
Figure 1.
(a) Schematic diagram of the fabrication route of the Cs3Bi2Br9/BiOBr heterojunction. (b-d) TEM and HRTEM images of the heterojunction.
Dual atom-bridge effect promoting interfacial charge transfer in 2D/2D Cs3Bi2Br9/BiOBr epitaxial heterojunction for efficient photocatalysis
Yuan Teng , Zichun Zhou , Jinghua Chen , Siying Huang , Hongyan Chen , Daibin Kuang

Activating the inert C(sp3)-H bond among the alkanes or aromatic hydrocarbons to the value-added oxygenates is of great significance for satisfying people’s daily life. Unfortunately, the traditional conversion strategies always suffered from the complex processes, poor product selectivity and toxic waste emission [1–5]. Therefore, it is an imperative requirement to develop effective techniques to convert the hydrocarbons to high-value chemicals. The photocatalytic C(sp3)-H activation using renewable solar energy is recognized as one of the promising strategies for fine chemicals and drug production [6–9]. However, the low solar-to-chemical conversion efficiency largely restricted its practical applications, which has been attracting a focus on exploring advanced photocatalyst materials. Among the reported photocatalysis materials, halide perovskites (HPVK) have rapidly been developed in recent years owing to their large extinction coefficient, excellent photoelectric properties and adjustable band alignment [10–16]. However, single HPVK usually underwent the limited photocatalytic activity and inferior photon conversion efficiency due to the unsatisfactorily spatial separation of photogenerated electron-hole pairs [17,18]. Hence, heterojunction engineering of HPVK-based photocatalysts has stimulated widely scientific interests given that it could either promote the charge carrier separation or manipulate the photocatalytic reaction dynamics [19–28]. Regrettably, most of the reported heterostructures formed by simple physically mixing or random connections, still suffered from their poor interface contact and unsatisfactory charge carriers transfer. Therefore, engineering HPVK-based heterojunctions purposely and fine-tuning its interfacial structure is highly attractive.
To this end, many efforts have been developed to tune the interfacial structures and properties of PVK-based hybrids, such as surface/interface functionalization [12,29–32] and introducing electrostatic or van der Waals interactions [33,34]. Peculiarly, the heterojunctions constructed by the epitaxial growth with matched lattice (a lattice mismatch < 5%) is preferable, which is advantageous in ensuring the superior interfacial contact between different components and providing fast charge transfer pathways [35–38]. However, conventional epitaxial growth usually requires a set of stringent synthetic conditions, which hampered its widespread applications. Very recently, in situ epitaxial growth by cosharing "atoms" without high temperature or vacuum may provide new opportunities for the preparation of high-quality heterojunctions, which can form effective charge channels to facilitate the transfer and separation of photogenerated carriers and improve atomic utilization efficiency [39–45]. A Cs2SnI6/SnS2 heterojunction with cosharing Sn atoms has firstly been synthetized by in situ growth of Cs2SnI6 perovskite quantum dots on the surfaces of SnS2 nanosheets, and obviously boosted interfacial charge transfer and remarkable photocatalytic activity toward CO2 reduction was acquired [41]. Moreover, Manna and co-workers reported that a CsPbX3-Pb4S3Br2 nanocrystal heterostructure with cosharing Pb sublattice could largely promote the spatial separation and transfer of photogenerated carriers [46]. Subsequently, Pradhan and co-workers found that the CsPbBr3-Pb4S3Br2 nanocrystal heterostructures exhibited superior photocatalytic CO2 reduction activity [47]. Recently, our group has in situ constructed CoxBi2-xO2CO3/Cs3Bi2Br9 direct Z-scheme heterojunction using the cosharing Bi atom bridge tactic and achieved a prime interface structure and efficient visible-light photocatalytic toluene oxidation performance [48]. All these results indicated that the in situ epitaxial growth by cosharingatoms should be a promising strategy to develop high-quality heterojunctions.
Motivated by these, here a 2D/2D Cs3Bi2Br9/BiOBr epitaxial heterojunction with cosharing Bi and Br atoms (dual atom-bridge effect) is successfully constructed by in situ growing Cs3Bi2Br9 nanosheets on the surface of BiOBr nanosheets (dozens of nanometers in thick and several micrometers in lateral size). The 2D/2D heterojunction with dual atom-bridge effect enables the formation of very closely contacted interfaces and effective charge transfer channels, which can not only broaden the light absorption range, but also effectively promote the Z-scheme spatial separation of photogenerated charges. Moreover, it can improve the interfacial electron structures to optimize the adsorption and activation of reactant species. As a proof of concept, when employing as photocatalysts for photooxidation of toluene, the 2D/2D Cs3Bi2Br9/BiOBr heterojunction exhibits the superhigh benzaldehyde amounts of 13397.1 µmol/g after continuous illumination for 3 h, significantly superior to those of pristine BiOBr and Cs3Bi2Br9 catalysts.
The preparation of Cs3Bi2Br9/BiOBr epitaxial heterojunction started from the hydrothermal synthesis of BiOBr nanosheets, followed by the acid etching solution process to in situ grow Cs3Bi2Br9 nanosheets on the surface of BiOBr, as illustrated in Fig. 1a. Here the BiOBr nanosheets with smooth surface and dozens of nanometers thick (Fig. S1 in Supporting information) served as the self-template to release Bi3+ during the acid treatment process, and the released Bi3+ would combine with Cs+ and Br− in the reaction solution to grow Cs3Bi2Br9, where the surface Br− may serve as anchor points to induce the subsequent nucleation and growth of Cs3Bi2Br9. As presented in Figs. S1 and S2 (Supporting information), a moderate amount of HBr led to the partial dissolution of BiOBr and the reduction of its crystallinity, causing the shallower edges of BiOBr microsheet. When proper amount of Cs+ was added, fast reaction of the superficial Bi3+ and Br− with Cs+ would happen, resulting epitaxial growth of Cs3Bi2Br9 nanosheets on the surface of parental BiOBr nanosheets. If superfluous HBr was added, a full conversion of BiOBr to Cs3Bi2Br9 can be found. The X-ray diffraction (XRD) patterns of heterojunction samples (Fig. S2) presented both the characteristic peaks of tetragonal BiOBr phase and hexagonal Cs3Bi2Br9 phase, indicating that the Cs3Bi2Br9/BiOBr heterojunction was synthetized successfully. Moreover, compared with the smooth surface of patent BiOBr, curled nanosheets emerged on the surface of BiOBr nanosheets (Fig. 1b). Further high-resolution TEM (HRTEM) images in Figs. 1c and d reveal that the lattice spacing of the curled nanosheets and background sheets is 0.201 and 0.124 nm, respectively, corresponding to the (204) plane of Cs3Bi2Br9 and (310) plane of BiOBr, where the close contact interface structure can be found. To further verify the distributions of Cs3Bi2Br9 on BiOBr surface, the energy dispersive X-ray (EDX) spectrum and the corresponding elemental mapping were conducted. As shown in Figs. S3 and S4 (Supporting information), the distribution of Cs element presented a sparse particle-like distribution on BiOBr, which further confirmed that the Cs3Bi2Br9 nanosheets were grown on BiOBr nanosheets. The Cs3Bi2Br9 obtained by a full conversion of BiOBr was nanosheets (Fig. S5 in Supporting information). The above results indicated that the Cs3Bi2Br9/BiOBr heterojunction has been successfully constructed by growing Cs3Bi2Br9 nanosheets on BiOBr nanosheets. Moreover, the proper amounts of HBr and Cs+ were found to favor the formation of the heterojunction with suitable components and morphology and light absorption properties (Figs. S5-S9 in Supporting information). Within certain range, increasing the amount of HBr and Cs+ can lead to more loading of Cs3Bi2Br9 without obviously sacrificing the nanosheet morphology of BiOBr. While the excessive HBr would break the BiOBr nanosheet and even fully convert BiOBr into Cs3Bi2Br9. Meanwhile, excess Cs+ would suppress the generation of Cs3Bi2Br9, leading to the formation of impurities. Except otherwise noted, the Cs3Bi2Br9/BiOBr sample discussed below was prepared with HBr of 100 µL and Cs+ of 0.0075 mmol.
As white powders, the light absorption region of BiOBr mainly located at the ultraviolet region with the bandgap energy (Eg) value of 2.80 eV (Fig. S10 in Supporting information). After hybridizing with Cs3Bi2Br9, the light absorption range was obviously broadened, due to the smaller Eg (2.56 eV) of Cs3Bi2Br9. The appropriate energy level configuration of different semiconductors is an important factor for achieving highly-effective photocatalytic performance. Therefore, the band spectroscopy valence band (XPS-VB) and Mott-Schottky (M-S) measurements were carried out. Fig. S11 (Supporting information) showed that the work function of Cs3Bi2Br9 was 4.27 eV. Combined with the VB value, it could be concluded that the valence band maximum (VBM) potential (EVB) and the conduction band minimum (CBM) potential (ECB) of Cs3Bi2Br9 was +2.19 and −0.37 V (vs. NHE, pH 0), respectively. On the other hand, the flat band potential (Vfb) of BiOBr was determined to be −0.62 V according to the M-S plots. Therefore, the ECB and EVB positions of BiOBr were calculated to be −0.72 and +2.08 V, respectively. Hence, a staggered band configuration can form when BiOBr got close contact with Cs3Bi2Br9, and an internal electric field (IEF) pointing from BiOBr to Cs3Bi2Br9 could be formed owing to Fermi levels alignment, as shown in Figs. 2a and b. It is generally recognized that a strong IEF is in favor of the formation of Z-scheme charge transfer channel, helping to effectively separate the photogenerated charge carriers spatially and preserving their strong redox abilities (Fig. 2c). Thus, the research task moved to distinguish whether Z-scheme or type-Ⅱ transfer route of photogenerated carriers formed in the hybrid.
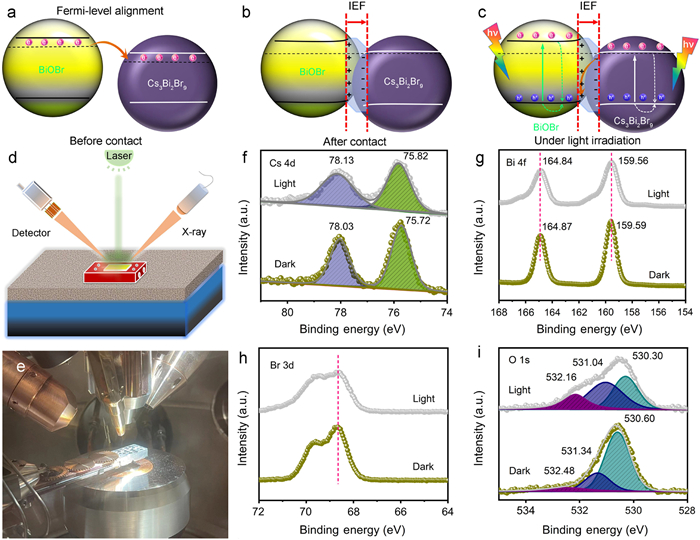
Given the fact that both Cs3Bi2Br9 and BiOBr can drive the formation of •O2− and •OH radicals according to their band positions, the electron paramagnetic resonance (EPR) tests cannot be used to judge the charge transfer direction among the Cs3Bi2Br9/BiOBr hybrid heterojunction. Alternatively, the in situ XPS spectra of the Cs3Bi2Br9/BiOBr heterojunction upon illumination and under dark were studied. As shown in Figs. 2d-i and Table S1 (Supporting information), the Cs 4d peaks locate at the binding energies (BEs) of 78.03 and 75.72 eV, corresponding to Cs 4d5/2 and Cs 4d3/2, respectively. The O 1s spectra exhibited three peaks at 530.60, 531.34, and 532.48 eV, assigned to the lattice oxygen, surface -OH species and adsorbed oxygen species, respectively [49–52]. When illuminated under 405 nm laser, the BEs of Cs 4d shifted toward higher values by ~0.10 eV relative to that of in dark. By contrast, the BEs of O 1s shifted toward lower values by ~0.30 eV. This opposite BEs shift direction indicated that the photogenerated electrons of Cs3Bi2Br9 transferred to BiOBr, following a unique Z-scheme route. Interestingly, the Bi 4f and Br 3d signal peaks exhibited no obvious BEs shifts before and after light irradiation, which might be due to the "bridging effect" of the cosharing Bi and Br elements. This dual atom-bridge concept is similar to the previous concept of cosharing atoms [41,48], where different components contain two cosharing atoms used as the bridge-like charge transfer channels to promote effective transfer and separation of photogenerated carriers and further enhance the atomic utilization efficiency.
What is more, Kelvin probe force microscopy (KPFM) was also utilized to study the spatial charge separation of the as-synthetized materials under in situ illumination, as displayed in Fig. S12 (Supporting information). It could be observed that both of the pristine BiOBr and Cs3Bi2Br9 samples exhibited no obvious surface photovoltage (SPV) changes before and after illumination, suggesting their relatively poor charge separation properties. By contrast, the as-constructed Cs3Bi2Br9/BiOBr heterojunction behaved a distinctive SPV change, and the largest SPV value could reach ~+60.0 mV, suggesting a more effective charge separation in the hybrid. These findings fully revealed that the elaborated heterojunction could accelerate the transfer and spacial separation of photoexcited charge carriers effectively, which was expected to drive the effective photocatalytic reaction. Notably, the positive SPV signals under light irradiation suggested that the photoinduced holes accumulated on the surface of the heterojunction. Since Cs3Bi2Br9 nanosheets epitaxially grew on the surface of BiOBr, this phenomenon also supported the Z-scheme charge transfer mechanism (photoelectron transfer from Cs3Bi2Br9 to BiOBr) rather than the type-Ⅱ pathway under the simulated sunlight illumination (Fig. 2c). In addition, transient photocurrent measurements (Fig. S13 in Supporting information) also found that the Cs3Bi2Br9/BiOBr heterojunction produced the highest photocurrent response (~−1.11 µA/cm2), which was about 3.83 and 6.53 times larger than those of Cs3Bi2Br9 (~−0.29 µA/cm2) and BiOBr (~−0.17 µA/cm2), respectively. Further EIS plots measured at 0.1 V vs. Ag/AgCl showed that the Cs3Bi2Br9/BiOBr behaved a drastically decreased semicircle compared to those of the individuals, implying a more facilitated charge transfer and transportation process. The obviously enlarged transient photocurrent and superior charge transfer property confirmed the more favorable photoexcited charge separation behaviors in Cs3Bi2Br9/BiOBr hybrid.
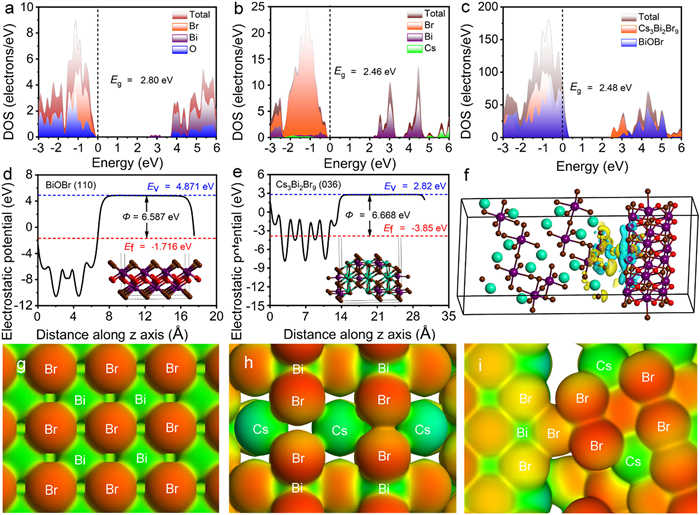
Furthermore, density functional theory (DFT) calculations were applied to study the energy band structure, theoretical work function (Φ), the interfacial structures and electronic properties of materials, as presented in Fig. 3 and Fig. S14 (Supporting information). The calculated projected density of states (PDOS) results (Figs. 3a-c) revealed that the VBM of BiOBr was mainly dictated by the Br 4p and O 2p orbitals, and the CBM consisted of the Bi 6p and O 2p states. As for the Cs3Bi2Br9, the VBM consisted of Br 4p and slight Bi 6p orbitals, and the CBM was mainly occupied by Bi 6p orbital. The calculated bandgap values of BiOBr and Cs3Bi2Br9 were 2.80 and 2.46 eV, respectively, which was in good agreement with the experimental values. The band calculations showed that both the CBM and VBM of BiOBr were higher than those of Cs3Bi2Br9, indicating that a stepped band alignment configuration was generated in the heterojunction (Fig. 3c). Further work function results (Figs. 3d and e) showed that the Cs3Bi2Br9 had lower Fermi levels than that of BiOBr, which were conducive to the flow of electrons from BiOBr to Cs3Bi2Br9 when coming to close contact with each other. The differential charge density results (Fig. 3f) validated the obvious charge redistribution at the interface with electron cloud density shifted from BiOBr to Cs3Bi2Br9, and the Bader charge was as high as 0.751 e (Table S2 in Supporting information). Moreover, the optimized electron density isosurface results (Figs. 3g-i) showed that the hybrid behaved a more prominent color difference relative to those of pristine BiOBr and Cs3Bi2Br9, which was an indicative of larger potential difference. These results suggested that strong interfacial electronic coupling interaction generated at the heterogeneous interfaces, which was expected to not only generate large IEF to drive the Z-scheme charge transfer, and also manipulate the surface electron structure for optimizing the adsorption and activation of reactants.
In view of the aforementioned results, a high-quality Cs3Bi2Br9/BiOBr epitaxial heterojunction with the strong interfacial interaction and favorable charge transfer and separation abilities has been obtained successfully. To confirm the superiority of this heterojunction, the photocatalytic toluene oxidation reaction under visible light illumination (AM1.5 G, λ ≥ 420 nm, 100 mW/cm2) was systematically studied, as displayed in Figs. S15-S17 (Supporting information). The single BiOBr catalyst had a low photocatalytic activity with the benzaldehyde yield rate of 71.7 µmol g−1 h−1, possibly because of its poor light absorption and charge separation ability. In contrast, the Cs3Bi2Br9 nanosheets showed a relatively high photocatalytic activity with the benzaldehyde yield rate of 1867.9 µmol g−1 h−1, owing to the suitable band alignment and better light absorption ability. After the in situ epitaxial growth of Cs3Bi2Br9 nanosheets on BiOBr nanosheets, obviously improved photocatalytic performances were achieved for all the hybrids prepared with different usages of HBr and Cs+. The best performances were achieved for the sample prepared with HBr of 100 µL and Cs+ of 0.0075 mmol, which exhibited the highest benzaldehyde yield rate of as high as 4465.7 µmol g−1 h−1 within 3 h, far exceeding those of the Cs3Bi2Br9 and BiOBr catalysts. For references, the photocatalytic performance of the acid-treated BiOBr was determined. Compared with the pristine BiOBr, slightly improved benzaldehyde yield rate was obtained, which might be resulted from the surface defects formed during the acid treatment which can expose a certain quantity of active sites and optimize the adsorption of reaction substrates. This result supported the conclusion that the formation of high-quality heterojunction contributed mainly to the superior photocatalytic performance of hybrid catalysts, including the rich active sites of the unique 2D/2D structure (Fig. 1b and Fig. S18 in Supporting information) and the formation of Z-scheme charge transfer mechanism which could promote the spacial separation of photoexcited carriers effectively (Fig. 2c).
To further figure out the underlying photocatalytic mechanism of Cs3Bi2Br9/BiOBr heterojunction, a series of control experiments have also been performed without light, O2, catalysts, or with various radical scavengers. As illustrated in Fig. 4a, no products were checked under dark or without catalyst, and the extremely poor photocatalytic performance was achieved without O2, suggesting that the light, photocatalyst and O2 were the important prerequisites for driving effective photocatalytic toluene oxidation. Further radical capture experiment results showed that no obvious performance decay was observed when t-butanol was introduced as the •OH captures. Similarly, the benzaldehyde yield rate just exhibited slight decay when the K2S2O8 was added as electrons (e−) scavenger. In stark contrast, significantly attenuated photocatalytic activity could be found when the benzoquinone (BQ) as •O2− scavenger and ammonium oxalate (AO) as the holes (h+) scavenger was introduced, respectively. The above results demonstrated that the oxidation of toluene on the surface of Cs3Bi2Br9/BiOBr heterojunction followed a radical process, and the •O2− and h+ species should be the main active species for achieving effective photocatalytic toluene oxidation while •OH radicals are unaffected [10,53,54]. In addition, in situ diffuse reflectance infrared Fourier transform spectroscopy (in situ DRIFTS) was utilized to monitor the species changes during the photocatalytic toluene oxidation reaction process. As presented in Fig. 4b, the main signal regions of toluene were located at between 3100 cm−1 and 2800 cm−1, which corresponded to the vibration of the aromatic ring and asymmetric vibration of C—H bond among the methyl, and the peak intensities gradually decreased with the extension of illumination time from 0 to 90 min. This should be resulted from the oxidation reaction of toluene molecules adsorbed on the surface of BiOBr/Cs3Bi2Br9. It was noted that the characteristic peaks of the carbonyl group of benzaldehyde occurred at ~1694 cm−1, and the peak intensity gradually increased with the irradiation time [55,56]. It indicated that toluene molecules adsorbed on the catalyst surface would react with h+ to produce benzyl radicals under illumination which would rapidly react with •O2 to form the benzaldehyde [57]. By contrast, no carbonyl group was observed under dark conditions, suggesting that the activation of C(sp3)-H bond was difficult to occur without light. These results illustrated that the objective products were originated from the introduced toluene molecules, and the toluene molecules absorbed on the surface of hybrid catalyst reacted rapidly with h+ and •O2− under visible-light irradiation.
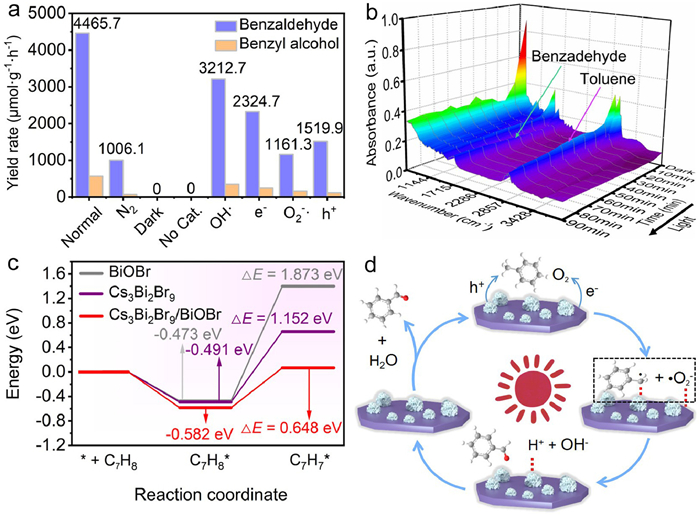
Furthermore, the energy changes (ΔE) of reaction molecule or the intermediate species produced during photocatalytic reaction were also studied by DFT calculation. As displayed in Fig. 4c, it could be clearly found that the toluene adsorption energy (Eads) values on the surface of BiOBr and Cs3Bi2Br9 was −0.473 and −0.491 eV, respectively, where more negative Eads values suggested that more favorable adsorption of toluene molecules. As expected, the Cs3Bi2Br9/BiOBr heterojunction exhibited the smallest Eads value (−0.582 eV), indicating an optimized toluene adsorption ability after heterojunction formation. Further ΔE (*C7H8 + h+ →*C7H7• + H+) tested results showed that the energy barrier of BiOBr and Cs3Bi2Br9 catalyst was 1.873 and 1.152 eV, respectively, supporting the relatively easier activation of C(sp3)-H bond and better photocatalytic performances of Cs3Bi2Br9. Interestingly, this value was lowered to 0.684 eV for Cs3Bi2Br9/BiOBr hybrid, indicating that the synergistic effect of Cs3Bi2Br9 and BiOBr facilitated the dissociation of C(sp3)-H bond. This might be attributed to the strong interfacial electronic coupling of such epitaxial heterojunction which optimized the surface electron structure of heterojunction catalyst, as revealed by the DFT analysis in Figs. 3f-i. Therefore, superior toluene photooxidation performances can be expected.
In view of the above results, we put forward a plausible reaction mechanism of photocatalytic toluene oxidation on the surface of 2D/2D Cs3Bi2Br9/BiOBr heterojunction (Fig. 4d). Upon photoexcitation, the VB electrons of Cs3Bi2Br9 and BiOBr jumped to their CBs. Then, the photoelectrons in the Cs3Bi2Br9 CB would recombine with the holes in the VB of BiOBr quickly with the help of strong IEF and bent band structure. In this case, the photogenerated electrons with strong reduction ability and holes with strong oxidation ability accumulated in the CB of BiOBr and the VB of Cs3Bi2Br9, respectively. These photoelectrons would reduce O2 molecules to •O2−, while the photoinduced holes on Cs3Bi2Br9 would oxidize toluene molecules to benzyl radicals (*C7H7•). Thereafter, the C7H7• and •O2− radicals reacted with each other to generate C7H7OO− species, which can transform into the benzaldehyde by dehydration, while a few C7H7• also reacted with O2 to form C7H7OO•, which would decompose into benzyl alcohol or benzaldehyde, etc. [58]. This unique Z-scheme heterostructure could promote the spacial separation of photoexcited electron-hole pairs and retain the redox ability of electrons in BiOBr CB and holes in Cs3Bi2Br9 VB, respectively, and thus contributing to the outstanding toluene photooxidation activity.
To sum up, a high-quality 2D/2D Cs3Bi2Br9/BiOBr heterojunction with the compact-contact interfaces and matched band structures has been successfully constructed by an in situ epitaxial growth strategy. Systematic researches including HRTEM, in situ XPS, KPFM, and DFT calculations reveal that epitaxial growth of heterojunction with cosharing dual atoms can ensure high-quality interfaces with strong interfacial electronic coupling and electron redistribution, which can actuate the Z-scheme transfer pathway to promote the spatial transfer and separation of photogenerated charges effectively without sacrificing their redox abilities; meanwhile, modulate their interfacial electronic structure to facilitate the adsorption of toluene molecules and lower the energy barrier of C(sp3)-H dissociation. As an encouraging result, the synergistic effect of BiOBr and Cs3Bi2Br9 contributes to a superhigh toluene oxidation activity under visible-light irradiation, and the benzaldehyde yield reaches to 13397.1 µmol/g within just 3 h, far surpass than those of the parental BiOBr and Cs3Bi2Br9 catalysts. The study not only highlights an interesting dual atom-bridge strategy to design high-quality Bi-based perovskite heterojunction photocatalysts, but also offers a new insight toward the design of advanced photocatalyst materials for organic synthesis.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yuan Teng: Writing – original draft, Investigation, Funding acquisition, Formal analysis, Data curation, Conceptualization. Zichun Zhou: Writing – review & editing, Methodology, Formal analysis, Data curation. Jinghua Chen: Writing – review & editing, Investigation, Formal analysis. Siying Huang: Investigation, Formal analysis. Hongyan Chen: Writing – review & editing, Writing – original draft, Supervision, Project administration, Conceptualization. Daibin Kuang: Writing – review & editing, Validation, Supervision, Project administration, Formal analysis, Conceptualization.
This study was financially supported by the National Natural Science Foundation of China (Nos. 22175202, 22005351, 22365016), the Guangdong Basic and Applied Basic Research Foundation (No. 2023A1515010180), Program of Guangzhou Science and Technology (No. 202201011591), the Science and Technology Innovation Program of Hunan Province (No. 2023RC3179), Scientific Research Start-up Fund of Jishou University (No. 1122003). The authors would like to thank the support of National Supercomputer Center in Sun Yat-sen University, Guangzhou.
Supplementary material associated with this article can be found, in the online version, at doi:
Y. Li, M. Lei, L. Gong, Nat. Catal. 2 (2019) 1016–1026. doi: 10.1038/s41929-019-0357-9
X. Li, C. Li, Y. Xu, et al., Nat. Energy 8 (2023) 1013–1022. doi: 10.1038/s41560-023-01317-5
L. Zhang, R.H. Li, X.X. Li, et al., Nat. Commun. 15 (2024) 537. doi: 10.1038/s41467-024-44833-y
J.H. Shen, R. Tang, Z.Y. Wu, et al., Trans. Tianjin Univ. 28 (2022) 236–244. doi: 10.1007/s12209-022-00333-y
P. Zhang, T. Wang, J.L. Gong, CCS Chem. 5 (2023) 1028–1042. doi: 10.31635/ccschem.023.202202643
G. Laudadio, Y. Deng, K. van der Wal, et al., Science 369 (2020) 92–96. doi: 10.1126/science.abb4688
L. Tan, X. Kong, M. Liu, et al., Chem. Sci. 14 (2023) 11761–11767. doi: 10.1039/D3SC00688C
M. Wang, Y. Huang, P. Hu, Science 383 (2024) 537–544. doi: 10.1126/science.adj9258
L.Q. Xiong, J.W. Tang, Adv. Energy Mater. 11 (2021) 2003216. doi: 10.1002/aenm.202003216
Y. Dai, C. Poidevin, C. Ochoa-Hernández, et al., Angew. Chem. Int. Ed. 59 (2020) 5788–5796. doi: 10.1002/anie.201915034
S. Chen, H.J. Yin, P. Liu, et al., Adv. Mater. 35 (2023) 2203836. doi: 10.1002/adma.202203836
S. Peng, Z. Yang, M. Sun, et al., Adv. Mater. 35 (2023) 2304711. doi: 10.1002/adma.202304711
Q. Jia, T. Shao, L. Tong, et al., Chin. Chem. Lett. 34 (2023) 107539. doi: 10.1016/j.cclet.2022.05.053
S. Park, W.J. Chang, C.W. Lee, et al., Nat. Energy 2 (2016) 16185. doi: 10.1038/nenergy.2016.185
C. Cai, Y. Teng, J.H. Wu, et al., Adv. Funct. Mater. 30 (2020) 2001478. doi: 10.1002/adfm.202001478
J. Wang, Y. Shi, Y. Wang, et al., ACS Energy Lett. 7 (2022) 2043–2059. doi: 10.1021/acsenergylett.2c00752
Y. Wu, P. Wang, X. Zhu, et al., Adv. Mater. 30 (2018) 1704342. doi: 10.1002/adma.201704342
N.N. Guo, Z.L. Liu, Y.F. Mu, et al., Chin. Chem. Lett. 33 (2022) 3039–3042. doi: 10.1016/j.cclet.2021.09.033
Y.F. Xu, M.Z. Yang, B.X. Chen, et al., J. Am. Chem. Soc. 139 (2017) 5660. doi: 10.1021/jacs.7b00489
H.L. Tan, F.F. Abdi, Y.H. Ng, et al., Chem. Soc. Rev. 48 (2019) 1255–1271. doi: 10.1039/C8CS00882E
A. Singh, B. Yuan, M.H. Rahman, et al., J. Am. Chem. Soc. 145 (2023) 19885–19893. doi: 10.1021/jacs.3c06127
P. Hu, G. Liang, B. Zhu, et al., ACS Catal. 13 (2023) 12623–12633. doi: 10.1021/acscatal.3c03095
X.B. Li, T. Han, Y.T. Zhou, et al., Sci. China Tech. Sci. 67 (2024) 1238–1252. doi: 10.1007/s11431-023-2604-x
L. Jiang, H.R. Du, L. Li, et al., Trans. Tianjin Univ. 29 (2023) 462–472. doi: 10.1007/s12209-023-00376-9
X.B. Li, J.Y. Liu, J.T. Huang, et al., Acta Phys. Chim. Sin. 37 (2021) 2010030.
X.B. Li, B.B. Kang, F. Dong, et al., Nano Energy 81 (2021) 105671. doi: 10.1016/j.nanoen.2020.105671
X.B. Li, H.S. Zhang, J.M. Luo, et al., Electrochim. Acta 258 (2017) 998–1007. doi: 10.1016/j.electacta.2017.11.151
W.W. Wang, X.B. Li, F. Deng, et al., Chin. Chem. Lett. 33 (2022) 5200–5207. doi: 10.1016/j.cclet.2022.01.058
M. Ou, W. Tu, S. Yin, et al., Angew. Chem. Int. Ed. 130 (2018) 13758–13762. doi: 10.1002/ange.201808930
W.Q. Wu, J.X. Zhong, J.F. Liao, et al., Nano Energy 75 (2020) 104929. doi: 10.1016/j.nanoen.2020.104929
J. Xi, J. Jiang, H. Duim, et al., Adv. Mater. 35 (2023) 2302896. doi: 10.1002/adma.202302896
S. Ma, X. Xue, K. Wang, et al., Adv. Energy Mater. 14 (2024) 2303193. doi: 10.1002/aenm.202303193
Y. Jiang, H.Y. Chen, J.Y. Li, et al., Adv. Funct. Mater. 30 (2020) 2004293. doi: 10.1002/adfm.202004293
H. Lai, Z. Lu, Y. Lu, et al., Adv. Mater. 35 (2023) 2208664. doi: 10.1002/adma.202208664
J. Chen, D.J. Morrow, Y. Fu, et al., J. Am. Chem. Soc. 139 (2017) 13525–13532. doi: 10.1021/jacs.7b07506
C. Zhu, A.L. Wang, W. Xiao, et al., Adv. Mater. 30 (2018) 1705516. doi: 10.1002/adma.201705516
P. Liu, Y. Xu, Y. Zhang, et al., Adv. Opt. Mater. 11 (2023) 2300440. doi: 10.1002/adom.202300440
Y. Cui, J. Wang, Y. Li, et al., Science 383 (2024) 212–219. doi: 10.1126/science.adk5947
Z. Ning, X. Gong, R. Comin, et al., Nature 523 (2015) 324–328. doi: 10.1038/nature14563
M. Liu, Y. Chen, C.S. Tan, et al., Nature 570 (2019) 96–101. doi: 10.1038/s41586-019-1239-7
X.D. Wang, Y.H. Huang, J.F. Liao, et al., J. Am. Chem. Soc. 141 (2019) 13434–13441. doi: 10.1021/jacs.9b04482
J. Zhang, X. Liu, P. Jiang, et al., Nano Energy 66 (2019) 104142. doi: 10.1016/j.nanoen.2019.104142
X. Zhang, X. Wu, X. Liu, et al., J. Am. Chem. Soc. 142 (2020) 4464–4471. doi: 10.1021/jacs.9b13681
Y. Tang, C.H. Mak, R. Liu, et al., Adv. Funct. Mater. 30 (2020) 2006919. doi: 10.1002/adfm.202006919
M.Y. Kuo, N. Spitha, M.P. Hautzinger, et al., J. Am. Chem. Soc. 143 (2021) 4969–4978. doi: 10.1021/jacs.0c10000
M. Imran, L. Peng, A. Pianetti, et al., J. Am. Chem. Soc. 143 (2021) 1435–1446. doi: 10.1021/jacs.0c10916
R. Das, A. Patra, S.K. Dutta, et al., J. Am. Chem. Soc. 144 (2022) 18629–18641. doi: 10.1021/jacs.2c08639
Y. Teng, J.H. Chen, Y.H. Huang, et al., Appl. Catal. B: Environ. 335 (2023) 122889. doi: 10.1016/j.apcatb.2023.122889
J. Wang, Y. Yu, L. Zhang, et al., Appl. Catal. B: Environ. 136 (2013) 112–121.
H. Wang, C. Cao, D. Li, et al., J. Am. Chem. Soc. 145 (2023) 16852–16861. doi: 10.1021/jacs.3c05237
X. Cao, A.J. Huang, C. Liang, et al., J. Am. Chem. Soc. 144 (2022) 3386–3397. doi: 10.1021/jacs.1c10112
J. Di, Y. Li, Y. Zhang, et al., Trans. Tianjin Univ. (29) (2023) 235–245. doi: 10.1007/s12209-022-00344-9
Z. Zhang, Y. Yang, Y. Wang, et al., Angew. Chem. Int. Ed. 59 (2020) 18136. doi: 10.1002/anie.202005495
D. Song, M. Li, F. Yang, et al., Chin. Chem. Lett. 35 (2024) 108591. doi: 10.1016/j.cclet.2023.108591
J. Wang, J. Li, W. Yang, et al., Appl. Catal. B: Environ. 297 (2021) 120489. doi: 10.1016/j.apcatb.2021.120489
H.H. Zhang, Z.C. Zhou, Y.J. Dong, et al., Sol. RRL 5 (2021) 2100559. doi: 10.1002/solr.202100559
Z.J. Bai, S. Tian, T.Q. Zeng, et al., ACS Catal. 12 (2022) 15157–15167. doi: 10.1021/acscatal.2c04652
Z. Xue, J. Yang, L. Ma, et al., ACS Catal. 14 (2024) 249–261. doi: 10.1021/acscatal.3c04484

Figure 1 (a) Schematic diagram of the fabrication route of the Cs3Bi2Br9/BiOBr heterojunction. (b-d) TEM and HRTEM images of the heterojunction.

Figure 2 (a-c) Schematic illustration of energy band arrangement of BiOBr and Cs3Bi2Br9 before and after contact, IEF-induced Z-scheme charge transfer and separation under visible-light irradiation. (d) Schematic illustration and (e) actual photo of the in situ XPS test for the Cs3Bi2Br9/BiOBr heterojunction: (f) Cs 4d, (g) Bi 4f, (h) Br 3d, and (i) O 1s, respectively.

Figure 3 (a-c) The calculated PDOS of BiOBr, Cs3Bi2Br9, and Cs3Bi2Br9/BiOBr heterojunction. (d, e) The calculated work function of BiOBr and Cs3Bi2Br9, respectively. (f) The charge density difference of Cs3Bi2Br9/BiOBr heterojunction, the yellow and blue area represent the electron accumulation and depletion, respectively. (g-i) The electron density isosurface of BiOBr, Cs3Bi2Br9, and Cs3Bi2Br9 (036)/BiOBr (110) surfaces.

Figure 4 (a) Control experiments of the heterojunction with different reaction conditions. (b) In situ DRIFTS spectra of Cs3Bi2Br9/BiOBr heterojunction under the dark and during toluene photooxidation. (c) The calculated ΔE of C7H8 → C7H7 + H+ reaction on BiOBr, Cs3Bi2Br9, and Cs3Bi2Br9(036)/BiOBr(110) surfaces. (d) Schematic diagram of charge transfer and photocatalytic toluene oxidation mechanism over the hybrid.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: