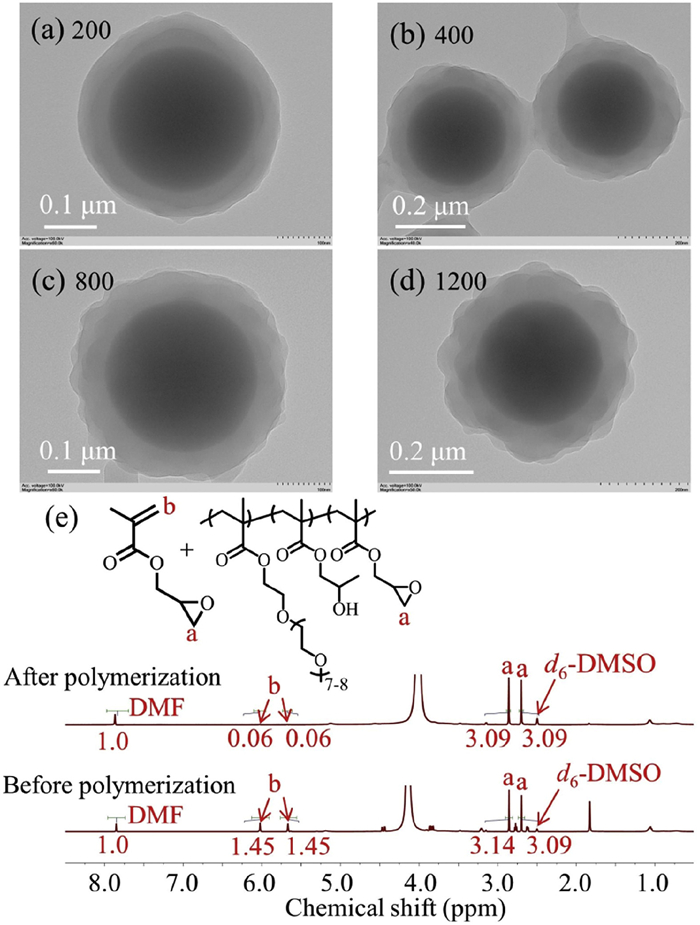
Aqueous surface-initiated photo-PISA of hydroxypropyl methacrylate (HPMA, 10% w/w) was then performed in the presence of SiO2-PPEGMA25.4—CTA and PPEGMA10.2—CTA with the [SiO2-PPEGMA25.4—CTA]/[PPEGMA10.2—CTA] ratio of 1/4. In each case, quantitative monomer conversion was achieved within 30 min of light irradiation as confirmed by 1H NMR spectroscopy. As shown in Figs. 1a-e, core-shell SiO2 particles with a polymer layer were formed at all investigated [HPMA]/[CTA] ratios. When the [HPMA]/[CTA] ratio reached 1000 or higher, concentric core–shell SiO2 particles were formed (Figs. 1c-e). This should be attributed to the high polymerization rate of surface-initiated photo-PISA and the weak dewetting effect of the SiO2-PPEGMA25.4—CTA particles [36]. To address this issue, a two-step approach was then developed. Specifically, core-shell SiO2 particles prepared by aqueous surface-initiated photo-PISA ([HPMA]/[CTA] = 800, Fig. 1b) were used as seeds for the subsequent photoinduced seeded RAFT dispersion polymerization of HPMA with various [HPMA]/[CTA] ratios. In each case, quantitative monomer conversion was achieved within 30 min of light irradiation. As shown in Figs. 1f-i and Fig. S6 (Supporting information), core–shell SiO2 particles with a uniform polymer layer were obtained. The polymer layer thickness was increased linearly with the [HPMA]/[CTA] ratio (Fig. 1j). For example, the polymer layer thickness was 43.6 nm for the core-shell SiO2 particles prepared at a [HPMA]/[CTA] ratio of 200 and this value was increased to 90.0 nm at a [HPMA]/[CTA] ratio of 1200. These results suggest that the combination of surface-initiated photo-PISA and seeded RAFT polymerization is a facile strategy to prepare core-shell particles with a well-defined polymer layer. It should be noted that the polymer layer on the SiO2 particles is a bilayer structure formed from the co-assembly of SiO2-PPEGMA25.4-PHPMAn and PPEGMA10.2-PHPMAn.
Figure 1
Figure 1.
(a-e) TEM images of core-shell SiO2 particles prepared by aqueous surface-initiated photo-PISA of HPMA using SiO2-PPEGMA25.4—CTA and PPEGMA10.2—CTA with different [HPMA]/[CTA] ratios. (f-i) TEM images of core-shell SiO2 particles prepared by aqueous photoinduced seeded RAFT dispersion polymerization of HPMA with various [HPMA]/[CTA] ratios using the core-shell SiO2 particles of Fig. 1b as seeds. (j) Evolution of the average polymer layer thickness of core-shell SiO2 particles prepared by the seeded RAFT dispersion polymerization with the [HPMA]/[CTA] ratio.
This approach was then expanded to photoinduced seeded RAFT emulsion polymerization at room temperature using glycidyl methacrylate (GlyMA) as the hydrophobic monomer and core–shell SiO2 particles of Fig. 1b as seeds. 1H NMR analysis confirmed that high GlyMA conversion (> 95%) was achieved within 30 min of light irradiation. In each case, core-shell SiO2 particles with a uniform polymer layer were formed (Figs. 2a-d). Similar to the case of HPMA, the polymer layer thickness also increased with the [GlyMA]/[CTA] ratio. Remarkably, core-shell SiO2 particles with a rougher surface were obtained as increasing the [GlyMA]/[CTA] ratio. This can be attributed to the incompatibility between the less hydrophobic PHPMA chain and the more hydrophobic PGlyMA chain [37–39], leading to the occurrence of nanoscale phase separation within the polymer layer (Scheme 1b). Due to the mild reaction conditions (e.g., room temperature, visible light), the hydrolysis of epoxy group can be greatly suppressed and > 98% epoxy group was retained as confirmed by 1H NMR spectroscopy (Fig. 2e). The epoxy group is a well-known reactive group that can react with amines, thiols, and proteins [38,40,41]. The obtained epoxy-functionalized core-shell SiO2 particles (prepared at the [GlyMA]/[CTA] ratio of 800) were then reacted with excess ethylene diamine (EDA) in water at 60 ℃ (Fig. 3a) and the morphology was unchanged as confirmed by TEM (Fig. 3b). FT-IR spectroscopy (Fig. 3c) verified the consumption of epoxy group (the decrease of bands at 850 and 909 cm−1) and the presence of primary amine group (the appearance of band at 1595 cm-1). The EDA-treated core-shell SiO2 particles were then dispersed in N,N-dimethylformamide (DMF) and the morphology was maintained (Fig. 3d), suggesting the polymer layer was cross-linked after the EDA treatment. In contrast, the polymer layer of the original epoxy-functionalized core-shell SiO2 particles disappeared after dispersing in DMF (Fig. S7 in Supporting information). The presence of primary amine group within the particles can be used to attach heavy metal nanoparticles (Fig. 3a). As a proof-of-concept experiment, silver (Ag) nanoparticles were generated in situ on the EDA-treated core-shell SiO2 particles via the reduction of AgNO3 with NaBH4. The color of the solution changed from milky white to yellow (inset images in Figs. 3b and e), indicating the formation of silver nanoparticles. Free Ag nanoparticles were separated via three centrifugation-redispersion cycles. TEM (Fig. 3e) and UV–vis spectroscopy (Fig. 3f) further confirmed the successful attachment of Ag nanoparticles on the EDA-treated core-shell SiO2 particles. The catalytic property of the Ag composites was investigated by reducing methylene blue using NaBH4. As shown in Fig. 3g, the characteristic absorption peaks of methylene blue (665 nm and 605 nm) were decreased with time, suggesting the successful decomposition of methylene blue in the presence of the Ag composites.
Figure 2
Figure 2.
(a-d) TEM images of core-shell SiO2 particles prepared by aqueous photoinduced seeded RAFT emulsion polymerization of GlyMA with various [GlyMA]/[CTA] ratios using the core-shell SiO2 particles of Fig. 1b as seeds. (e) 1H NMR spectra of reaction mixtures for aqueous photoinduced seeded RAFT emulsion polymerization of GlyMA ([GlyMA]/[CTA] = 800) before and after polymerization.
Figure 3.
(a) Schematic illustration of the reaction of epoxy-functionalized core-shell SiO2 particles with EDA and the subsequent preparation of Ag composites via the in situ reduction of AgNO3 using NaBH4. (b) TEM image of the EDA-treated core-shell SiO2 particles (inset: digital photograph of EDA-treated core-shell SiO2 particles solution). (c) FT-IR spectra of the epoxy-functionalized core-shell SiO2 particles before and after the treatment with EDA. (d) TEM image of the EDA-treated core-shell SiO2 particles after dispersing in DMF. (e) TEM image of Ag composites prepared by the in situ reduction of AgNO3 using NaBH4 on EDA-treated core-shell SiO2 particles (inset: digital photograph of Ag composites solution). (f) UV–vis spectra of EDA-treated core-shell SiO2 particles and the obtained Ag composites in water. (g) UV–vis spectra of methylene blue reduced by NaBH4 using the Ag composites as a catalysis (inset: digital photograph of methylene blue solution before and after reduction).
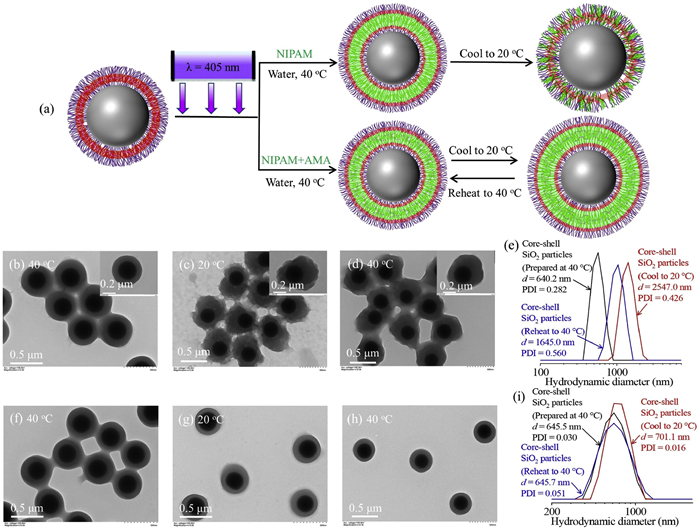
Poly(N-isopropylacrylamide) (PNIPAM) is the most studied thermo-responsive polymer that can be dissolved in water at a temperature below the lower critical solution temperature (LCST, ~32 ℃) and becomes water-insoluble at a temperature above the LCST [42]. The distinct nature of PNIPAM enables one to prepare various environment-responsive colloidal particles that find applications in many fields [7,43,44]. Herein, we also utilized the thermo-responsive nature of PNIPAM to tune the structure of core-shell SiO2 particles (Fig. 4a). Firstly, aqueous surface-initiated photo-PISA of HPMA ([HPMA]/[CTA] = 800) in the presence of 3.5 mol% allyl methacrylate (AMA) was performed to obtain core-shell SiO2 particles with a cross-linked polymer layer (Fig. S8 in Supporting information). Photoinduced seeded RAFT dispersion polymerization of NIPAM ([NIPAM]/[CTA] = 1200) was then performed in water at 40 ℃ using the core-shell SiO2 particles of Fig. S8 as seeds. After the polymerization, core-shell SiO2 particles with a smooth surface were obtained (Fig. 4b). Moreover, the polymer layer thickness was increased from 23 nm to 101 nm, suggesting the successful incorporation of PNIPAM into the polymer layer. After cooling the sample to 20 ℃, the core-shell SiO2 particles became rough (Fig. 4c). This can be attributed to the migration of PNIPAM chains from the PHPMA layer to the surface after decreasing the temperature to below LCST. Dynamic light scattering (DLS) measurement also verified the change of morphology (Fig. 4e). X-ray photoelectron spectroscopy (XPS) also confirmed the migration of PNIPAM chains from the PHPMA layer to the surface based on the difference in the nitrogen content (Fig. S9 in Supporting information). This migration process is not reversible since the morphology was unchanged after heating the sample again to 40 ℃ (Fig. 4d). In contrast, the migration of PNIPAM can be suppressed via the cross-linking of PNIPAM chains by adding AMA during the seeded RAFT polymerization. The morphology of the core-shell SiO2 particles did not change after cooling the sample to 20 ℃ (Figs. 4f and g) and reheating the sample to 40 ℃ (Fig. 4h). It should be noted that the DLS diameter was increased from 645.5 nm at 40 ℃ to 701.1 nm at 20 ℃ and decreased to 645.7 nm after heating back to 40 ℃ (Fig. 4i).
Figure 4
Figure 4.
(a) Schematic illustration for the preparation of core-shell SiO2 particles via photoinduced seeded RAFT dispersion polymerization of NIPAM at 40 ℃ with or without AMA. (b) TEM image of core-shell SiO2 particles prepared by aqueous photoinduced seeded RAFT dispersion polymerization of NIPAM at 40 ℃ with the [NIPAM]/[CTA] ratio of 1200 using the core-shell SiO2 particles of Fig. S8 as seeds. (c) TEM image of the sample of (b) after decreasing the reaction temperature to 20 ℃. (d) TEM image of the sample of (c) after reheating to 40 ℃. (e) DLS distributions of samples of (b–d) in water. (f) TEM image of core-shell SiO2 particles prepared by aqueous photoinduced seeded RAFT dispersion polymerization of NIPAM and AMA at 40 ℃ with the [NIPAM]/[CTA] ratio of 1200 using the core-shell SiO2 particles of Fig. S8 as seeds. (g) TEM image of the sample of (f) after decreasing the reaction temperature to 20 ℃. (h) TEM image of the sample of (g) after reheating to 40 ℃. (i) DLS distributions of samples of (f–h) in water.
In summary, we have successfully prepared a diverse set of core-shell colloidal particles with a well-defined polymer layer by a two-step process combining aqueous surface-initiated photo-PISA of HPMA and photoinduced seeded RAFT polymerization. The polymer layer thickness could be controlled by changing the feed [monomer]/[CTA] ratio. Surface structures of the core-shell particles could be tuned by using different types of monomers during seeded RAFT polymerization. This study provides a facile platform for the rational preparation of various core-shell colloidal particles with a well-defined polymer layer.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
The authors acknowledge support from the Science and Technology Program of Guangzhou (No. 2024A04J2821), the National Natural Science Foundation of China (Nos. 52222301, 22171055), and the Guangdong Natural Science Foundation for Distinguished Young Scholar (No. 2022B1515020078). We would like to thank Wei Song of Analysis and Test Center of Guangdong University of technology for DLS measurement.
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.110431.
[1]
J.O. Zoppe, N.C. Ataman, P. Mocny, et al., Chem. Rev. 117 (2017) 1105–1318. doi: 10.1021/acs.chemrev.6b00314
Scheme 1
(a) Schematic illustration for the synthesis of core-shell SiO2 particles by surface-initiated photo-PISA of HPMA using SiO2-PPEGMA25.4—CTA and PPEGMA10.2—CTA. (b) Schematic illustration for the synthesis of core-shell SiO2 particles by aqueous seeded RAFT dispersion polymerization of HPMA (left) or aqueous seeded RAFT emulsion polymerization of GlyMA (right).
Figure 1
(a-e) TEM images of core-shell SiO2 particles prepared by aqueous surface-initiated photo-PISA of HPMA using SiO2-PPEGMA25.4—CTA and PPEGMA10.2—CTA with different [HPMA]/[CTA] ratios. (f-i) TEM images of core-shell SiO2 particles prepared by aqueous photoinduced seeded RAFT dispersion polymerization of HPMA with various [HPMA]/[CTA] ratios using the core-shell SiO2 particles of Fig. 1b as seeds. (j) Evolution of the average polymer layer thickness of core-shell SiO2 particles prepared by the seeded RAFT dispersion polymerization with the [HPMA]/[CTA] ratio.
Figure 2
(a-d) TEM images of core-shell SiO2 particles prepared by aqueous photoinduced seeded RAFT emulsion polymerization of GlyMA with various [GlyMA]/[CTA] ratios using the core-shell SiO2 particles of Fig. 1b as seeds. (e) 1H NMR spectra of reaction mixtures for aqueous photoinduced seeded RAFT emulsion polymerization of GlyMA ([GlyMA]/[CTA] = 800) before and after polymerization.
Figure 3
(a) Schematic illustration of the reaction of epoxy-functionalized core-shell SiO2 particles with EDA and the subsequent preparation of Ag composites via the in situ reduction of AgNO3 using NaBH4. (b) TEM image of the EDA-treated core-shell SiO2 particles (inset: digital photograph of EDA-treated core-shell SiO2 particles solution). (c) FT-IR spectra of the epoxy-functionalized core-shell SiO2 particles before and after the treatment with EDA. (d) TEM image of the EDA-treated core-shell SiO2 particles after dispersing in DMF. (e) TEM image of Ag composites prepared by the in situ reduction of AgNO3 using NaBH4 on EDA-treated core-shell SiO2 particles (inset: digital photograph of Ag composites solution). (f) UV–vis spectra of EDA-treated core-shell SiO2 particles and the obtained Ag composites in water. (g) UV–vis spectra of methylene blue reduced by NaBH4 using the Ag composites as a catalysis (inset: digital photograph of methylene blue solution before and after reduction).
Figure 4
(a) Schematic illustration for the preparation of core-shell SiO2 particles via photoinduced seeded RAFT dispersion polymerization of NIPAM at 40 ℃ with or without AMA. (b) TEM image of core-shell SiO2 particles prepared by aqueous photoinduced seeded RAFT dispersion polymerization of NIPAM at 40 ℃ with the [NIPAM]/[CTA] ratio of 1200 using the core-shell SiO2 particles of Fig. S8 as seeds. (c) TEM image of the sample of (b) after decreasing the reaction temperature to 20 ℃. (d) TEM image of the sample of (c) after reheating to 40 ℃. (e) DLS distributions of samples of (b–d) in water. (f) TEM image of core-shell SiO2 particles prepared by aqueous photoinduced seeded RAFT dispersion polymerization of NIPAM and AMA at 40 ℃ with the [NIPAM]/[CTA] ratio of 1200 using the core-shell SiO2 particles of Fig. S8 as seeds. (g) TEM image of the sample of (f) after decreasing the reaction temperature to 20 ℃. (h) TEM image of the sample of (g) after reheating to 40 ℃. (i) DLS distributions of samples of (f–h) in water.

 DownLoad:
DownLoad:



 下载:
下载:






 下载:
下载:
