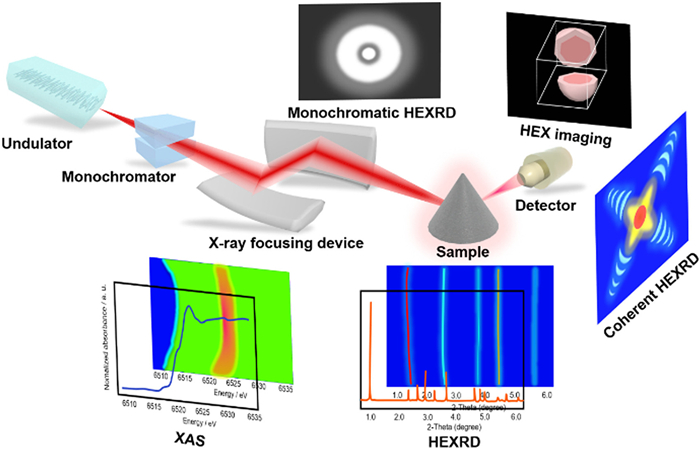
Figure 1.
Schematic model of the multiple technique space-resolved characterizations based on synchrotron X-ray.

With the large-scale applications of sustainable energies such as solar, wind, and water, the demands for high-efficiency energy storage devices are surging [1-3]. Lithium ion batteries (LIBs), with their high energy density and long lifespan, have captured the major energy storage market in the past decade [4-7]. Nevertheless, the scarce reserve and uneven distribution of Li resources cause inevitable issues concerning to price and sustainability, which cannot support the long-term development of energy storage market in a smooth manner [8-10]. Hence, researching alternative high-electrochemical-performance energy storage devices for LIBs is becoming more and more urgent. Sodium ion batteries (SIBs), working in a similar “rocking-chair” electrochemical reaction mechanism to that of LIBs, have attracted widespread attentions because of the natural abundance, facile extraction, and the worldwide distribution of Na resources, receiving large amount of investigations in the past few years [11-14]. Generally, the energy densities of SIBs are lower than those of LIBs owing to the lower standard electrochemical potential and higher mass of Na ion than that of Li ion, however, the low cost of electrode materials and the using of Al current collector enable SIBs to present evident cost advantage, which is much more suitable for grid-scale energy storge systems [15-17]. Actually, relative efforts on the commercial applications of SIBs have been thoroughly performed in the past decades [18-20].
As a crucial segment of SIBs, the cathode materials paly a determined role in the electrochemical properties of SIBs, e.g., cycling life, rate capability, and energy/power densities [21,22]. Up to now, several excellent electrode materials have been studied to serve as the cathodes of SIBs, such as sodium layered transition metal oxides (NaxTMO2), prussian blue analogues, polyanionic compounds, and organics [23-27]. Among them, the NaxTMO2 (x is the stoichiometric content of Na, TM is transition metal element, such as Fe, Mn, Ni, Mg, Zn, and Co) layered oxides present extensive application prospects for commercial SIBs owing to their facile synthesis methods, superior ionic/electronic conductivity, low cost, and high theoretical capacity [28,29]. Furthermore, the large-scale applications of their Li analogues in commercial LIBs enable NaxTMO2 much easier to industrialization for SIBs [30-32]. Among the NaxTMO2 polytypes, the two representative phase structures, P2 and O3 types, are considered to be the most promising candidates for practical applications because of their generous redox couples and high theoretical capacity [33-35]. However, the phase transformations of P2-, O3-, and P2/O3-type crystal configurations during desodiation/sodiation usually accompany with structural degradations due to TM dissolution, Jahn-Teller distortion, and oxygen loss, etc. Additionally, the NaxTMO2 layered oxides commonly suffer from sluggish desodiation/sodiation kinetics and air instability, leading to discontented electrochemical properties [36-38]. Therefore, in the past serval decades, a variety of effective strategies had been carried out to optimize the phase structures and electrochemical performances of NaxTMO2, such as element doping/substitutions, high-entropy designs, and surface modifications [39-41]. Generally, the element doping/substitutions and high-entropy designs could significantly suppress irreversible phase transformations through enhancing the chemical bond strength, optimizing Na+/vacancy distribution, stabilizing surface/bulk structure, etc. [42-44]. While the surface reconstruction strategies utilizing high-stable oxides (spinel/post-spinel oxides, tunnel oxides, etc.) could efficiently buffer against the stress and strain in crystal lattice during Na ion extraction/insertion, and then improve the lifespan of NaxTMO2 polytypes. To date, although prominent progresses associating with NaxTMO2 polytypes have been attained, it is still unable to fulfill the demands of practical applications owing to the unsatisfactory cycle life and specific capacity [45-47]. In order to build high-performance NaxTMO2 for SIBs, it is essential to comprehensively analyze and deeply understand the thermodynamical and kinetical processes about phase transitions and lattice parameter changes upon desodiation/sodiation [48-50].
The synchrotron photons emitting from the charged particles with relativistic speed and changing direction show continuously tunable photon energy in a wide frequency range and high flux [51,52]. Actually, the synchrotron photon energy locates within the energy range from around 1 eV to 10 keV, which crosses the infrared to hard X-ray region. It is worth mentioning that the synchrotron photon energy originating from modern third-generation synchrotron could reach up to the order of a few GeV, exhibiting significant advantages for high-sensitivity material characterizations [53]. Specially, the synchrotron-based X-ray with sensitive responses to the materials present high inspection efficiency for phase transitions and the valence evolutions of NaxTMO2 layered oxides during charge/discharge processes. To date, synchrotron radiation techniques with state-of-the-art synchrotron X-ray source, detecting based on synchrotron radiation photon, had been widely applied to reveal the electrochemical reaction mechanisms of NaxTMO2 polytypes by identifying the lattice configurations, electronic structures and geometric information, chemical environments and phase transformations during electrochemical reactions relying on its high temporal and spatial resolution, such as high energy X-ray diffraction (HEXRD), X-ray adsorption (fine/near edge) spectroscopy (XAS), high energy X-ray (HEX) imaging, coherent HEXRD, X-ray fluorescence microscopy, and monochromatic HEXRD (Fig. 1) [51,54-57].
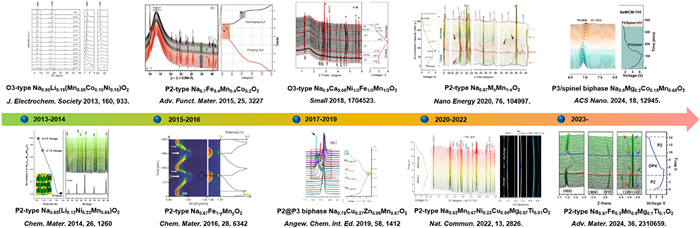
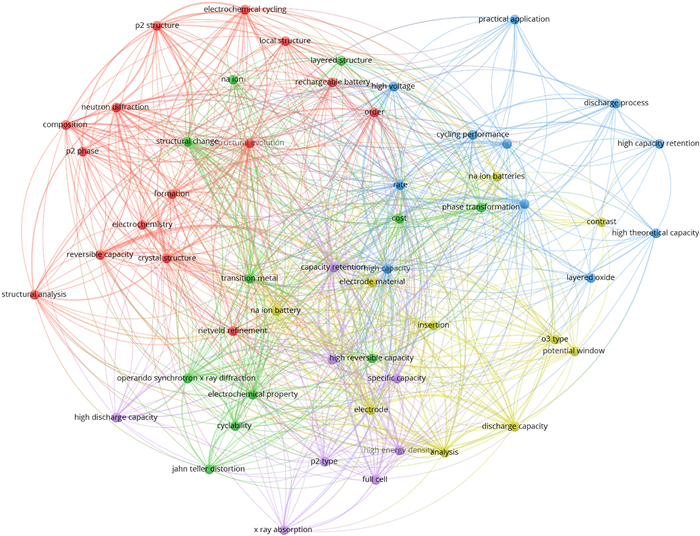
X-ray diffraction (XRD) is an indispensable technology to discern the atomic-scale structure of materials for predicting and comprehending the relevances between components, structures, and electrochemical performances of the electrode materials [55,58-60]. In addition, it can also be used to analyze the microstructure information (e.g., strain, grain size, texture) of powders and particles [61]. In order to clarify the electrochemical reaction mechanisms of electrode materials, detailed phase transitions during charge/discharge processes are needed, however, the laboratory XRD, generally originating from Cu K radiation source (λ = 0.154 nm), cannot give integral diffraction patterns and even presents diffuse components owing to its weak diffraction intensity and untunable photon energy [62]. While for the HEXRD, which based on synchrotron radiation facilities, with its high intensity, high resolution, high penetration depth, and low beam divergence, has become a powerful probe to investigate the crystal structures of NaxTMO2 without nothing material structure destruction during measurement. Furthermore, since its high-resolution diffraction, the crystal lattice parameters, strains, and stresses of electrode materials can also be accurately calculated by combining with Rietveld refinement [63,64]. Additionally, compared with conventional XRD, HEXRD also presents tunable energy as well as fast recording for diffraction and scattering data, which is more suitable to detect the structural details of NaxTMO2 during Na ion insertion/extraction in real time. Nowadays, in-situ and ex-situ HEXRD modes are widely applied to distinguish the physical and chemical transitions of NaxTMO2 taking place in short time-scales and then to unveil the evolutions of lattice parameters during desodiation/sodiation [65,66]. Fig. 2 depicts the applications of HEXRD technique in NaxTMO2 layered oxide cathodes in the past decade. The phase evolution behaviors of Ni/Mn and Fe/Mn-based layered oxides with different dopants and crystal structures were systematically researched for Na ion storage at about ten years ago. Subsequently, multiphase symbiosis NaxTMO2 cathodes, such as P2/O3, P2@P3, and P3/spinel, were built to enhance the structural stability of layered oxides. In recent years, high-entropy NaxTMO2 polytypes with its multi-element coordination were studied for high-performance cathodes in detail. The HEXRD patterns collecting in in-situ and ex-situ modes could present the phase structure evolution behaviors of NaxTMO2 polytypes concomitant with Na ion migration in succession, which are much help for revealing the phase stability and then clarifying the phase structure optimizing mechanisms [67-69]. In addition, the optimal working voltage ranges of NaxTMO2 cathodes without detrimental phase transformations could also be accurately delimited according to the recorded in-situ HEXRD patterns at different Na ion contents. The research hotspots in the past decade about NaxTMO2 layered oxide cathodes for SIBs charactering by in-situ and ex-situ HEXRD technique are displayed in Fig. 3. The NaxTMO2 cathodes with different components/phase structures have been examined by using HEXRD to deeply comprehend the enhancement mechanisms of cycling stability, rate performance, and specific capacity at the level of lattice structure.
Herein, we first describe the advantages of HEXRD technique and then summarize recent progresses in the composition regulations and phase structure optimizations of P2-, O3-, and P2/O3-type NaxTMO2 layered oxides. Meanwhile, the influences of material components/structures on their electrochemical properties and phase evolution behaviors characterizing via in-situ HEXRD are discussed. Finally, we prospect the challenges faced by NaxTMO2 layered oxide cathodes and the research directions in the road to propel their practical applications. We believe that this review will inspire more insights in revealing reaction mechanisms by means of in-situ HEXRD as well as supply more guidelines for developing high-performance NaxTMO2 cathodes, and then promote the commercial applications of SIBs.
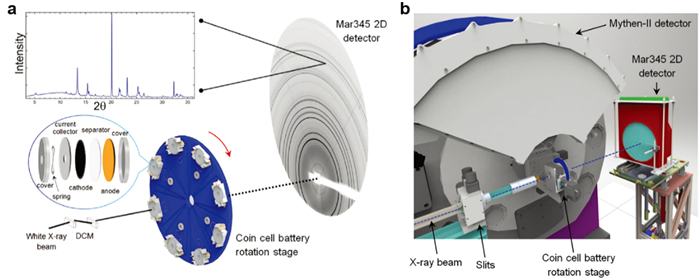
Exploring NaxTMO2 layered oxide cathodes with long-term cycling life, wide operating voltage window, superior rate capability, and high specific capacity, remains a great challenge for practical applications [70-72]. Undoubtedly, revealing and deeply understanding of the phase evolution mechanisms of P2 and O3-type NaxTMO2 upon charge/discharge are of primary works to design high-performance layered oxide cathodes [73-77]. Considering the advantages of HEXRD on diagnosing the phase structures and lattice parameters of NaxTMO2 layered oxides, tremendous efforts were performed to reveal the phase evolution behaviors of P2-, O3-, and P2/O3-type structures during desodiation/sodiation utilizing in-situ HEXRD technique. The in-situ HEXRD equipment using for crystal structure detection are shown in Fig. 4. Detailedly, the NaxTMO2 cathode material coating on thin Al current collector is coupled with anode to assemble a coin cell battery (Fig. 4a) [78]. The synchrotron X-rays would penetrate Al current collector and then irradiate into cathode materials to examine the crystal structure transitions during electrochemical reactions. Additionally, the in-situ HEXRD facility could equip with multi detection positions on the rotation stage to improve the implementation efficiency (Fig. 4b).
The P2-type layered structure with an oxygen stacking sequence of ABBA is one of a classic crystal phase of NaxTMO2 layered oxides, which have attracted numerous interests due to its high electronic/ionic conductivity and rapid kinetics [79-81]. Nevertheless, they also face the challenges of detrimental structure evolutions during repeated charge/discharge cycle concerning to slab gliding, oxygen wastage, and Jahn-Teller distortions especially at high voltage states due to the hyperactive TM ions and the inhomogeneous distributions of Na+/vacancy ordering in the Na-deficient systems, which bring about dramatical performance degradations [82-85]. In the past few years, lots of optimization methods were attempted to improve the battery properties of P2-type NaxTMO2 by inhibiting irreversible phase transitions and electrochemical side reactions [86-88].
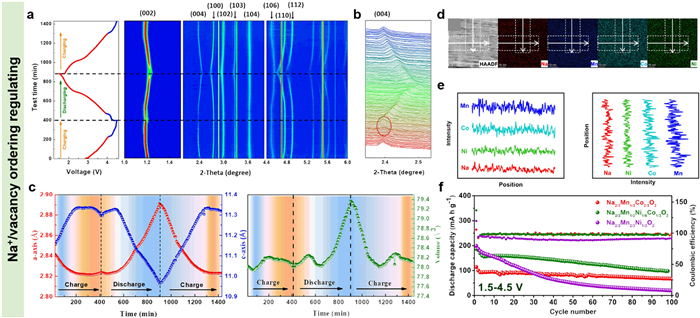
In order to construct high-performance P2-type layered oxides with excellent cycling stability within a wide voltage window range, Liu and co-workers had researched the influences of Na+/vacancy distributions in the Na layers on the structural stability of P2-type Na2/3MnxNix-1/3Co4/3-2xO2 (1/3 ≤ x ≤ 2/3) [89]. The dynamic phase structure transitions of the P2-type Na2/3Mn1/2Ni1/6Co1/3O2 during charge/discharge characterizing by in-situ HEXRD shown that there were not new peaks emerging in the high voltage part and the P’2 phase generating in the low voltage area. All of the diffraction peaks went through a reversible process during Na ions extraction and insertion (Fig. 5a). The second charge/discharge cycle displayed that such P2-type phase structure had been further reinforced after the first cycle (Fig. 5b). Furthermore, the unit cell volume exhibited a percent change of only 1.9% in a large voltage range of 1.5-4.5 V (Fig. 5c), much lower than that of the phase structure transformation of P2-O2 (23.2% volume change). Commonly, the P2-O2 phase transition with large unit cell volume change is very pernicious to the structural stability of P2-type NaxTMO2 cathodes, which is not completely reversible and desirable to be avoided [90,91]. Herein, the stable crystal lattice structure of P2-type Na2/3Mn1/2Ni1/6Co1/3O2 further characterizing via scanning transmission electron microscopy-high angle annular dark field (STEM-HAADF) was ascribed to the uniformly distributed Na ions in the Na layers even there was only 0.17 Na ion residual (Figs. 5d and e). The evenly distributed Na ions allowed layered structures to maintain large space and then suppress detrimental phase transformation like of P2-O2. While the P2-type Na2/3Mn2/3Ni1/3O2 exhibited poor cycling stability because of that the Na vacancies more easily assembled around Ni ions in the lattice and caused unhomogeneous stress distribution in the Na slabs (Fig. 5f).
Some non-metal dopants, such as B and F, can significantly enhance the structural stability of NaxTMO2 layered oxides due to their unique electronic structure and outstanding electronegativity. Furthermore, such light-weight elements are beneficial to improving the mass energy density of hosts. For the B doped NaxTMO2 layered oxides, the light-weight B atoms in the lattices are located in the TM slabs, which could build stronger covalent B-O bonds and donate more electrons to O atoms and then suppress the irreversible oxygen loss in high-voltage regions [39]. Liu et al. had regulated the electronic structure and cycling stability of P’2-type Na0.637B0.038MnO2 by B doping [92]. Herein, the boron atoms in the crystal configuration were used to construct robust B-O-Mn bonds, which could prominently suppress the Jahn-Teller effect of Mn3+, mitigate uneven contraction, and inhibit slab gliding in the lattice (Fig. 6a). For the P’2-type Na2/3MnO2, it underwent a phase transition pathway of P’2(Ⅰ)-OP4-P’2(Ⅰ)-P’2(Ⅱ). The emergence of P’2(Ⅱ) phase suggested noticeable Jahn-Teller effect of Mn3+ in the crystal lattice upon deep sodiation. Conversely, the P’2-type Na0.637B0.038MnO2 exhibited reversible P’2(Ⅰ)-OP4-P’2(Ⅰ) phase transition during the first cycle in the voltage range from 1.5 V to 4.3 V (Fig. 6b). Thence, an improved capacity retention (81%) was obtained for P’2-type Na0.637B0.038MnO2 after 100 cycles, higher than that of the P’2-type Na2/3MnO2 (52%). The F doping is also a trenchant way to enhance the structural stability of NaxTMO2 layered oxides via its outstanding electronegativity. Previous researches indicated that the strong TM-F bonds could greatly suppress TM ions migration and stabilize oxygen redox chemistry. Furthermore, for the ions with strong Jahn-Teller effect, such as Mn3+ and Fe4+, the TM-F bonds could break electronic symmetry and alleviate Jahn-Teller distortions [93].

Metal element substitutions in Na and/or TM slabs have been widely used to restrain pernicious phase transformations, reducing lattice deformations, and ameliorating electronic environments of NaxTMO2 layered oxides relying on their rich diversities and a variety of electronic structures [15,94-96]. The Fe/Mn-based NaxTMO2 layered oxides are promising cathodes for SIBs owing to their low cost, environmental friendliness, active redox couples, and abundant resource in the earth’s crust [71,91]. However, the Mn3+ and Fe4+ in the high spin state will cause Jahn-Teller distortions and TM ion migrations, which usually result in poor crystal structure stability. In order to address these issues, single/multi-metal-element doping is meticulously studied. Indris et al. had synthesized P2-type Na2/3Mn7/12Fe1/3Ti1/12O2 layered oxides by substituting Fe with Mn and Ti in the TM layer (Figs. 6c and d) through solid-state reaction [97]. The in-situ HEXRD patterns of P2-type Na2/3Mn1/2Fe1/2O2 presented a complex phase evolution route of P2-P2/Z-Z-Z/P2-P2-P’2 (Fig. 6e). In sharp contrast, the P2-type Na2/3Mn7/12Fe1/3Ti1/12O2 displayed a single P2 phase in a large voltage range of 1.5-4.3 V (Fig. 6f). Further characterizations demonstrated that the Mn substitution could suppress P2-Z phase transformation in the high-voltage regions, while the Ti substitution could restrain the generation of P’2 phase in the low voltage parts. Compared to the P2-type Na2/3Mn1/2Fe1/2O2, the Mn and Ti co-substituted P2-type Na2/3Mn7/12Fe1/3Ti1/12O2 exhibited higher rate performance in the voltage ranges of 1.5-4.3 V and 1.5-4.5 V. Additionally, the substituted sample displayed excellent capacity retention of 80% after 50 cycles at 0.1 C in 1.5-4.3 V and 83.4% after 30 cycles at 0.1 C in 1.5-4.5 V, much higher than those of the unsubstituted counterpart of 64.9% and 71.1%, respectively. Xiao et al. successfully prepared P2-type Na0.65Li0.08Cu0.08Fe0.24Mn0.6O2 via co-substituting Fe sites with Li and Cu at high temperature [98]. As shown in Figs. 6g and h, the HEXRD patterns of P2-type Na0.65Li0.08Cu0.08Fe0.24Mn0.6O2 displayed a single P2 phase throughout the first cycle and there is only an ultralow cell volume variation of 0.7% during desodiation (Figs. 6i and j), meaning almost solid solution reaction and zero strain. Conversely, the P2-type Na0.65Fe0.4Mn0.6O2 displayed obvious P2-Z phase transition in the range of 2.5-4.2 V, suggesting remarkable TM slab sliding. As a result, the P2-type Na0.65Li0.08Cu0.08Fe0.24Mn0.6O2 exhibited a capacity retention of 88.2% after 500 cycles at 0.1 C, much higher than that of the P2-type Na0.65Fe0.4Mn0.6O2 (22.7%).
Except for the TM site doping in the crystal lattices, element doping/substitution in the alkali metal site of NaxTMO2 layered oxides also exhibit remarkable outcomes. Xiong et al. had investigated the effects of Li substitution sites in P2-type Na0.67Ni0.33Mn0.67O2 (Fig. 7a) on the structure stability and electrochemical properties [99]. Both the Li0.1Na0.57Ni0.33Mn0.67O2 (Fig. 7b) and Li0.1Na0.67Ni0.33Mn0.67O2 (Fig. 7c) exhibited typical P2 phase structure, however, the Li substituted site were different. For the Na-deficient P2-type Li0.1Na0.57Ni0.33Mn0.67O2, the Li ions were mainly located in the prismatic Na sites (Fig. 7d). While for the fully sodiated samples, the Li ions distributed both in the vacancies of Na layers and the octahedral sites in the TM slabs. Such different substitution positions could all improve the specific capacity and cycling stability (Fig. 7e) of P2-type Na0.67Ni0.33Mn0.67O2. However, compared to the dual-site co-substituted counterpart, single-site substituted P2-type Li0.1Na0.57Ni0.33Mn0.67O2 exhibited the best cycling lifespan. The in-situ HEXRD patterns of the P2-type Na0.67Ni0.33Mn0.67O2 and P2-type Li0.1Na0.57Ni0.33Mn0.67O2 exhibited similar phase evolution behaviors in the range of 1.5-4.0 V (Figs. 7f and g). While corresponding lattice parameters refining with the Rietveld refinement presented that the single-site substituted sample displayed smaller c-, a-axis, and unit cell volume changes than those of the unsubstituted P2-type Na0.67Ni0.33Mn0.67O2 during desodiation/sodiation (Fig. 7h).
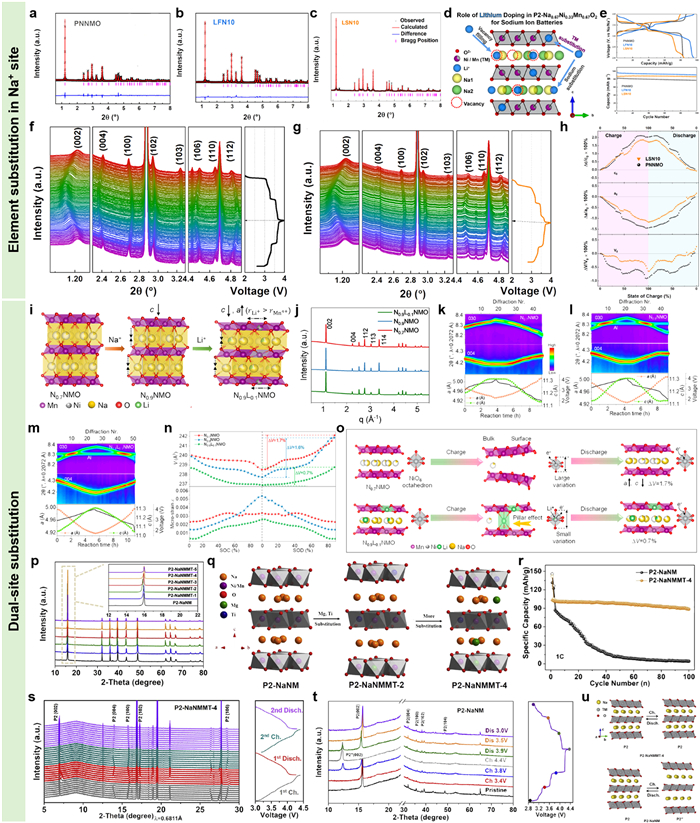
Reasonable dual-site substitutions could optimize the chemical environments of Na and TM layers and then reinforce the phase structures of P2-type NaxTMO2 cathodes by integrating the advantages of different active sites [100,101]. Hua et al. had synthesized a dual-site substituted P2-type Na0.8Li0.024[Li0.065Ni0.22Mn0.66]O2 with high Na content through solid state reaction (Figs. 7i and j) [102]. Compared to the counterparts of P2-type Na0.7Ni0.25Mn0.75O2 and P2-type Na0.9Ni0.25Mn0.75O2.15, the Rietveld refinements of in-situ HEXRD patterns (Figs. 7k-n) at the first cycle displayed that such dual-site substituted P2-type Na0.8Li0.024[Li0.065Ni0.22Mn0.66]O2 exhibited the slightest percentage variations in a-, c-axis, and the unit-cell volume (0.7%) in the range of 2-4 V, suggesting zero-strain phase transformation. Further characterizations proved that the Li ions occupying on the Na and TM sites served as a pillar (Fig. 7o), which could greatly mitigate the Na+/vacancy rearrangement and alleviate detrimental P2-O2 phase transformation. As a result, the dual-site substituted P2-type Na0.8Li0.024[Li0.065Ni0.22Mn0.66]O2 yielded the highest specific capacity (92 mAh/g at 0.1 C) and rate capability (86%, 10 C) compared to its counterparts. Additionally, it also exhibited enhanced structural stability in contrast to other two samples.
Except for Li ion, Mg ion had also been doped into the Na and TM layers to improve the structural/electrochemical stability of NaxTMO2 cathodes due to its appropriate ion radius and inert electrochemical activity. In 2020, Huang and co-works synthesized a series of P2-type Na2/3Ni1/3-xMgxMn2/3-yTiyO2 cathode materials (Fig. 7p) and studied the correlations between Ti and Mg and the structural/electrochemical stability [103]. In this P2 phase structure, the Mg ions occupied transition metal sites as well as Na ions sites (Fig. 7q), while the Ti ion located in the TM layer. Benefitting from the symbiotic advantages, such P2-type Na2/3Ni0.25Mg0.83Mn0.55Ti0.117O2 displayed remarkably improved cycling stability (87% after 100 cycles) in contrast to the P2-type Na2/3Ni1/3Mn2/3O2 (3%) (Fig. 7r). The phase evolution pathways of P2-type Na2/3Ni0.25Mg0.83Mn0.55Ti0.117O2 examining via in-situ HEXRD exhibited a single P2 phase structure throughout Na ion extraction/intercalation processes in the voltage window of 3-4.4 V (Fig. 7s). While for the P2-type Na2/3Ni1/3Mn2/3O2, apparent characteristic peak belonging to P2′’ phase was identified when charging to 3.8 V (Figs. 7t and u), suggesting distinct expansion and gliding of TM slabs. The enhanced structure stability of P2-type Na2/3Ni0.25Mg0.83Mn0.55Ti0.117O2 was ascribed to that the Mg ions locating in Na sites could reduce Na layer spacing due to its smaller ion radius (0.72 Å) than that of Na ion (1.02 Å). Besides, the Ti ions in the TM layers could efficiently suppress cathodic charge order and reinforce structural strength of TM layers because of the formation of stronger Ti-O bonds.
O3-type NaxTMO2 layered oxides with the features of three-layer (ABCABC) oxygen stacking and octahedral Na bonding environment are promising candidates for SIBs because of their sufficient Na ion content in the layered structure, which commonly exhibits higher specific capacity [104-106]. However, the seriously structural degradations in air would increase carbon footprint and manufactural energy cost, which enable them hardly to meet the commercial applications [107-109]. Additionally, the O3-type layered oxide cathodes would experience cumbersome multiphase transitions across Na ions extraction/intercalation together with TM dissolutions, cathode interface reactions, and oxygen losses, leading to mechanical cracks, poor cycling stability and/or thermal runway especially at deeply desodiated states [110-112]. Therefore, exploring valid phase structure reinforce strategies by aliovalent doping, superstructure designing, surface reconstructing etc., are of greatly significant to promote the applications of O3-type cathodes for SIBs [113-115]. Necessarily, in-situ HEXRD had been utilized to deeply comprehend the O3 phase evolution behaviors during Na ions desertion/intercalation in the previous works.
The O3-type layered oxides commonly undergo detrimental phase transformations and oxygen loss especially at deeply desodiatated states, which result in rapid capacity decay. Reducing the cutoff voltage had been performed to inhibit the structure degradation of O3-tpye NaxTMO2 cathodes [116-118]. Chung et al. had studied the capacity degradation mechanisms of O3-type NaFeO2 with different cutoff voltages by using in-situ HEXRD and in-situ XRD techniques [119]. As seen from the in-situ HEXRD patterns (Fig. 8a), which had been converted to a Cu K source patterns for comparisons, the O3-type NaFeO2 went through O3H-O3H/O3′H-O3′H-O3′H/O3′m-O’3m/O3′’H-O’3m phase transitions, herein, the H and m in the subscript represented hexagonal phase and monoclinic structure, respectively. It is worth mentioning that the O3′’H phase can barely identify by using laboratory XRD in previous works [120], further corroborating the high sensitivity and low detection limit of HEXRD technique. Correspondingly, the CO2 gas began to release at around 3.8 V, no oxygen gas was generated throughout the first cycle, and there were only 0.04 Na insertion during the subsequent discharge, which suggested irreversible electrolyte decompositions and structure destructions in the large voltage range. Further in-situ XRD characterization proved that the O3′H-O’3m phase transition was reversible and the O3-type NaFeO2 could deliver steady cycling in a narrow potential window of 2.5-3.5 V (Fig. 8b).
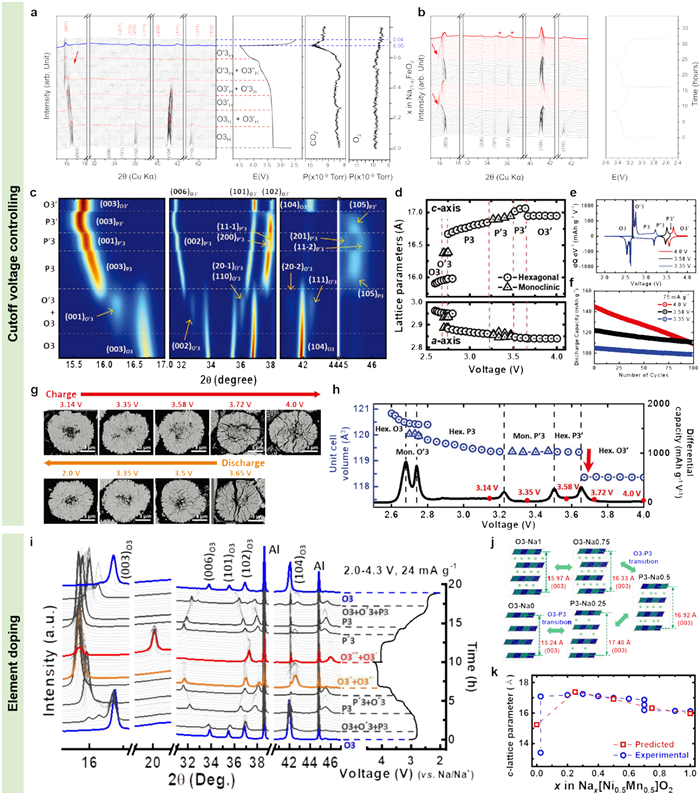
Although narrowing the operating voltage window can sharply extend the cycling lifespan of NaxTMO2 cathodes, their specific capacities are generally lower. Similar to P2-type NaxTMO2 layered oxides, element doping/substitution with inactive alkali, alkaline-earth, and TM ions in different active sites are also demonstrated to be a valid way to ameliorate the structure stability of O3-type NaxTMO2 in a large voltage window [121-123]. For instance, Sun and co-workers had explored the phase evolution pathways and structural variations of O3-type NaNi0.5Mn0.5O2 cathodes during desodiation/sodiation [124]. The pristine hexagonal O3 underwent multiple phase transitions of O3-O’3-P3-P’3-P3′-O3′ as the gradual extraction of Na ions at 1st cycle (Fig. 8c). The a-axis progressively shrank in the whole charging process, while the c-axis continuously expanded before 3.6 V and suddenly shrank after that. Such phenomenon indicated significant degradation of the crystal structure in c-axis direction inducing by accumulated mechanical stress (Fig. 8d). Therefore, in order to extend the cycling lifespan of O3-type NaNi0.5Mn0.5O2, avoiding harmful P3′-O3′ phase variations by shrinking the working voltage window is effective (Fig. 8e). When the P3′ to O3′ phase transformation was inhibited by reducing the cutoff voltage to 3.58 V, a long-term cycling life could be realized (Fig. 8f). Such detrimental structure evolution could also be intuitively observed from the sectional SEM images (Fig. 8g), the P3′-O3′ phase transition caused appreciable microcracks in the secondary particles as a result of the anisotropic nature of such phase structures and immense cell volume contraction (Fig. 8h). However, the O3-type NaNi0.5Mn0.5O2 operating in such a low cutoff voltage exhibited low capacity (125.4 mAh/g) and would lead to unsatisfactory energy density. In order to improve the cycling stability of O3-type NaNi0.5Mn0.5O2 in a large working voltage range, they also explored the structural changes of Ca doped O3-type Na1-2xCaxNi0.5Mn0.5O2 upon desodiation/sodiation [125]. As seen, the O3-type Na0.98Ca0.01Ni0.5Mn0.5O2 underwent a reversible phase evolution pathway of O3-O’3-P3-P’3-O3′-O3′’ in a wide voltage range of 2-4.3 V (Fig. 8i). The phase evolution behaviors (Fig. 8j) and the variations in lattice parameters (Fig. 8k) of O3-type Na0.98Ca0.01Ni0.5Mn0.5O2 during desodiation/sodiation were in good agreement with the calculated results according to the first-principle and presented moderate transitions among multiple metastable structures. Compared with the O3-type NaNi0.5Mn0.5O2, the improved cycling stability had been attributed to that the Ca ion with a smaller ion radius (~1 Å) locating in the Na layer could reinforce the Ca-O bond strength, which could notably suppress structural degradations. Thence, a high initial capacity (209 mAh/g) and an improved cycling lifespan (75% after 100 cycles) were obtained in the voltage range of 2-4.3 V.
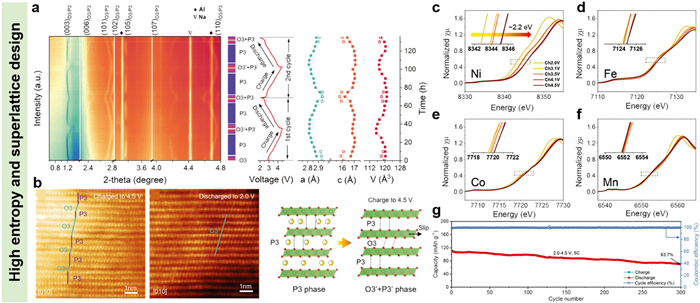
To overcome the structural degradations of O3-type layered oxides, high-entropy design coupling with element doping/substitution had been applied to synthesize high-performance O3-type cathodes for SIBs by integrating the symbiosis effect of different elements [126-128]. For the high-entropy NaxTMO2 layered oxides, there are five or more elements in the TM slabs, which serve as diverse roles for the structural stability and electrochemical properties of hosts. Generally, the Cu, Mg, and Ti with inactive redox couples can stabilize the host structures of layered oxides, the Ni, Mn, and Co elements are able to provide abundant charge compensations, the Li and Sn can alleviate lattice distortion and improve the operating voltage, respectively [114,129,130]. Xin et al. had prepared a high-entropy O3-type Na2/3Li1/6Fe1/6Co1/6Ni1/6Mn1/3O2 layered oxide through solid state method, which also exhibited a superlattice structure with Li/transition metal ordering [131]. The dynamic phase structure changes were detected by utilizing in-situ HEXRD. As shown in Fig. 9a, the pristine Na2/3Li1/6Fe1/6Co1/6Ni1/6Mn1/3O2 displayed a typical O3-type layered structure. When the charging voltage reached to 3 V, the P3-type phase began to appear in the first charging procedure, and the O3 phase completely transferred into P3 phase structure accompanying with continuous extraction of Na ions. As the voltage risen to the cut-off value of 4.5 V, the O3′ and P3′ phases were generated, such mixture phase was the stacking fault structure of P3 and O3 phases, which could be visually observed from its high-resolution transmission electron microscopy (HR-TEM) images (Fig. 9b). During the discharging process, the P3 phase reappeared and then evolved into O3 phase when discharged to 2 V. The HR-TEM image at the end of discharge displayed typical O3 phase structure, suggesting superior cycling reversibility of the O3-type Na2/3Li1/6Fe1/6Co1/6Ni1/6Mn1/3O2. Such stable crystal structure had been ascribed to the high-entropy component and the firm ordering structure owing to that the Li ion with small ion radius could effectively reduce electrostatic repulsion in the adjacent TM layers. The XANES spectra (Figs. 9c-f) shown that the Ni2+/Ni4+ and Fe2+/Fe4+ couples supplied charge compensation upon Na ions desertion/intercalation, while the Co and Mn were inactive. Profiting from the structural advantages, such O3-type Na2/3Li1/6Fe1/6Co1/6Ni1/6Mn1/3O2 exhibited an excellent capacity of 171.2 mAh/g and a superior capacity retention of 63.7% after 300 cycles (Fig. 9g). The O3-P3 phase transformation had also been reported for other O3-type NaxTMO2 layered oxides, such as Na0.83Li0.1Ni0.25Co0.2Mn0.15Ti0.15Sn0.15O2−δ [132], NaLi0.18Co0.23Ru0.59O2 [110], and NaNi1/3Fe1/3Mn1/3-0.02Zr0.02O2 [133], which was a reversible phase evolution route for O3 phase and benefitted to realizing long-term cycling life [134].
In previously reported works, multiphase synergy had been performed to optimize the electrochemical performances, structural stability, and air and moisture tolerability of NaxTMO2 layered oxides by combining the merits of different phase structures [135-141]. Considering the advantages of P2 and O3 phases, P2/O3 biphase layered oxides had been deeply researched to construct outstanding cathode materials for SIBs by integrating the fast ion transition kinetics of P2 phase and the high Na ion content of O3 phase [106,142-145]. Additionally, the biphasic structure could prominently inhibit detrimental phase evolutions of P2 and O3 phases upon desodiation/sodiation, e.g., O2, O’3, Z phase [146,147]. Kim et al. reported a P2/O3 biphase Na0.67Li0.2Fe0.2Mn0.6O2 by substituting TM site with various Li contents [148]. The Li contents in the host phase plays a critical role in the final crystal structure of the as-prepared sample, when the fraction of Li ion reached to 20% (Fig. 10a), a Li honeycomb ordering P2/O3-type hybrid architecture was generated. Similar phenomenon had also been found for the Na1-xLixNi0.5Mn0.5O2+d system, the P2/O3 biphasic structure emerged when the percentage content of Li exceeded 10% [149]. The phase transition pathways during Na ions desertion/intercalation characterizing via in-situ HEXRD presented that the P2-type Na0.67Fe0.25Mn0.75O2 exhibited obvious P2-Z phase transition in high-voltage region and P2-P’2 phase transformation in low-voltage area (Fig. 10b), conforming the occurrences of slab gliding and Jahn-Teller disordering, respectively, while the P2/O3-type Na0.67Li0.2Fe0.2Mn0.6O2 shown a reversible phase transformation between P2/O3 and P2 even in a large voltage window of 1.5-4.5 V (Fig. 10c). The density-functional-theory-calculation (DFT) results proved that the Na ions in the interlayer space preferred to locate near the Li ions to form the stable phase structures (Figs. 10d and e) on account of the enhanced repulsion between adjacent cations in TM layer in deeply charging states. The shortest distances of Li-Na-Li and Li-vacancy-Na pairs propelled the lowest system energy state, which prevented further depletion of Na ions and structural degradations. Benefitting from the synergetic effects of P2 and O3 phases and the reinforced interlayer structure, such biphasic Na0.67Li0.2Fe0.2Mn0.6O2 exhibited a remarkable enhanced capacity retention of 83.5% than that of the P2-type Na0.67Fe0.25Mn0.75O2 (5.4%) after 60 cycles.

For P2/O3 biphase NaxTMO2 layered oxides, besides the element species and contents in the multiphase structure, the constitutions of different crystal lattices are also a nonnegligible factor to the structure stability and electrochemical properties of the as-synthesized products. The appropriate proportion of P2 and O3 phases can reconcile the ion transmission kinetics and the active Na ion content in the biphasic systems, which is beneficial to stimulating the best performance of P2/O3-type NaxTMO2 layered oxides. Therefore, it is critical to interpret the influences of crystal lattice components on the electrochemical/structural stability of P2/O3-type NaxTMO2. Li et al. had prepared a P2/O3-type NaxCu0.1Co0.1Ni0.25Mn0.4Ti0.15O2 layered oxide with different P2 and O3 phase ratios by adjusting the Na contents in the layered TM frameworks [150]. With the increase of Na ion content in the systems, the pristine materials progressively transformed from P2 phase (x = 0.62) to O3 phase (x = 0.84) and exhibited gradually elevated specific capacitances and similar capacitance retentions after 200 cycles in a voltage range of 2-4.2 V (Fig. 10f). While for the samples in the extended voltage window of 2-4.5 V, the capacity and cycling stability of the samples gradually increased when the Na ion content below 0.76 (115.5 mAh/g, 85.6% after 200 cycles), and dramatically declined at x = 0.80 (86.5 mAh/g, 70.1%). The contour maps of in-situ XRD patterns presented that the P2/O3-type Na0.76Cu0.1Co0.1Ni0.25Mn0.4Ti0.15O2 underwent reversible P2/O3-P2/P3-OP4/P3-P2/P3-P2/O3 phase transitions in the voltage range from 2 V to 4.5 V in the initial cycle, while for the high Na ion content samples characterizing by in-situ HEXRD, the phase evolution pathways of P2/O3-P2/P3-OP2/OP4-P2/OP2-P2/P3-P2/O3 and O3-P3/O3-P3-OP2-P3-P3/O3 for P2/O3-type Na0.8Cu0.1Co0.1Ni0.25Mn0.4Ti0.15O2 (Fig. 10g) and O3-type Na0.84Cu0.1Co0.1Ni0.25Mn0.4Ti0.15O2 were identified, respectively. Both of them displayed obvious angle shifts in the low-angle region. Accordingly, reasonable phase ratio in P2/O3-type NaxTMO2 layered oxides is what activates synergistic effect, otherwise it is counterproductive.
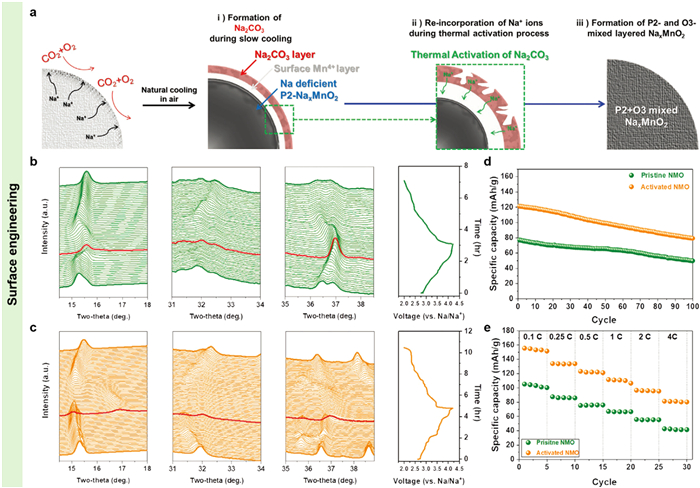
In order to govern the P2 and O3 phase ratio in the P2/O3-type NaxTMO2 layered oxides, except for composition regulations, the thermal treatment conditions during solid-state reactions also play a determined role in the lattice configurations of final products, such as the calcination temperature, holding time, and cooling rate [151-153]. Ji and co-authors explored the phase evolution routes of the P2/O3-type Na0.7Ni0.2Cu0.1Fe0.2Mn0.5O2–δ during heating treatment and desodiation/sodiation [33]. It was found that the phase transformation of O3-P2 at high temperature (1273 K) with low energy barrier and rapid phase transition rate was a thermodynamic driving procedure. While the O3/P3-P2 phase transformation was an ion-diffusion-kinetics growth process at a relative low temperature (1123 K), which required to consume a long time for total phase transition (Figs. 10h and i). The ex-situ HEXRD patterns revealed that such biphasic layered oxide underwent reversible P2-"Z" and O3-P3-O3′ phase transformations during charge/discharge cycling within a voltage window of 1.5-4.2 V (Fig. 10j). Compared to single P2- and O3-type counterparts, the P2/O3-type Na0.7Ni0.2Cu0.1Fe0.2Mn0.5O2–δ exhibited the highest specific capacity, the best rate performance, and the optimal cycling stability (Figs. 10k and l) relying on the synergetic effects. Generally, the P2 and O3-type layered oxides react easily with CO2 and moisture in the air to generate residual alkali, such as Na2CO3, NaOH, and NaHCO3, which dramatically deteriorates the long-term stability of NaxTMO2 layered oxides and reduces the active Na ions content in the lattice [46,154]. In order to address the above issue, Kang et al. fabricated P2/O3-type NaMnO2 through solid-state reaction and subsequent thermal activation [155]. The Na2CO3 species on the surface of P2-type NaMnO2 was converted to electrochemical active Na ions in the crystal lattice through thermal activation and then the P2 and O3 mixed cathode was generated (Fig. 11a). The structural transformations of P2-type NaMnO2 across Na ions desertion/intercalation characterizing by in-situ HEXRD displayed a reversible solid solution reaction. While the P2/O3 biphasic NaMnO2 shown reversible P2/O3-O2 phase transition (Figs. 11b and c) at the first cycle. Typically, the O3 phase presents poor cycling stability and bad rate capability than that of P2 phase. Surprisingly, such P2/O3 biphasic NaxMnO2 shown significantly improved specific capacitance and rate performance, and a similar cycling stability to that of the pristine P2-type NaMnO2 (Figs. 11d and e).
In the past few decades, the NaxTMO2 layered oxide cathodes underwent substantial development and their electrochemical properties had been improved to a certain extern. Nevertheless, the unsatisfactory cycling life, low specific capacity, and poor air stability of NaxTMO2 layered oxide cathodes seriously hinder their commercialization applications for SIBs as before. Researching high-performance NaxTMO2 layered oxides will be a highly active research field of SIBs for a long time in the future.
As has been shown in this review, the phase evolution behaviors of P2-, O3-, and P2/O3-type NaxTMO2 layered oxides relating to Na ion extraction/insertion characterizing by in-situ HEXRD are comprehensively overviewed. The most common phase transitions of NaxTMO2 are P-O structural variation, which generally accompanying with large unit cell volume changes, remarkable structure degradations, and capacity fading. In order to enhance the electrochemical properties of NaxTMO2 cathodes, multi strategies have been carried out in the previous researches. Even though reducing cutoff voltage could avoid detrimental phase transition in deeply charging and discharging states and then improve the cycling life of NaxTMO2 layered oxides, it comes at the cost of sacrificing specific capacity, which go against to develop high-energy-density SIBs. Component and lattice configuration regulations, such as high-entropy and superlattice designs, element substitutions/doping, are trenchant methods to build target phase structures and to suppress irreversible phase transformations of NaxTMO2 cathodes by reinforcing chemical bond energy, inhibiting TM migration and dissolution, and/or buffering Jahn-Teller effect. However, the roles of doped ions are not one-size-fits-all mechanisms, especially in the high-entropy NaxTMO2 layered oxide systems. Moreover, the doping sites in the lattice configurations significant manipulate the Na ions transmission kinetics and the structural stability as well. Unfortunately, the collaborative principles among the TM ions, alkali metal ions, and the phase structure enhancement mechanisms are still unclearable, which should be comprehensively explored in the future. It is worth mentioning that, considering the costs and energy densities of NaxTMO2 layered oxides, the Mn and/or Fe-based NaxTMO2 cathodes may more suitable for practical applications of SIBs, while the precious TM elements such as Li, Ni, Co, Ir, and Ru are more proper to be used as dopants.
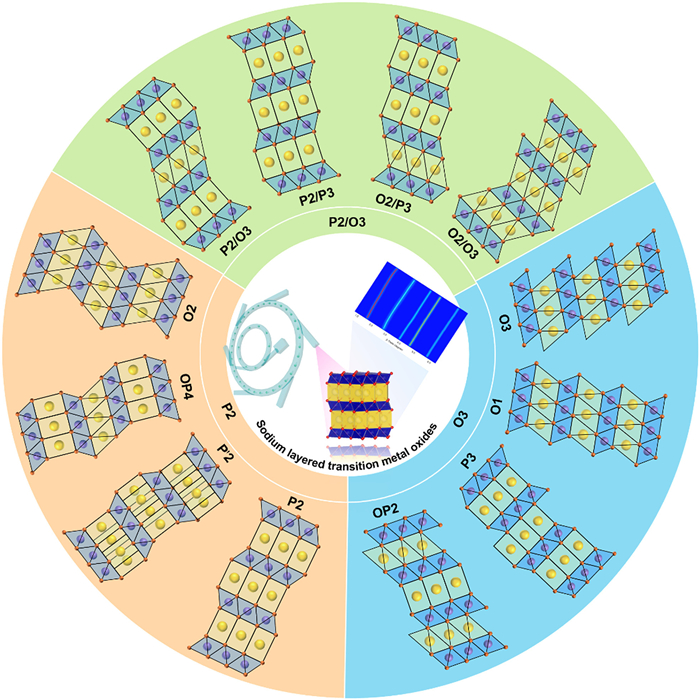
It should be noted that the crystal structures of NaxTMO2 layered oxides with variously structural optimization strategies in the previous works typically undergo lattice slipping to form a series of metastable phases (Fig. 12), such as OP4, O2, OP2, O1 and P2/P3. Especially, the metastable phases with deficient-Na crystal lattices would slowly induce unrecoverable crystal structure deformations, TM ion dissolutions, and electrochemical side reactions during repeated Na ions extraction and intercalation. Therefore, exploring NaxTMO2 layered oxide cathodes with reversible phase transitions like of solid solution reaction are still major research goals in the future. In addition to the above, the composite phase structures by integrating the high ion kinetics P phase with Na-rich O phase can endow them superior electrochemical properties relying on the synergistic effects between different phase structures. Exploring P2, O3, and P3 biphase and multiphase layered oxide cathodes for SIBs are of meaningful works. In particular, the constructions of multiphase NaxTMO2 with certain phase compositions and spatial distributions are the main challenges for this strategy. The element species and ratios in the precursors and the thermal treatment conditions are all crucial linkages to the target phase structures. Previous studies show that the P3 phase NaxTMO2 layered oxides could be generated at a relative low temperature of about 700 ℃, while the P2 and O3 phases emerge at above 900 ℃ since the different formation energies, giving the possibility to design multiphase layered oxides. Considering the elaborate structure-activity relationships and high cost to research NaxTMO2 layered oxides with diverse phase structures and element compositions, machine learning, based on state-of-the-art algorithms and large datasets, is a powerful tool to predict the crystal configurations, structural stability, and electrochemical properties of materials, which could remarkably reduce the workload and repaid screen specific range with a desired characteristic of layered oxides in the road to develop high-performance NaxTMO2 cathodes.
Characterization techniques are extremely important for deeply excavating and profoundly understanding the electrode materials of SIBs. The HEXRD technology operating in the modes of in-situ/operando and ex-situ based on synchrotron radiation is a potent probe to investigate the dynamic crystal structure transformations of electrode materials and supply fundamental insights for revealing the electrochemical reaction mechanisms of SIBs. However, nowadays, in order to obtain integral pictures of NaxTMO2 cathodes, it is exceedingly insufficient to capture the totally information by utilizing in-situ HEXRD technique alone due to its monotonous unique attribution. Collocating with other scattering and spectroscopy techniques, such as in-situ and/or ex-situ Raman, XAS, Mass spectroscopy (MS), and electron paramagnetic resonance (EPR), can allow for more full-scale dynamic phase structure evolution behaviors during Na ion desertion/intercalation. Additionally, the microscopic techniques are indispensable to intuitively observe the real-time variations of the morphology and crystal structure of NaxTMO2 layered oxides causing by ion migration, such as scanning electron microscopy (SEM) and scanning transition electron microscopy (STEM). Furthermore, the microscopy/spectroscopy combination techniques, such as TEM-energy dispersive X-ray spectrometry and STEM-electron energy loss spectroscopy, can simultaneously monitor the variations of morphology, crystal lattice configurations, and elemental compositions during charge/discharge processes, which are valuable for unveiling the electrochemical reaction mechanisms of NaxTMO2 cathodes comprehensively. Besides, the theoretical calculations according to the first-principle, such as the average Bader charge, Gibbs free energy, and the phase formation energy of NaxTMO2 systems, are essential to clarify the intrinsic properties of NaxTMO2 layered oxides in depth. Undoubtedly, collecting the crystal structure and composition information of the electrode materials of commercial SIBs in real time are extremely meaningful works for understanding and controlling the operations of batteries. Even though the HEXRD technique exhibits remarkable advantages and availability for the phase evolution behaviors of NaxTMO2 cathodes, however, for the commercial SIBs and pouch cells, the HEXRD technique is impotent to accurately identify the crystal structure transformations of electrode materials in real time because of the diverse electrode sequences (such as stacking and winding) and elaborate electrochemical environments in the battery systems. Hence, some state-of-the-art characterization techniques are required to be integrated with HEXRD, such as HEX imaging (computed tomography), coherent HEXRD, and neutron diffraction. Above all, the comprehensive applications of state-of-the-art spectroscopy and microscopy characterization technologies and integrating with theoretical calculations should be combinedly used to design and construct high-performance NaxTMO2 layered oxides for SIBs.
The authors declare no competing financial interest.
Yan-Jiang Li: Writing – original draft. Shu-Lei Chou: Supervision, Funding acquisition. Yao Xiao: Validation, Supervision.
This work was supported by the State Grid Corporation Science and Technology Project (No. 5419-202158503A-0-5-ZN).
Q. Liu, Z. Hu, W. Li, et al., Energy Environ. Sci. 14 (2021) 158–179. doi: 10.1039/D0EE02997A
B. Peng, G. Wan, N. Ahmad, et al., Adv. Energy Mater. 13 (2023) 2370117. doi: 10.1002/aenm.202370117
T. Zhou, Y. Chen, Chem. Eng. J. 490 (2024) 151731. doi: 10.1016/j.cej.2024.151731
H. Yang, D. Wang, Y. Liu, et al., Energy Environ. Sci. 17 (2024) 1756–1780. doi: 10.1039/D3EE02934D
M.M. Rahman, F. Lin, Matter 4 (2021) 490–527. doi: 10.1016/j.matt.2020.12.004
S. Sun, S. Liu, Y. Chen, et al., Adv. Funct. Mater. 33 (2023) 2213711. doi: 10.1002/adfm.202213711
Z.Q. Li, Y.F. Liu, H.X. Liu, et al., Chem. Sci. 15 (2024) 11302–11310. doi: 10.1039/D4SC02754J
W. Zuo, A. Innocenti, M. Zarrabeitia, et al., Acc. Chem. Res. 56 (2023) 284–296. doi: 10.1021/acs.accounts.2c00690
S. Chu, S. Guo, H. Zhou, Chem. Soc. Rev. 50 (2021) 13189–13235. doi: 10.1039/D1CS00442E
X. Xu, F. Li, D. Zhang, et al., Carbon Neutral. 2 (2022) 54–62.
N. Ortiz-Vitoriano, N.E. Drewett, E. Gonzalo, et al., Energy Environ. Sci. 10 (2017) 1051–1074. doi: 10.1039/C7EE00566K
L. Gan, X.G. Yuan, J.J. Han, et al., Adv. Funct. Mater. 33 (2022) 2209026.
H. Mao, S. Yang, Y. Yang, et al., Carbon Neutral. 3 (2024) 673–688. doi: 10.1002/cnl2.146
Y. Gao, H. Zhang, J. Peng, et al., Carbon Energy 6 (2024) 464. doi: 10.1002/cey2.464
X. Zhang, W. Zuo, S. Liu, et al., Adv. Mater. 36 (2024) 2310659. doi: 10.1002/adma.202310659
C. Lin, J. Zhang, Y.V. Lim, et al., Carbon Neutral. 1 (2022) 224–232. doi: 10.1002/cnl2.26
T. Zhou, B. Zhang, S. He, et al., J. Mater. Chem. A 12 (2024) 19422–19439. doi: 10.1039/D4TA03313B
K. Kubota, T. Asari, S. Komaba, Adv. Mater. 35 (2023) 2300714. doi: 10.1002/adma.202300714
Y. Zhao, Y. Kang, J. Wozny, et al., Nat. Rev. Mater. 8 (2023) 623–634. doi: 10.1038/s41578-023-00574-w
Y. Su, B. Johannessen, S. Zhang, et al., Adv. Mater. 35 (2023) 2305149. doi: 10.1002/adma.202305149
H. Xu, Q. Yan, W. Yao, et al., Small Struct. 3 (2022) 2100217. doi: 10.1002/sstr.202100217
Y. Niu, Y. Zhao, M. Xu, Carbon Neutral. 2 (2023) 150–168. doi: 10.1002/cnl2.48
X. Liang, J.Y. Hwang, Y.K. Sun, Adv. Energy Mater. 13 (2023) 2301975. doi: 10.1002/aenm.202301975
S. Chu, D. Kim, G. Choi, et al., Angew. Chem. Int. Ed. 135 (2023) 202216174. doi: 10.1002/ange.202216174
X.H. Liu, W.H. Lai, J. Peng, et al., Carbon Neutral. 1 (2022) 49–58. doi: 10.1002/cnl2.6
C. Guo, Y. Gao, S.Q. Li, et al., Adv. Funct. Mater. 34 (2024) 2314851. doi: 10.1002/adfm.202314851
L. Wang, J. Wang, L. Wang, et al., ACS Nano 18 (2024) 10863–10873. doi: 10.1021/acsnano.4c00764
K. Mathiyalagan, D. Shin, Y.C. Lee, J. Energy Chem. 90 (2024) 40–57. doi: 10.1016/j.jechem.2023.10.023
T. Song, C. Wang, C.S. Lee, Carbon Neutral. 1 (2022) 68–92. doi: 10.1002/cnl2.7
W. Zuo, Y. Yang, Acc. Chem. Res. 3 (2022) 709–720.
W. Dou, M. Zheng, W. Zhang, et al., Adv. Funct. Mater. 33 (2023) 2305161. doi: 10.1002/adfm.202305161
Y. Zhang, B. Wu, J. Bi, et al., Carbon Energy 6 (2024) 480. doi: 10.1002/cey2.480
X. Gao, H. Liu, H. Chen, et al., Sci. Bull. 67 (2022) 1589–1602. doi: 10.1016/j.scib.2022.06.024
J. Wang, D. Zhou, X. He, et al., ACS Appl. Mater. Interfaces 12 (2020) 5017–5024. doi: 10.1021/acsami.9b18109
S. Jamil, F. Mudasar, T. Yuan, et al., ACS Appl. Mater. Interfaces 16 (2024) 14669–14679. doi: 10.1021/acsami.3c15667
Y. Huang, W. Zhang, Y. Zhou, et al., ACS Nano 18 (2024) 13106–13116. doi: 10.1021/acsnano.4c01962
Y.F. Liu, K. Han, D.N. Peng, et al., InfoMat 5 (2023) 12422. doi: 10.1002/inf2.12422
B. Xiao, Y. Wang, S. Tan, et al., Angew. Chem. Int. Ed. 133 (2021) 8339–8348. doi: 10.1002/ange.202016334
Y.J. Guo, P.F. Wang, Y.B. Niu, et al., Nat. Commun. 12 (2021) 5267. doi: 10.1038/s41467-021-25610-7
H. Gao, J. Li, F. Zhang, et al., Adv. Energy Mater. 14 (2024) 2304529. doi: 10.1002/aenm.202304529
Y.N. Zhou, Z. Xiao, D. Han, et al., J. Mater. Chem. A 11 (2023) 2618–2626. doi: 10.1039/D2TA09277H
T. Cui, L. Liu, Y. Xiang, et al., J. Am. Chem. Soc. 146 (2024) 13924–13933. doi: 10.1021/jacs.4c01787
H. Fang, H. Ji, J. Zhai, et al., Small 19 (2023) 2301360. doi: 10.1002/smll.202301360
X. Gao, X. Zhang, X. Liu, et al., Small Methods 7 (2023) 2300152. doi: 10.1002/smtd.202300152
X. Xia, T. Liu, C. Cheng, et al., Adv. Mater. 35 (2022) 2209556.
L.Y. Kong, H.X. Liu, Y.F. Zhu, et al., Sci. China Chem. 67 (2024) 191–213. doi: 10.1007/s11426-022-1550-2
J. Wang, Y.F. Zhu, Y. Su, et al., Chem. Soc. Rev. 53 (2024) 4230–4301. doi: 10.1039/D3CS00929G
Y. Xiao, Y.F. Zhu, H.R. Yao, et al., Adv. Energy Mater. 9 (2019) 1803978. doi: 10.1002/aenm.201803978
Y. Li, K.A. Mazzio, N. Yaqoob, et al., Adv. Mater. 36 (2024) 2309842. doi: 10.1002/adma.202309842
L. Qiu, M. Zhang, Y. Song, et al., Carbon Energy 5 (2022) 298.
F. Lin, Y. Liu, X. Yu, et al., Chem. Rev. 117 (2017) 13123–13186. doi: 10.1021/acs.chemrev.7b00007
X. Wang, H. Zhou, Z. Chen, et al., Energy Storage Mater. 49 (2022) 181–208. doi: 10.1016/j.ensm.2022.04.012
S. Mobilio, F. Boscherini, C. Meneghini, Synchrotron Radiation, Springer-Verlag Berlin An, Berlin, 2016.
S. Chen, S. Jiao, Q. Liang, et al., Anal. Chem. 96 (2024) 8021–8035. doi: 10.1021/acs.analchem.4c01399
L. Mino, E. Borfecchia, J. Segura-Ruiz, et al., Rev. Mod. Phys. 90 (2018) 025007. doi: 10.1103/RevModPhys.90.025007
G. Qian, J. Wang, H. Li, et al., Natl. Sci. Rev. 9 (2022) 146.
L. Wang, J. Wang, X. Zhang, et al., Nano Energy 34 (2017) 215–223. doi: 10.1016/j.nanoen.2017.02.046
Z. Shadike, E. Zhao, Y.N. Zhou, et al., Adv. Energy Mater. 8 (2018) 1702588. doi: 10.1002/aenm.201702588
W. Zhang, J. Zheng, Z. Ren, et al., Adv. Mater. 36 (2024) 2310347. doi: 10.1002/adma.202310347
K. Wang, Y. Du, Y. Li, et al., Carbon Energy 5 (2022) 264.
C.C. Lin, H.Y. Liu, J.W. Kang, et al., Energy Storage Mater. 51 (2022) 159–171. doi: 10.1016/j.ensm.2022.06.035
V. Petkov, Mater. Today 11 (2008) 28–38.
Q. Gu, J.A. Kimpton, H.E.A. Brand, et al., Adv. Energy Mater. 7 (2017) 1602831. doi: 10.1002/aenm.201602831
Z. Yang, Y. Lu, X. Liu, et al., Nano Res. 16 (2023) 9954–9967. doi: 10.1007/s12274-023-5630-1
H. Su, G. Guo, Y. Ren, et al., Energy Environ. Sci. 13 (2020) 4371–4380. doi: 10.1039/D0EE02313B
F. Fu, X. Liu, X. Fu, et al., Nat. Commun. 13 (2022) 2826. doi: 10.1038/s41467-022-30113-0
N. Yabuuchi, R. Hara, M. Kajiyama, et al., Adv. Energy Mater. 4 (2014) 1301453. doi: 10.1002/aenm.201301453
R. Kataoka, T. Mukai, A. Yoshizawa, et al., J. Electrochem. Soc. 160 (2013) 933–939. doi: 10.1149/2.125306jes
Y.H. Jung, A.S. Christiansen, R.E. Johnsen, et al., Adv. Funct. Mater. 25 (2015) 3227–3237. doi: 10.1002/adfm.201500469
M. Yuan, H. Liu, F. Ran, Mater. Today 63 (2023) 360–379. doi: 10.1016/j.mattod.2023.02.007
N. Hong, J. Li, H. Wang, et al., Adv. Funct. Mater. 34 (2024) 2402398. doi: 10.1002/adfm.202402398
Z. Chen, Y. Deng, J. Kong, et al., Adv. Mater. 36 (2024) 2402008. doi: 10.1002/adma.202402008
S.Y. Zhang, Y.J. Guo, Y.N. Zhou, et al., Small 17 (2021) 2007236. doi: 10.1002/smll.202007236
X. Yang, S. Wang, H. Li, et al., Electron 2 (2024) 18. doi: 10.1002/elt2.18
L. Yang, L.Y. Kuo, J.M. López del Amo, et al., Adv. Funct. Mater. 31 (2021) 2102939. doi: 10.1002/adfm.202102939
Z. Wu, Y. Ni, S. Tan, et al., J. Am. Chem. Soc. 145 (2023) 9596–9606. doi: 10.1021/jacs.3c00117
J. Chen, G. Adit, L. Li, et al., Energy Environ. Mater. 6 (2023) 12633. doi: 10.1002/eem2.12633
R. Kataoka, T. Mukai, A. Yoshizawa, et al., J. Electrochem. Soc. 162 (2015) 553–558. doi: 10.1149/2.0181504jes
H. Liu, W. Deng, X. Gao, et al., Nano Select 1 (2020) 200–225. doi: 10.1002/nano.202000030
L. Gan, X.G. Yuan, J.J. Han, et al., Carbon Neutral. 2 (2023) 235–244. doi: 10.1002/cnl2.53
D. Dai, X. Lai, X. Wang, et al., Chin. Chem. Lett. 35 (2024) 109405. doi: 10.1016/j.cclet.2023.109405
C. Wang, L. Liu, S. Zhao, et al., Nat. Commun. 12 (2021) 2256. doi: 10.1038/s41467-021-22523-3
J. Jin, Y. Liu, Q. Shen, et al., Adv. Funct. Mater. 32 (2022) 2203424. doi: 10.1002/adfm.202203424
Y. Tang, Q. Zhang, W. Zuo, et al., Nat. Sustain. 7 (2024) 348–359. doi: 10.1038/s41893-024-01288-9
H.W. Li, J.Y. Li, H.H. Dong, et al., Small 20 (2023) 2306690.
S.M. Kang, D. Kim, K.S. Lee, et al., Adv. Sci. 7 (2020) 2001263. doi: 10.1002/advs.202001263
H. Xu, C. Cheng, S. Chu, et al., Adv. Funct. Mater. 30 (2020) 2005164. doi: 10.1002/adfm.202005164
W. Zuo, J. Qiu, X. Liu, et al., Nat. Commun. 11 (2020) 3544. doi: 10.1038/s41467-020-17290-6
Z. Liu, J. Shen, S. Feng, et al., Angew. Chem. Int. Ed. 133 (2021) 21128–21137. doi: 10.1002/ange.202108109
J. Huang, J. Gao, N. Hong, et al., Nano Energy 126 (2024) 109676. doi: 10.1016/j.nanoen.2024.109676
H. Jiang, G. Qian, R. Liu, et al., Sci. China Mater. 66 (2023) 4542–4549. doi: 10.1007/s40843-023-2617-5
T. Yang, Q. Li, Z. Liu, et al., Adv. Mater. 36 (2023) 2306533.
X. Song, R. Liu, J. Jin, et al., Energy Storage Mater. 69 (2024) 103377. doi: 10.1016/j.ensm.2024.103377
P. Zou, L. Yao, C. Wang, et al., Angew. Chem. Int. Ed. 135 (2023) 202304628. doi: 10.1002/ange.202304628
Q.C. Wang, Z. Shadike, X.L. Li, et al., Adv. Energy Mater. 11 (2021) 2003455. doi: 10.1002/aenm.202003455
D. Mikhailova, L. Haase, H.B.A. Nguyen, et al., Adv. Funct. Mater. 34 (2024) 2406384. doi: 10.1002/adfm.202406384
J. Peng, A. Sarapulova, Q. Fu, et al., Chem. Mater. 36 (2024) 4107–4120. doi: 10.1021/acs.chemmater.3c01552
R. Qi, M. Chu, W. Zhao, et al., Nano Energy 88 (2021) 106206. doi: 10.1016/j.nanoen.2021.106206
Y. Xie, E. Gabriel, L. Fan, et al., Chem. Mater. 33 (2021) 4445–4455. doi: 10.1021/acs.chemmater.1c00569
Z. Cheng, B. Zhao, Y.J. Guo, et al., Adv. Energy Mater. 12 (2022) 2103461. doi: 10.1002/aenm.202103461
X.B. Jia, J. Wang, Y.F. Liu, et al., Adv. Mater. 36 (2024) 2307938. doi: 10.1002/adma.202307938
X. Yang, S. Wang, H. Li, et al., ACS Nano 17 (2023) 18616–18628. doi: 10.1021/acsnano.3c07625
Y. Huang, Z. Yan, W. Luo, et al., Energy Storage Mater. 29 (2020) 182–189. doi: 10.1016/j.ensm.2020.04.012
H. Ren, Y. Li, Q. Ni, et al., Adv. Mater. 34 (2022) 2106171. doi: 10.1002/adma.202106171
X. Ma, C. Yang, Z. Xu, et al., Nanoscale 15 (2023) 14737–14753. doi: 10.1039/D3NR02373G
B. Xiao, X. Liu, M. Song, et al., Nano Energy 89 (2021) 106371. doi: 10.1016/j.nanoen.2021.106371
G. Brugnetti, C. Triolo, A. Massaro, et al., Chem. Mater. 35 (2023) 8440–8454. doi: 10.1021/acs.chemmater.3c01196
Z. Ma, H. Xu, Y. Liu, et al., J. Mater. Chem. A 10 (2022) 24216–24225. doi: 10.1039/D2TA06951B
L. Wang, S. Fang, H. Wang, et al., Carbon Energy 6 (2024) e632. doi: 10.1002/cey2.632
Y. Tian, Y. Cai, Y. Chen, et al., Adv. Funct. Mater. 34 (2024) 2316342. doi: 10.1002/adfm.202316342
N. Li, W. Yin, B. Wang, et al., Energy Environ. Mater. 7 (2023) 12671.
B. Wang, J. Ma, K. Wang, et al., Adv. Energy Mater. 14 (2024) 2401090. doi: 10.1002/aenm.202401090
Y. Yoda, K. Kubota, K. Kuroki, et al., Small 16 (2020) 2006483. doi: 10.1002/smll.202006483
X.Y. Zhang, H.Y. Hu, X.Y. Liu, et al., Nano Energy 128 (2024) 109905. doi: 10.1016/j.nanoen.2024.109905
T. Song, L. Chen, D. Gastol, et al., Chem. Mater. 34 (2022) 4153–4165. doi: 10.1021/acs.chemmater.2c00522
S. Mariyappan, T. Marchandier, F. Rabuel, et al., Chem. Mater. 32 (2020) 1657–1666. doi: 10.1021/acs.chemmater.9b05205
Q. Wang, S. Mariyappan, J. Vergnet, et al., Adv. Energy Mater. 9 (2019) 1901785. doi: 10.1002/aenm.201901785
J. Deng, W.B. Luo, X. Lu, et al., Adv. Energy Mater. 8 (2017) 1701610.
D. Susanto, M.K. Cho, G. Ali, et al., Chem. Mater. 31 (2019) 3644–3651. doi: 10.1021/acs.chemmater.9b00149
Y. Li, Y. Gao, X. Wang, et al., Nano Energy 47 (2018) 519–526. doi: 10.1016/j.nanoen.2018.03.007
J.E. Wang, W.H. Han, K.J. Chang, et al., J. Mater. Chem. A 6 (2018) 22731–22740. doi: 10.1039/C8TA06159A
S. Huang, Y. Sun, T. Yuan, et al., Carbon Neutral. 3 (2024) 584–596. doi: 10.1002/cnl2.136
S. Zhao, Q. Shi, W. Feng, et al., Chinese Chem. Lett. 35 (2024) 108606. doi: 10.1016/j.cclet.2023.108606
T.Y. Yu, H.H. Ryu, G. Han, et al., Adv. Energy Mater. 10 (2020) 2001609. doi: 10.1002/aenm.202001609
T.Y. Yu, J. Kim, J.Y. Hwang, et al., J. Mater. Chem. A 8 (2020) 13776–13786. doi: 10.1039/D0TA04847J
Y. Liu, D. Wang, H. Li, et al., J. Mater. Chem. A 10 (2022) 3869–3888. doi: 10.1039/D1TA10329F
K. Sada, S. Kmiec, A. Manthiram, Angew. Chem. Int. Ed. 63 (2024) 202403865. doi: 10.1002/anie.202403865
F. Wu, S. Wu, X. Ye, et al., Chin. Chem. Lett. (2024), doi: 10.1016/j.cclet.2024.109851.
X.Y. Du, Y. Meng, H. Yuan, et al., Energy Storage Mater. 56 (2023) 132–140. doi: 10.1016/j.ensm.2023.01.010
H. Hou, J. Qiu, B. Li, et al., Chinese Chem. Lett. 34 (2023) 108810. doi: 10.1016/j.cclet.2023.108810
L. Yao, P. Zou, C. Wang, et al., Adv. Energy Mater. 12 (2022) 2201989. doi: 10.1002/aenm.202201989
H. Wang, X. Gao, S. Zhang, et al., ACS Nano 17 (2023) 12530–12543. doi: 10.1021/acsnano.3c02290
C. Jiang, Y. Wang, Y. Xin, et al., Carbon Neutral. 3 (2024) 233–244. doi: 10.1002/cnl2.115
H.Y. Hu, H. Wang, Y.F. Zhu, et al., ACS Nano 17 (2023) 15871–15882. doi: 10.1021/acsnano.3c03819
C. Hakim, N. Sabi, I. Saadoune, J. Energy Chem. 61 (2021) 47–60. doi: 10.1016/j.jechem.2021.02.027
E. Gabriel, C. Ma, K. Graff, et al., eScience 3 (2023) 100139. doi: 10.1016/j.esci.2023.100139
J.Y. Li, H.Y. Hu, H.W. Li, et al., ACS Nano 18 (2024) 12945–12956. doi: 10.1021/acsnano.4c00966
Q. Wang, D. Zhou, C. Zhao, et al., Nat. Sustain. 7 (2024) 338–347. doi: 10.1038/s41893-024-01266-1
Y. Su, N.N. Zhang, J.Y. Li, et al., ACS Appl. Mater. Interfaces 15 (2023) 44839–44847. doi: 10.1021/acsami.3c07164
Y.F. Liu, H.Y. Hu, Y.F. Zhu, et al., Chem. Commun. 60 (2024) 6496–6499. doi: 10.1039/D4CC02166E
J. Wang, Q.Q. Sun, J. Yu, et al., Compos. Part B-Eng. 284 (2024) 111664. doi: 10.1016/j.compositesb.2024.111664
Z. Cheng, X.Y. Fan, L. Yu, et al., Angew. Chem. Int. Ed. 134 (2022) 202117728. doi: 10.1002/ange.202117728
A.K. Paidi, W.B. Park, P. Ramakrishnan, et al., Adv. Mater. 34 (2022) 2270218. doi: 10.1002/adma.202270218
Y.F. Zhu, Y. Xiao, W.B. Hua, et al., Angew. Chem. Int. Ed. 59 (2020) 9299–9304. doi: 10.1002/anie.201915650
Z. Liu, Y. Song, S. Fu, et al., Microstructures 4 (2024) 2024036.
J.H. Stansby, M. Avdeev, H.E.A. Brand, et al., Dalton Trans. 50 (2021) 1357–1365. doi: 10.1039/D0DT03351K
L. Mu, S. Xu, Y. Li, et al., Adv. Mater. 27 (2015) 6928–6933. doi: 10.1002/adma.201502449
J.E. Wang, H. Kim, Y.H. Jung, et al., Small 17 (2021) 2100146. doi: 10.1002/smll.202100146
E. Lee, J. Lu, Y. Ren, et al., Adv. Energy Mater. 4 (2014) 1400458. doi: 10.1002/aenm.201400458
R. Liu, W. Huang, J. Liu, et al., Adv. Mater. 36 (2024) 2401048. doi: 10.1002/adma.202401048
X. Liu, G. Zhong, Z. Xiao, et al., Nano Energy 76 (2020) 104997. doi: 10.1016/j.nanoen.2020.104997
G.L. Xu, X. Liu, X. Zhou, et al., Nat. Commun. 13 (2022) 436. doi: 10.1038/s41467-022-28052-x
H.Y. Hu, J.Y. Li, Y.F. Liu, et al., Chem. Sci. 15 (2024) 5192–5200. doi: 10.1039/D3SC06878A
J. Li, H. Hu, J. Wang, et al., Carbon Neutral. 1 (2022) 96–116. doi: 10.1002/cnl2.19
J. Yang, J.M. Lim, M. Park, et al., Adv. Energy Mater. 11 (2021) 2102444. doi: 10.1002/aenm.202102444

Figure 1 Schematic model of the multiple technique space-resolved characterizations based on synchrotron X-ray.

Figure 2 The developments relating to the applications of HEXRD in NaxTMO2 cathodes in the past decade.

Figure 3 Research hotspots of NaxTMO2 cathodes for SIBs characterizing by HEXRD technique from 2014 to August 2024 (the data were collected using Web of Science, August 2024).

Figure 4 (a) Schematic diagram of in-situ HEXRD detection with a coin cell battery structure, Australian synchrotron. (b) In-situ HEXRD equipment with an eight position modified coin cell battery rotation stage. Reprinted with permission [63]. Copyright 2017, Wiley-VCH.

Figure 5 Contour plot of (a) in-situ HEXRD patterns and (b) (004) peak profile, and (c) the changes in lattice parameters of a-, c-axis, and unit cell volume for P2-type Na2/3Mn1/2Ni1/6Co1/3O2 during charge/discharge. (d) STEM-HAADF images, element maps, and (e) line scan analysis of P2-type Na2/3Mn1/2Ni1/6Co1/3O2. (f) Cycling stability of the samples. Reprinted with permission [89]. Copyright 2021, Wiley-VCH.

Figure 6 (a) Schematic illustrations for the deleterious and expected phase transitions without and with B doping, respectively. (b) Contour maps of in-situ HEXRD patterns and corresponding charge/discharge curves at 1st cycle for P’2-type Na0.637B0.038MnO2. Reprinted with permission [92]. Copyright 2023, Wiley-VCH. HEXRD patterns and Rietveld refinement plots, schematic illustrations of crystal structures, and SEM images of (c) Na2/3Mn1/2Fe1/2O2 and (d) Na2/3Mn7/12Fe1/3Ti1/12O2. Contour maps of in-situ HEXRD patterns and corresponding charge/discharge profiles, variations in lattice parameters of a- and c-axis for (e) P2-type Na2/3Mn1/2Fe1/2O2 and (f) P2-type Na2/3Mn7/12Fe1/3Ti1/12O2. Reprinted with permission [97]. Copyright 2024, American Chemical Society. Powder XRD patterns and Rietveld refinement plots, schematic illustrations of crystal structures for (g) P2-type Na0.65Fe0.4Mn0.6O2 and (h) P2-type Na0.65Li0.08Cu0.08Fe0.24Mn0.6O2. (i) Contour plots of in-situ HEXRD patterns collected at 0.1 C during the first charge/discharge cycle and (j) the enlarged (002) and (101) peaks, corresponding lattice parameters of in-plane, average interlayer distance, and unit cell volume of P2-type Na0.65Li0.08Cu0.08Fe0.24Mn0.6O2. Reprinted with permission [98]. Copyright 2020, Elsevier.

Figure 7 Rietveld refinements of HEXRD patterns for (a) Na0.67Ni0.33Mn0.67O2, (b) Li0.1Na0.67Ni0.33Mn0.67O2, and (c) Li0.1Na0.67Ni0.33Mn0.67O2. (d) Schematic illustration of the crystal structure of Li0.1Na0.67Ni0.33Mn0.67O2. (e) Charge/discharge plots in the first cycle and cycling stability of the samples. Waterfall plots of in-situ HEXRD patterns for (f) Na0.67Ni0.33Mn0.67O2 and (g) Li0.1Na0.57Ni0.33Mn0.67O2 during the first charge/discharge cycle. (h) Changes in lattice parameters of c-, a-axis, and unit cell volume for Na0.67Ni0.33Mn0.67O2 and Li0.1Na0.57Ni0.33Mn0.67O2. Reprinted with permission [99]. Copyright 2021, American Chemical Society. (i) Schematic illustration of the crystal structures and (j) HEXRD patterns of Na0.7Ni0.25Mn0.75O2, Na0.9Ni0.25Mn0.75O2.15, and Na0.8Li0.024[Li0.065Ni0.22Mn0.66]O2. Contour maps of in-situ HEXRD patterns and variations in the lattice parameters of a- and c-axis during the first cycle for (k) Na0.7Ni0.25Mn0.75O2, (l) Na0.9Ni0.25Mn0.75O2.15, and (m) Na0.8Li0.024[Li0.065Ni0.22Mn0.66]O2. (n) Changes in the unit cell volume and micro-strain of the samples in the first cycle. (o) Schematic illustration of the pillar effect of Li ions in the Na layer for Na0.7Ni0.25Mn0.75O2 and Na0.8Li0.024[Li0.065Ni0.22Mn0.66]O2. Reprinted with permission [102]. Copyright 2021, American Chemical Society. (p) XRD patterns of P2-type Na2/3Ni1/3-xMgxMn2/3-yTiyO2 with various compositions. (q) Schematic illustration of the crystal structures of Na2/3Ni1/3Mn2/3O2, Na2/3Ni0.3Mg0.033Mn0.6Ti0.067O2, and Na2/3Ni0.25Mg0.083Mn0.55Ti0.0117O2. (r) Cycling stability of Na2/3Ni1/3Mn2/3O2 and Na2/3Ni0.25Mg0.083Mn0.55Ti0.0117O2. (s) Waterfall plots of in-situ HEXRD patterns and corresponding charge/discharge plots of P2-type Na2/3Ni0.25Mg0.083Mn0.55Ti0.0117O2. (t) Ex-situ XRD patterns and corresponding charge/discharge curves of the 1st cycle for P2-type Na2/3Ni1/3Mn2/3O2. (u) Schematic phase transitions of P2-type Na2/3Ni0.25Mg0.083Mn0.55Ti0.0117O2 and P2-type Na2/3Ni1/3Mn2/3O2 during charge/discharge cycle. Reprinted with permission [103]. Copyright 2020, Elsevier.

Figure 8 (a) In-situ HEXRD patterns and corresponding charge/discharge curves, CO2 and O2 gas evolutions, and (b) in-situ XRD patterns and corresponding charge/discharge plots in the voltage window of 2.5-3.5 V of O3-type NaFeO2. Reprinted with permission [119]. Copyright 2019, American Chemical Society. (c) Contour maps of in-situ HEXRD patterns and (d) variations in the lattice parameters of c- and a-axis in the first charging to 4 V. (e) CV curves and (f) cycling stability plots at different cutoff voltages for O3-type NaNi0.5Mn0.5O2. (g) The cross-sectional SEM images at different charge/discharge states and (h) the unit cell volume vs. dQ/dV profile during charging for of O3-type NaNi0.5Mn0.5O2. Reprinted with permission [124]. Copyright 2020, Wiley-VCH. (i) Waterfall plots of in-situ HEXRD patterns during the first cycle of Na0.98Ca0.01Ni0.5Mn0.5O2. (j, k) The predicted structure evolutions, variations in layer space during desodiation according to Rietveld refinement based on in-situ HEXRD patterns and the first-principle calculations, respectively. Reprinted with permission [125]. Copyright 2020, Royal Society of Chemistry.

Figure 9 (a) Contour maps of in-situ HEXRD patterns, corresponding charge/discharge profiles, and the changes in lattice parameters of a-, c-axis, and unit cell volume for O3-type Na2/3Li1/6Fe1/6Co1/6Ni1/6Mn1/3O2. (b) HR-TEM images in charging 4.5 V and discharging 2 V states and schematic illustration of the phase transformation pathway for Na2/3Li1/6Fe1/6Co1/6Ni1/6Mn1/3O2 upon cycling. XANES spectra of the (c) Ni K-edge, (d) Fe K-edge, (e) Co K-edge, and (f) Mn K-edge during cycling. (g) Cycling stability of O3-type Na2/3Li1/6Fe1/6Co1/6Ni1/6Mn1/3O2 at 5 C. Reprinted with permission [131]. Copyright 2022, Wiley-VCH.

Figure 10 (a) XRD patterns of P2-type Na0.67Fe0.25Mn0.75O2, P2-type Na0.67Li0.1Fe0.2Mn0.6O2, and P2/O3-type Na0.67Li0.2Fe0.2Mn0.6O2. In-situ HEXRD patterns and corresponding phase evolution pathways and charge/discharge plots of (b) P2-type Na0.67Fe0.25Mn0.75O2 and (c) P2/O3-type Na0.67Li0.2Fe0.2Mn0.6O2. (d) The total energy of P2/O3-type Na0.67Li0.2Fe0.2Mn0.6O2 system as a function of the distance between Na ion and Li ion. (e) Schematic illustration of the Li-vacancy-Na and Na-Li-Na pairs in the layered structure with the most stable state. Reprinted with permission [148]. Copyright 2021, Wiley-VCH. (f) Capacity retentions of P2/O3-type NaxCu0.1Co0.1Ni0.25Mn0.4Ti0.15O2 with different P2 and O3 phase ratios within the voltage ranges of 2-4.2 V and 2-4.5 V. (g) Contour maps of in-situ HEXRD patterns and corresponding charge/discharge profiles at 1st cycle for P2/O3-type Na0.8Cu0.1Co0.1Ni0.25Mn0.4Ti0.15O2. Reprinted with permission [150]. Copyright 2024, Wiley-VCH. (h) The XRD patterns of Na0.7Ni0.2Cu0.1Fe0.2Mn0.5O2–δ quenching from 1273 K after holding for 1 h and 5 h, and (i) sintering at 1123 K for various holding times. (j) Ex-situ HEXRD patterns of P2/O3-type Na0.7Ni0.2Cu0.1Fe0.2Mn0.5O2–δ upon desodiation/sodiation and corresponding charge/discharge profiles. (k) Rate capability and (l) cycling stability of Na0.7Ni0.2Cu0.1Fe0.2Mn0.5O2–δ with different phase structures. Reprinted with permission [33]. Copy Right 2022 Elsevier.

Figure 11 (a) Schematic illustration of the formation of Na2CO3 species and P2/O3-type NaMnO2 through thermal activation. In-situ HEXRD patterns and corresponding charge/discharge curves in the first cycle at 0.25 C of (b) P2-type NaMnO2 and (c) P2/O3-type NaMnO2. (d) Comparisons of the cycling stability and (e) rate capability of P2-type NaMnO2 and P2/O3-type NaMnO2. Reprinted with permission [155]. Copyright 2021, Wiley-VCH.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: