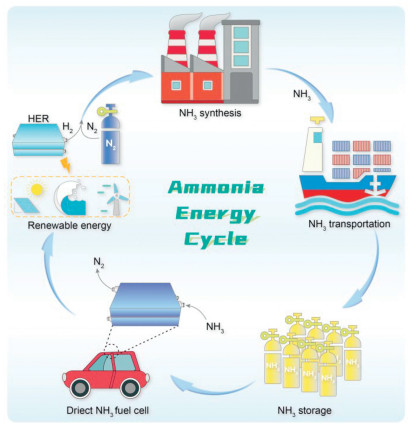
Figure 1.
Schematic illustration of the renewable ammonia energy cycle.
Advances in platinum-based materials for electrocatalytic ammonia oxidation: Mechanisms and research progress
Youpeng Wang , Yuan Ji , Chengbo Li , Zhaoyang Chen , Xu Li , Tingting Zheng , Qiu Jiang , Chuan Xia

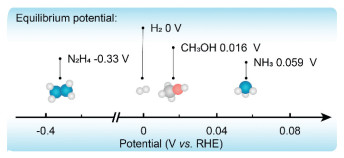
As economic growth intensifies, the issues associated with fossil fuels, including non-renewability and environmental pollution, are becoming increasingly evident. Consequently, developing alternative energy sources to replace traditional fossil energy has emerged as a crucial strategy for addressing the energy crisis and mitigating environmental challenges [1, 2]. Compared with the combustion of traditional fossil fuels, fuel cells, an efficient energy conversion technology not constrained by the Carnot cycle, have garnered significant interest over the years. Notably, proton exchange membrane fuel cell (PEMFC), which utilizes hydrogen as a fuel source, has achieved particular maturity [3, 4]. However, the application of PEMFC is significantly constrained by the high costs associated with the storage and transportation of hydrogen under high pressure [5]. In response to these limitations, researchers have explored alternative solutions. A promising approach involves the development of carbon- and nitrogen-based energy sources such as ammonia, methanol, isopropanol, and hydrazine, which aim to serve as viable substitutes for hydrogen energy [6-11]. In terms of the theoretical oxidation potential, all of these small molecules (ammonia, methanol, isopropanol and hydrazine) are closely aligned with the hydrogen oxidation potential and possess sufficiently low oxidation potentials for use as anodes in fuel cells, which utilize an oxygen reduction reaction (ORR) on the other side of the cathode. However, there are specific challenges with some of these alternatives. Methanol, as a representative carbon-based fuel, leads to CO2 emissions. Alternatively, carbon-free carbon-based materials, such as isopropyl alcohol, can undergo dehydrogenation to produce acetone in fuel cells to release energy. However, these materials have a lower volumetric energy density and high cost. While hydrazine, another alternative, as a representative of nitrogen-based carbon-emission-free fuel, is prohibitively expensive, diminishing its practicality as a fuel. To overcome these hurdles, ammonia has been proposed as an alternative small molecule fuel. It offers the dual benefits of being carbon-emission-free and cost-effective, making it a more viable and sustainable option for fuel cell technologies [12]. The use of ammonia as a fuel offers several distinct advantages that increase its appeal for energy applications (Fig. 1). First, ammonia is easily stored, requiring only approximately 10 atm of pressure for liquefaction at room temperature [7]; Second, it boasts a high volumetric energy density of 12.92–14.4 MJ/L [13]; Third, there is a well-established industry chain for its preparation, transportation, and storage, particularly in terms of sea transport and land pipe-line transport [14]. Furthermore, ammonia does not emit a carbon footprint during use [15], and the synthesis of ammonia is progressively moving toward non-polluting methods, such as electrochemical synthesis and the offshore production of green ammonia. These innovations provide favorable conditions for the use of ammonia as a fuel over other energy carriers (Fig. 2 and Table 1) [16-18]. As a result, such developments pave the way for a carbon-free cycle with ammonia as its core energy carrier, highlighting its significant potential as a sustainable energy solution.
 DownLoad:
CSV
DownLoad:
CSV
The key reaction in an ammonia fuel cell is the ammonia oxidation reaction (AOR). Although research into AOR commenced relatively early, it has not reached a sufficient depth, primarily owing to the earlier immaturity of ammonia synthesis methods and the developmental stage of characterization technologies. Consequently, guidance on catalyst design remains limited. To overcome these challenges and optimize the performance of ammonia fuel cells, more in-depth analytical research is imperative. Currently, the reported catalysts for AOR are categorized into noble metal-based and non-noble metal-based catalysts [3]. Among these, non-noble metal catalysts normally exhibit a high overpotential, which prevents them from forming an effective ammonia fuel cell anode. Consequently, their use is confined primarily to applications in water pollution treatment, where their high overpotential does not impede the desired outcome [19].
Among noble metal catalysts, platinum (Pt)-based catalysts demonstrate exceptional activity and are considered the most promising options for electrocatalytic ammonia oxidation. However, even with the use of Pt as a catalyst, the AOR still has an overpotential greater than 0.4 V. This significant overpotential indicates that while Pt-based catalysts are effective, there remains substantial room for improvement to reduce energy loss and enhance the overall efficiency of the reaction process [5]. In addition, Pt-based catalysts also suffer from serious toxic effects that impede their long-term functionality. Currently, the prevailing view is that Pt-based catalysts have excessive adsorption energy for *N intermediates from ammonia oxidative dehydrogenation, leading to the occupation of Pt active sites covered by *N, thus causing catalyst deactivation. This toxicity hampers the effectiveness and durability of these catalysts, rendering them unsuitable for prolonged use. Consequently, to fully harness the potential of clean ammonia energy, in-depth research into the mechanisms of AOR is imperative. This includes the development of more robust and less toxic catalysts and the assembly of more efficient devices [20, 21]. In particular, there is a critical need for more advanced methods to characterize the complex 6e- transfer process in AORs [22].
In this review, we present a comprehensive overview of the reaction mechanisms and critical bottlenecks in electrocatalytic AORs using Pt-based catalysts. Given that mechanistic studies necessitate the capture of signals from AOR intermediates as direct evidence, we delineate methods for capturing these intermediates and summarize the advantages and disadvantages of various characterization techniques. As a result, recent advancements in the optimization of catalysts tailored to specific reaction pathways are summarized. This review covers the evolution from Pt-based monometallic catalysts to Pt-alloyed multimetallic variants, highlighting how modifications in catalyst morphology, dimensions, and carrier structures significantly influence their activity. Additionally, we present a summary of the various testing conditions employed for electrocatalytic AORs and propose a standardized testing procedure to ensure the consistency and reliability of the experimental results. This review concludes with perspectives on the current challenges facing the field and potential future directions for research on electrocatalytic AORs, emphasizing the need for further advancements in catalyst development and system design to increase the viability of ammonia as a clean energy source.
Research on the AOR of Pt-based metals can be traced back to 1905, when it was first found that Pt could catalyze the oxidation of ammonia to generate nitrogen in an alkaline solution [23]. Over the years, as the understanding of catalysts and reactors has advanced, Pt-based alkaline ammonia fuel cells with Pt-based catalysts serving as anodes have gradually developed. The reaction mechanism of the AOR increased significantly over time. In 1963, Oswin and Salomon proposed a detailed mechanism for the stepwise dehydrogenation of ammonia, known as the Oswin-Salomon (O-S) mechanism [24]. According to their findings, the reaction begins with the adsorption of *NH3 molecules on the catalyst surface. This is followed by sequential dehydrogenation to form *NH2, *NH, and *N intermediates. This process culminates with the coupling of two *N intermediates to form N2 molecules, which then desorb from the catalyst surface (Fig. 3). A few years later, the O-S mechanism was further refined by Gerischer and Mauerer in 1970 [25]. They proposed modifications to the initial dehydrogenation steps, suggesting that coupling between *NHx and *NHy intermediates occurs, forming an *N2Hx+y complex. This complex then undergoes further dehydrogenation before finally desorbing to N2. This adjustment provided deeper insight into the complex interactions and transformations occurring during the ammonia oxidation process. A significant further advancement occurred in 2006 when Pérez et al. utilized surface-enhanced Raman spectroscopy (SERS) to analyze the reaction and identified the N3- intermediate in the reaction process, marking a crucial development in the understanding of the reaction mechanism for AOR [26]. This discovery not only substantiated earlier theoretical models but also expanded the knowledge of the intermediate species involved, contributing to a more comprehensive understanding of the electrocatalytic AOR.
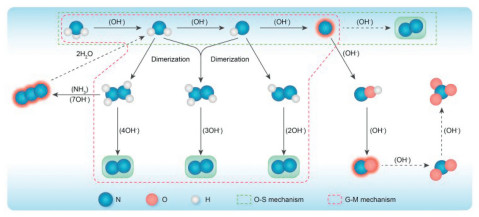
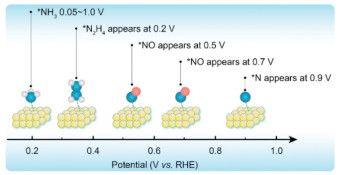
Since the beginning of the 21st century, the development of various spectroscopic techniques has led to the identification of additional intermediate species involved in the electrocatalytic oxidation of ammonia. As illustrated in Fig. 4, these advancements have enabled researchers to observe the differences in intermediate species formed at various electrode potentials. In 2015, Matsui et al. obtained adsorption signals for *NH3 molecules in the potential range of 0.05 V-1 VRHE, adsorption signals for *N2H4 were obtained at 0.2 and 0.4 VRHE, and an adsorption signal for *NO was obtained at 0.5 VRHE [27]. In 2016, Katayama et al. observed a signal for *NO at 0.7 VRHE. Although the pathway of ammonia oxidation has become increasingly clear with the identification of intermediates one by one, progress in the development of AOR has been hindered [28]. The primary challenges identified include: (1) Catalyst deactivation. Differential electrochemical mass spectrometry (DEMS) and SERS have shown that during the first step of the reaction, *NH3 is adsorbed onto the active sites of the catalyst. After complete dehydrogenation, the resulting single *N species may fail to couple with another *N species to form N2. Instead, this *N species remains bound to the active site, effectively blocking it and leading to catalyst deactivation [29]. Additionally, at higher potentials, the presence of *NOx species has been detected. These species also contribute to the deactivation of the catalyst. The formation of *NOx not only occupies active sites but also may lead to degradation or poisoning of the catalyst, further diminishing its effectiveness and durability [30]; (2) High overpotential. Currently, even the most active catalyst, the PtIr alloy, has an overpotential exceeding 400 mV. This high overpotential results in a lower output voltage and reduced power for fuel cells that utilize the AOR at the anode and the ORR at the cathode. Such inefficiencies significantly hamper the practical application and scalability of these fuel cells in real-world settings [31].
Capturing intermediate signals in the electrochemical AOR of Pt-based catalysts presents unique challenges compared with other electrochemical processes. These challenges include faster reaction kinetics, short intermediate residence times, and interference from other gas products [30]. Despite these difficulties, researchers have made significant progress in recent decades (Table 2). They successfully obtained signals from various adsorbed species via both ex-situ and in-situ spectroscopic characterization techniques. These advancements have provided valuable insights into the dynamics and mechanisms of the ammonia oxidation process, enhancing our understanding and guiding further improvements in catalyst design and performance.
DownLoad:
CSV
DEMS is a sophisticated spectroscopic characterization tool that integrates an electrochemical reaction cell with mass spectrometry. This combination allows real-time detection of gasses or volatiles produced during electrochemical reactions. DEMS is particularly valuable because it enables the direct monitoring of reaction byproducts as they are formed, providing insights into reaction mechanisms, kinetics, and the nature of transient species within the electrochemical environment. Since the ammonia oxidation reaction involves the generation of N2 and NOx gasses, the use of DEMS to detect the AOR is an appropriate choice. The first application of DEMS for the detection of AOR was performed by Wasmv et al. [32]. Researchers employing Pt black as a catalyst have conducted experiments on AOR in a 0.5 mol/L KOH solution using ammonia, nitrite, and hydroxylamine as reaction nitrogen sources. The findings from these tests revealed several key insights into the reaction dynamics: (1) Ammonia oxidizes at a potential of 0.7 VRHE to produce N2, and NOx at potentials greater than 0.8 VRHE. NOx generation occurs in the Pt-O adsorption potential region, and the adsorbed *NO can be reduced in the low potential region. (2) The adsorptive oxidation of ammonia occurs concurrently with the precipitation of N2. This observation suggested that the adsorptive intermediates do not consist of triplebonded nitrogen atoms; (3) Ammonia-oxidized intermediates are capable of being oxidized at potentials lower than 0.2 VRHE; (4) Hydrolyxamine follows electroreduction and electrooxidation pathways similar to those of ammonia, except that the reaction potentials are different; (5) Adsorbed nitrite can be reduced to nitrogen oxides. In 1998, by detecting N2, Gootzen et al. stripped out the charge amount that did not produce N2 [29]. Based on the different electron transfer numbers of the intermediate NHx (x = 0, 1, 2) species, they compared the charge amount to indirectly indicate that *N is the final adsorbed species, that is, the toxic species. In 2008, Moran et al. prepared PtIr alloy catalysts with an atomic ratio of 70:30 by electrodeposition and compared them with pure Pt metal catalysts [33]. DEMS revealed that when PtIr alloy was used as a catalyst, the N2 signals exhibited the same trend as those of ammonia oxidation LSV up to 0.7 VRHE, indicating 100% N2 selectivity, but when the potential was greater than 0.8 VRHE, new mass spectrometry signals were detected, indicating NOx generation in the high potential interval. On Pt, the difference is that there is more NOx generation in the high potential interval. For both Pt and PtIr, in the potential range of 0.4–0.7 VRHE, researchers have observed current generation but no corresponding signals in the mass spectrum. This phenomenon indicates that this specific potential interval is associated with the formation of intermediates resulting from the dehydrogenation of ammonia. In 2011, Peng et al. investigated the products of AOR in non-aqueous media [34].
The ammonia oxidation products in non-aqueous media without oxygenated species were only N2 according to DEMS tests and were able to sustain the reaction compared with those in aqueous media. The authors deduced that there are 3 reasons for this: (1) There is a retardation of the AOR in aqueous media due to the competitive adsorption of oxygenated species on the surface with NH3; (2) The binding between the oxygenated species and nitrogenous substances on the surface leads to a slow catalytic process; (3) The ammonia oxidation in aqueous media is not rapid enough, resulting in the catalyst surface being passivated. In 2015, Finkelstein et al. conducted an in-depth study of the ammonia oxidation process on Pt (100) facets [35]. By employing the technique of electrodepositing Pt (100) films and recording cyclic voltammograms (CVs) at different potential intervals, researchers combined these data with previously reported findings from DEMS and in situ attenuated total reflectance infrared spectroscopy (ATR-IR). From this integrated approach, a mechanism was proposed to explain the generation of NOx during ammonia oxidation: after complete dehydrogenation of ammonia, it continues to bind OH-, which is then dehydrogenated to form *NO adsorbates, which then dehydrogenate to form NOx- by binding OH-. In 2018, Kim et al. chemically treated three representative oxygenated nitrogen species (NO, NO2- and NO3-) onto a Pt surface [36]. Their research aimed to compare these species with the AOR process to determine their effects on the electrode. The findings revealed that all these oxygenated nitrogen species could poison the electrode, thereby impacting its efficiency and functionality. Using DEMS, researchers collected gas signals during the AOR process. They observed that NOx production began at potentials greater than 0.8 VRHE, aligning with the previously understood pathway for NOx- production [29, 32, 35]. This consistency underscores the complex interactions at the electrode surface and highlights the detrimental impact of oxygenated nitrogen species on electrode performance during ammonia oxidation. In 2020, Kim et al. investigated the reasons for the poor stability of Pt during AOR. On-line EFC/ICP-MS analyses revealed that both the anodic and cathodic dissolution of Pt (mainly originating from its oxidation state transition) are significantly enhanced by the presence of NH3 in the electrolyte [37]. The presence of intermediate NOx species, as identified by DEMS, complicates the reversion of dissolved Pt back to the Pt surface during the reduction potential. This issue arises because NOx species contribute to both structural deformations, such as dissolution and particle shedding, and surface poisoning of the catalyst. In these studies, Katsounaros et al. also reported that AOR can generate N2O at 0.95 VRHE. They obtained this by performing an online DEMS test of AOR on a Pt (100) facet, and similarly, N2 and NO were also detected [38]. On the basis of this, to address these challenges, the authors investigated a suitable range through which the reduction potential could mitigate both structural deformation and surface poisoning. However, this approach is limited by the limitations of DEMS, which is primarily capable of detecting gaseous substances. Consequently, DEMS cannot provide direct evidence of NHx intermediates, which are crucial for a complete understanding of the reaction mechanisms. Moreover, this technique does not account for factors such as the adsorption configuration of species at the active site, which are essential for understanding catalyst behavior and optimizing reaction conditions.
SERS has a better signal than normal Raman, which facilitates the detection of N-related intermediates in ammonia oxidation reactions. In 2001, de Vooys et al. detected SERS signals during the AOR on Au and Pd electrodes [39]. On the Au electrode, where H2O and D2O were used as solvents for in situ SERS, adsorption from NH3 molecules was detected in the range of the 365–385 cm-1 band because of a blueshift of the peak position caused by isotopes. On Pd, on the other hand, the 455–465 cm-1 band did not shift with the isotope effect, which was attributed to the stretching mode of metal-N. This result further confirms that *N is a toxic species in the ammonia oxidation reaction. Vi-dal-Iglesias et al. compared the SERS features of azides with the signals obtained from the ammonia oxidation test, and they obtained the same positive Stark-tuning slope (30 cm-1 V-1) feature, identifying the 432–435 cm-1 band belonging to Pt-N3- during the ammonia oxidation test. However, they did not explain the trend of variation in this species with potential [26].
ATR-FTIR is a simple method for analyzing the chemical composition of object surfaces and is well suited for the detection of ammonia oxidation intermediates because of its sensitivity and simplicity of testing. In 2015, Matsui et al. investigated the ammonia oxidation behavior on a Pt electrode under alkaline conditions using in-situ ATR-FTIR [27]. By replacing H2O with D2O and comparing the ATR-FTIR feature spectra of N2H4, the presence of N2H4 intermediates was directly confirmed for the first time, and the plausibility of the G-M mechanism was also confirmed. In addition, the NH3 adsorption signal and the NOx signal were observed at low potentials, which is consistent with the results obtained by other detection methods. In 2016, Katayama et al. investigated the role of CeO2 modified Pt in ammonia oxidation catalysis [40]. In-situ ATR-FTIR tests revealed that the OH- adsorbed on the Pt surface was continuously consumed during ammonia oxidation, which may have resulted in a potential dependent ammonia oxidation reaction. The modification of CeO2 promoted the adsorption capacity of OH- on the surface of Pt compared with that of pure Pt, which improved the activity of CeO2 modified Pt for ammonia oxidation catalysis. In addition, Katayama et al. studied the role of Y2O3 modified Pt in ammonia oxidation catalysis [28]. Under in-situ monitoring by ATR-FTIR, N2H4 signals cannot be detected in Pd during ammonia oxidation because of the low activity of Pd for electrocatalytic ammonia oxidation. After the Y2O3 was modified with Pd, N2H4 signals were clearly detected, which indicates that the Y2O3 modification promoted ammonia. The signal area of N2H4 increases significantly on Y2O3 modified Pt, which also indicates the high activity of Y2O3 modified Pt for ammonia oxidation.
In 2005, RRDE was applied to the detection of ammonia oxidation intermediates [41]. Endo et al. reported that the current gradually decreased with increasing rotational speed of the RRDE tested in AOR, and after excluding the effects of temperature, NOx, O2 and other gasses dissolved, further by comparing the changes in current with and without rotation, it was found that the current was suppressed when the electrode was rotated and recovered significantly during the stationary period. Considering that Ni does not have ammonia oxidation properties in the low potential interval, the Pt ring was further replaced with Ni, and a low potential was applied to the Ni ring for the oxidation of intermediates, as a result of which the current signal was still detected on the Ni ring. On this basis, the authors concluded that this phenomenon was caused by the escape of some intermediate substances (NHx, N2Hy, and NH2OH) from the surface during rotation. However, this method cannot distinguish actual intermediates and cannot detect non-desorption intermediates, so it has limited significance for the mechanism analysis of ammonia oxidation. To detect the existence of intermediate N2H4 more directly, in 2021, small molecules were used to capture N2H4 in situ on the catalyst surface [42]. Siddharth et al. mixed 4-(1, 2, 2-triphenylvinyl) benzaldehyde (TPE-CHO) with a Pt/C catalyst and dropped it on an electrode for the capture of intermediates in the ammonia oxidation process. The molecule is able to form the dimerization product (TPE-CN)2 in the presence of N2H4, which is unique in that TPE-CHO has a fluorescence response, whereas (TPE-CN)2 formed by the N2H4 linkage does not have a fluorescence response, and TPE-CHO is chemically stable in the AOR potential window, where other molecules, such as NH3, are not able to induce the reaction of TPE-CHO to form (TPE-(CN)2. Owing to the above properties, researchers have successfully detected the signal of N2H4 during ammonia oxidation, and the intensity of this signal varies linearly with the amount of catalyst over a range of intervals. In addition, many studies have been devoted to predicting the reaction path of ammonia oxidation via theoretical calculations by calculating the ΔG values of different intermediates and products at different potentials, so as to infer the most effective path.
The development of Pt-based catalysts for ammonia oxidation has a long history since 1905, when the catalytic production of N2 on the surface of Pt was first observed [23]. However, early development of these catalysts was hampered by the limited capabilities of characterization technologies, making it challenging to understand the molecular level phenomena that cause poisoning in Pt as an ammonia oxidation catalyst. In the 21st century, with significant improvements in characterization and synthetic methods, researchers now have the ability to characterize and synthesize catalysts at the molecular and atomic levels. As a result, despite being a traditional choice, Pt remains the most promising material for ammonia oxidation applications [3]. The design and development of Pt-based catalysts have evolved from monometallic Pt to multimetallic Pt alloys. Innovations in the selective exposure of facets, the engineering of different sizes or morphologies, and modifications in support structures have led to the emergence of a number of high performance catalysts. These advancements reflect a sophisticated understanding of catalyst architecture and its impact on activity and durability, paving the way for more effective and robust applications in ammonia oxidation (Fig. 5).
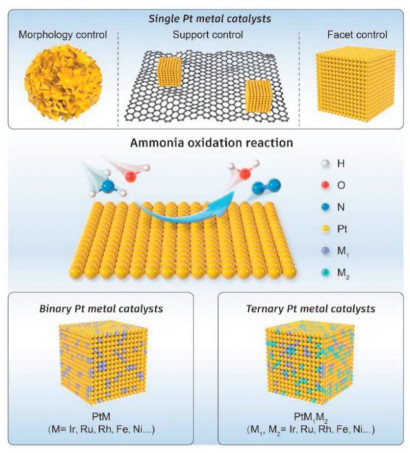
The catalytic activity of metallic Pt largely derives from its high proficiency in facilitating dehydrogenation reactions. The ability to regulate the activity of monometallic Pt has been a focus of research, particularly in terms of the influence of different crystallographic faces. Pt crystals exhibit a variety of facets, each with distinct atomic arrangements that can significantly impact their chemical reactivity. Initially, researchers concentrated on how these different crystal faces affect catalytic AOR behavior, given that the surface structure of a catalyst is crucial for its interaction with reactants. For example, specific crystal faces can provide varying degrees of electronic density and geometric sites, which can enhance or inhibit AOR catalytic activity. For example, Vidal-Iglesias et al. deposited single crystal Pt with CV and tested the ammonia oxidation activity of single crystal Pt in 0.1 mol/L NaOH and 0.5 mmol/L (NH4)2SO4 solutions [43]. They reported that the Pt (100) facet had higher ammonia oxidation activity than did the Pt (110) and Pt (111) facets. Subsequent theoretical calculations revealed that the higher activity of the Pt (100) facet may be because the am-monia oxidation intermediate *NH2 is more stable on the (100) facet than on the other facets, and *N is less likely to remain on the (100) facet. On the basis of previous research results, many researchers have tried various methods for the synthesis of Pt (100)-faceted catalysts. Fu et al. employed a multi-step approach to control the growth and morphology of Pt particles [44]. They utilized l-lysine coordination with Pt(Ⅱ) to regulate the nucleation and growth of Pt particles, effectively controlling their size and shape. Additionally, polyvinylpyrrolidone (PVP) was employed to further modulate the size of the Pt particles, ensuring precise control over their dimensions. Furthermore, researchers have adopted a hydrothermal reduction method in which Pt(Ⅱ) is reduced with formaldehyde under specific hydrothermal conditions. This synthesis technique enabled the preparation of nanosized Pt particles with fixed morphologies and facets. The results obtained from transmission electron microscopy (TEM) and X-ray diffraction (XRD) analysis revealed that the synthesized Pt particles are predominantly composed of (100) facets. In the electrochemical test in 1 mol/L KOH and 0.1 mol/L NH4, with a sweep rate of 10 mV/s, the synthesized Pt nanocuboids exhibited a mass activity of 300.0 mA/mg, which is much greater than that of commercial Pt black (81.3 mA/mg). Compared with that of Pt black, there is also a 25 mV correction for the onset potential. The higher activity of the Pt nanocuboids is attributed to two factors: (1) The low *N adsorption energy of the Pt (100) facet increases the inclination of the catalyst toward N‒N coupling, and (2) the synthesized Pt nanocuboids (3.3 nm) are smaller than Pt black (8.5 nm). In another study, He et al. synthesized Pt particles enriched with (100) facets by ionic liquid-assisted controlled synthesis [45]. Both TEM and XRD characterization revealed that the structure of the synthesized Pt particles (Pt-NCs) was dominated by (100) crystalline facets. With 1 mol/L KOH and 0.1 mol/L NH3·H2O at a sweep rate of 50 mV/s, the synthesized Pt-NCs exhibited a mass activity of 169.86 A/g, which was much greater than that of the Pt control (52.05 A/g) synthesized without an ionic liquid. The current-time (i-t) test demonstrated that the Pt-NCs exhibited superior stability, which can be attributed to the construction of the Pt (100) facet facilitated by the presence of the ionic liquid. Martınez-Rodrıguez et al. also synthesized cubic platinum nanoparticles via another water-in-oil microemulsion method. The shape and surface structure of the synthesized Pt particles were controlled by modulating the type and concentration of acid [46]. The results showed that the Pt nanoparticles obtained with 25% hydrochloric acid were enriched with (100) facets. The number of (100) crystalline facets was characterized by hydrogen adsorption/desorption deconvolution and irreversible germanium adsorption. Pt particles synthesized with 25% HCl exhibited an activity of 1.96 mA/cm2 under 0.2 mol/L NaOH + 0.1 mol/L NH3 at a scanning rate of 10 mV/s, surpassing that of Pt particles synthesized with other acid concentrations. This superior activity can be attributed to the presence of Pt (100) facets in the synthesized particles.
In addition to facet engineering, the morphology of catalyst particles also significantly influences their catalytic activity. Liu et al. demonstrated this by obtaining Pt catalysts with diverse morphologies, including nanosheets and dendrites, through electrochemical deposition techniques [47]. These catalysts exhibited higher catalytic activity than commercial Pt/C, with activity levels nearly double. This enhanced activity can be attributed to several factors: Pt catalysts with nanosheets and dendritic morphologies possess larger surface areas and higher surface-to-volume ratios, leading to increased exposure of active sites and more efficient utilization of Pt atoms for catalytic reactions. Additionally, defects formed during synthesis provide additional active sites and alter the adsorption energies of reaction intermediates, contributing to a more favorable intermediate state adsorption energy and further enhancing the catalytic activity.
The modulation of different supports also has a great effect on the ammonia oxidation activity of the catalysts. Zhou et al. anchored Pt clusters on N-doped porous graphene aerogel by electrodeposition, and the obtained catalyst exhibited a mass activity of 1.77 mA/µg Pt and a specific activity of 0.64 mA/cm2 electrochemical surface area (ECSA) in a 0.1 mol/L NH3, 1 mol/L KOH solution [48]. This is mainly attributed to the uniformly dispersed Pt nanocluster-like morphology, high electrical conductivity, three-dimensional porous network, and N-doped structure in the graphene framework. Moreover, in addition to carbon substrates, metal substrates have also been used to support Pt. Pt monolayers (PtML) supported on nanoparticles with different compositions (i.e., Ru, Rh, Pd, Ir, and Au) were synthesized by Liu et al. [49]. Specifically, Cu was electrodeposited on different noble metal carriers, and Pt was uniformly dispersed on the noble metal surfaces by substitution. The process does not involve the influence of surfactants and binders, and can intuitively reflect the effect of the substrate on Pt. The catalytic activity of ammonia oxidation under 1 mol/L KOH + 0.1 mol/L NH3 at 50 mV/s was in the following order: PtML/Au nanoparticles > PtML/Ir nanoparticles ≈ PtML/Rh nanoparticles > PtML/Ru nanoparticles ≈ PtML/Pd nanoparticles. The authors concluded that the change in the activity of ammonia oxidation comes from the strain effect of the substrate on Pt. As the surface strain of the nanoparticle support on PtML changes from tensile to compressive strain, the specific activity of the PtML electrocatalyst decreases. In addition to the use of precious metals as substrates, non-precious metals have also been investigated. Yang et al. electrodeposited flower-like Pt onto Ni mesh using CV scanning for comparison with Pt particles obtained using constant potential scanning [50]. Under the conditions of 1 mol/L NH3 + 5 mol/L KOH, and a 20 mV/s sweep rate, the catalytic activity of CV-Pt was greater than that of CP-Pt. In addition, the authors also proposed a well-established procedure involving operando gas chromatography and reported that CV-Pt exhibited 90% Faraday efficiency in the range of −0.25~0.35 VAg/AgCl.
The aforementioned monometallic Pt catalysts represent some of the typical examples reported recently, showing advancements in catalytic performance. The performances of these diverse monometallic Pt catalysts are summarized in Table 3 [51-64], which provides a comprehensive overview of their catalytic activities and properties.
DownLoad:
CSV
| Catalyst | Electrolyte | Scan rate (mV/s) | Onset potential | Peak current density (mA/cm2) | Ref. |
| Pt | 0.05 mol/L KOH + 0.1 mol/L NH3 | 20 | – | 4 | [51] |
| Pt/C | 0.2 mol/L NH4OH + 1.0 mol/L KOH | 15 | – | 0.920 | [52] |
| Pt nanoparticle | 0.1 mol/L NH3 + 1.0 mol/L KOH | 10 | −0.4 VHg/HgO | 58 | [53] |
| Pt/C | 1.0 mol/L NH3 + 1.0 mol/L KOH | 20 | −0.46 VSHE | 6.150 | [54] |
| Pt cubesH-S | 0.5 mol/L NH4OH + 0.5 mol/L KOH | 100 | 0.450 VRHE | 5.240 | [55] |
| PtR | 1 mol/L NH3 + 1 mol/L KOH | 10 | – | 0.400 | [56] |
| Colloidal Pt | 0.1 mol/L NH3 + 0.2 mol/L NaOH | 10 | – | 1.35 | [57] |
| er-GOx/Pt hydrid materials | 1.0 mol/L NH4OH | 10 | −0.3 VAg/AgCl | 0.255 | [58] |
| Pt decorated Ni | 0.1 mol/L NH3 + 1.0 mol/L KOH | 10 | – | 0.75 | [59] |
| Pt nanoparticles | 0.1 mol/L NH3 + 1.0 mol/L KOH | 10 | – | 0.8 | [60] |
| Ptseed | 0.1 mol/L NaOH +0.1 mol/L NH3 | 20 | – | 0.53 | [61] |
| Dendritic Pt | 0.1 mol/L NaOH +0.03 mol/L NH3 | 50 | −0.55 VSCE | – | [62] |
| Pt/NGA-800 | 1 mol/L NH3 + 1 mol/L KOH | – | – | 27.5 | [63] |
| Pt/ITO | 0.1 mol/L NH3 + 1.0 mol/L KOH | 10 | – | – | [64] |
After resolving the mechanism of ammonia oxidation by monometallic Pt as a model catalyst, the adoption of a second metal to modulate the electronic structure of Pt to achieve superior catalytic activity for ammonia oxidation has become the choice of many researchers. Vooys et al. obtained the adsorption energy of different precious metals for *N by density functional theory (DFT) calculations [65]. According to theoretical calculations, composites composed of Ir and Pt exhibit lower adsorption energies for *N species than do pure Pt catalysts [66]. This observation positions these Ir-Pt alloys at the peak of the volcano curve, indicating their optimal catalytic performance for AOR. As a result, by meticulously adjusting the atomic ratios of Pt and Ir, Moran et al. discovered that a Pt: Ir ratio of 70:30 indeed results in a lower initial potential than that of a monometallic Pt catalyst. This observation indicates that the incorporation of Ir into the Pt catalyst can significantly enhance its intrinsic activity [33]. In addition to Ir, researchers have also tested other metals. Endo et al. prepared PtIr, PtRu, and PtNi alloys, and reported that the incorporation of Ir and Ru resulted in a lower onset potential of the AOR; in contrast, the addition of Ni did not significantly improve the activity. This difference in catalytic behavior was attributed to the role of Ir and Ru in facilitating the dehydrogenation of NH3 on Pt, making the reaction more favorable and resulting in improved catalytic performance [67]. In another study, Sacre et al. deposited a layer of PtIr atoms on a MgO substrate by laser pulsing at 600 ℃ [68]. Adjusting the atomic ratios of Ir and Pt has led to the development of an alloy with an optimal 25% atomic ratio of Ir, which exhibits the best AOR performance. This strategic incorporation of Ir into Pt results in a 0.7% reduction in the in-plane lattice parameter of the Pt (100) surface structure. Such a contraction of the lattice significantly lowers the energy barrier for N—N coupling during ammonia oxidation. In another study, Assumpcão et al. synthesized PtRh alloys with different atomic ratios (90:10, 70:30, and 50:50) using the sodium borohydride reduction method [69]. The XRD results revealed the formation of PtRh alloys, whereas the TEM results revealed particle sizes between 4.1 nm and 4.5 nm. The experimental results for catalysts tested for electrocatalytic AOR demonstrated that doping with Rh effectively lowered the onset potential. However, it was observed that the peak current of the catalysts decreased with an increasing proportion of Rh atoms. This phenomenon can be attributed to the lower potential required for the dehydrogenation of ammonia on Rh than on Pt and Ir. Additionally, the adsorption energy of the *N intermediate on Rh is considerably greater than that on Pt, which might hinder the overall reaction kinetics despite the initial lower onset potential. Using the same method, Silva et al. obtained PtAu alloys with different atomic ratios (50:50 and 70:30) [70]. XRD analyses confirmed the formation of these PtAu alloys, whereas TEM revealed particle sizes ranging from 5.8 nm to 6.4 nm. In particular, the PtAu/C alloy at a 70:30 ratio exhibited a notably high peak current during AOR. This superior performance is likely due to the inertness of Au toward the *N intermediate, which when alloyed with Pt, optimizes the electronic environment of Pt, enhancing its interaction with *N and thereby catalyzing a more effective AOR.
Since Ir, Ru, and Au are also noble metals, researchers have turned their attention to cheaper transition metals. Chan et al. synthesized Pt-M (M = Fe, Co, Ni, Zn) by a wet chemistry method [71]. The order of ammonia oxidation activity in the 0.1 mol/L NH3 + 0.1 mol/L KOH solution was PtZn > PtFe > PtCo > PtNi > Pt/C. The authors concluded that the doping of transition metals reduced the oxophilicity of Pt, which weakened the adsorption of hydroxyl groups on the catalyst surface, providing favorable conditions for Pt adsorption of NH3 for oxidation.
Recent reports have highlighted the emergence of more typical binary Pt alloy catalysts. The performances of these additional binary Pt alloy catalysts are detailed in Table 4 [72-87].
DownLoad:
CSV
| Catalyst | Electrolyte | Scan rate (mV/s) | Onset potential | Peak current density (mA/cm2) | Ref. |
| PtIr/C | 1 mol/L NH3 | 10 | 0.470 VRHE | – | [72] |
| Pt/Ir/MWCNT nanoparticle | 0.1 mol/L NH3 + 1.0 mol/L KOH | 10 | – | – | [73] |
| Pt90Ru10/C | 1.0 mol/L NH4OH + 1.0 mol/L KOH | 20 | – | 0.92 | [74] |
| Pt7Pd3 | 0.5 mol/L NH4OH + 1.0 mol/L KOH | 30 | −0.490 VRHE | 7 | [75] |
| PtIr | – | 5 | – | 110 | [76] |
| RGO/PtIr | 1.0 mol/L NH4OH | 10 | − 0.4 VAg/AgCl | 20 | [77] |
| PtRu | 0.1 mol/L NH3 + 1.0 mol/L KOH | 20 | 0.3 VRHE | 1.8 | [78] |
| Pt90Ir10 | 0.08 mol/L NH3 + 1.0 mol/L KOH | 20 | – | 0.4 | [79] |
| SnO2-Pt/C | 0.1 mol/L NH3 + 0.1 mol/L KOH | 20 | 0.45 VRHE | 1.62 | [80] |
| PtNi | 0.1 mol/L NH3 + 1.0 mol/L KOH | 10 | – | 1.1 | [81] |
| PtNi | 0.1 mol/L NH3 + 1.0 mol/L KOH | 5 | – | 0.8 | [82] |
| Pt0.8Ir0.2 | 0.1 mol/L NH3 + 1.0 mol/L KOH | 1 | – | 0.038 | [83] |
| Pt6Ru-NCs | 1.0 mol/L NH3 + 1.0 mol/L KOH | 50 | 0.5 VRHE | – | [84] |
| Pt-Zn/C nanocubes | 0.1 mol/L NH3 + 1.0 mol/L KOH | 50 | – | – | [85] |
| PtxIr100-x/MgO | 0.1 mol/L NH3 + 0.2 mol/L NaOH | 20 | 0.53 VRHE | 1 | [86] |
| Pt/PBI/MWNT-CeO2 | 0.1 mol/L NH3 + 1.0 mol/L KOH | 20 | – | – | [87] |
The incorporation of a second metal into Pt catalysts has been shown to significantly enhance catalytic activity. Motivated by these improvements, researchers are increasingly exploring the addition of multiple metals to form complex alloys with Pt. Lin et al. prepared hyperbranched concave octahedral dendrites of PtIr-Cu (HCOND) with high-index crystalline facets using a solvent thermal method [88]. The effects of the surfactant (CTAB) and solvent ratio (DMA and OAM) on the morphology were explored to determine the exposure of different crystalline facets. The experimental results show that CTAB adsorbs more easily on the low-index facet, thus guiding crystal growth toward the high-index facet. Under the conditions of 0.1 mol/L NH3 and 1.0 mol/L KOH at 5 mV/s, PtIrCu HCOND has a peak current density of 122.9 A/gPtIr, which is higher than that of PtIrCu NPs (109.8 A/gPtIr), PtIr NCs (61.5 A/gPt), PtCu NCs (26.1 A/gPt) and commercial Pt/C (53.1 A/gPt) and has an extremely low onset potential of 0.35 VRHE, which is 60 mV, 80 mV, 150 mV and 160 mV lower than that of PtIrCu NPs, PtIr NCs, PtCu NCs and commercial Pt/C. The authors concluded that the lattice distortion induced by Ir and Cu doping resulted in smoother ammonia oxidative dehydrogenation and thus improved the catalytic activity. Wu et al. synthesized PtIrNi nanoparticles (PtIrNi/SiO2CNT-COOH) uniformly dispersed on a binary composite carrier consisting of porous silica (SiO2) and carboxylated functionalized carbon nanotubes (CNT-COOH) by an ultrasound-assisted method [89]. TEM characterization revealed that the ternary PtIrNi alloy was homogeneously dispersed on SiO2, and that SiO2 was tightly bound to the carboxylated carbon nanotubes. The CNT-SiO2 support has a large specific surface area and an abundant mesoporous structure, which is conducive to increasing the number of active sites and mass transfer. XPS characterization revealed the electronic interactions between Pt, Ir, and Ni and the electron transfer between SiO2 and CNTs and Pt. In multiple aspects, PtIrNi1/SiO2CNT-COOH has a ~0.40 V onset potential and a peak current density of 124.0 A/g in 1.0 mol/L KOH and 0.1 mol/L NH3 at 5 mV/s and 900 rpm. The authors also modulated other variables, such as the replacement of SiO2 and CNT-COOH carriers with CeO2 and XC-72, the atomic ratios of different Pt, Ir, and Ni, and the comparison of the performances illustrated the importance of the modifications of SiO2, CNT-COOH, Ir, and Ni for the electronic modulation of Pt. DFT calculations further confirmed that the increase in the AOR activity of the PtIrNi alloys is due to the lower energy barrier for dehydrogenation from *NH2 to *NH, whereas dimerization of *NH is thermodynamically and kinetically more favorable. Electronic structure analysis revealed that the energy of the d-orbital population of the density of states shifts upward (−1.97 to −1.87 eV) when Ni is introduced to the Pt-Ir surface. Following previous work, Wu's group prepared ternary PtIrZn nanoparticles with an average particle size of 2.3 ± 0.2 nm by an ultrasound-assisted method that were highly dispersed on a binary composite support consisting of porous CeO2 and carboxyl-rich carbon nanotubes derived from ZIF-8 [90]. Compared with CNT-COOH as a support, the carbon support derived from ZIF-8 has a greater specific surface area, and CeO2, rather than SiO2, disperses the metal nanoparticles more homogeneously. The synthesized catalyst (PtIrZn/CeO2-ZIF-8) has a peak current density of 31.8 A/g and an onset potential of 0.345 V under the test conditions of 1.0 mol/L KOH with 0.1 mol/L NH3 at 900 rpm and 5 mV/s, which are superior to those of commercially available PtIr/C (jp = 25.1 A/g and Eonset = 0.428 V) under the same conditions. From their calculations, the authors concluded that the addition of Zn atoms to the PtIr alloy modulates the electronic structure of Pt-Ir on the (100) facet, and facilitates the dehydrogenation of NH3. Although Ir is recognized as a good metal for modifying Pt, Ir is more costly than Pt. Therefore, some studies have focused on the development of Ir-free catalysts for ammonia oxidation. Xin et al. employed machine learning techniques to construct ternary Pt alloys and predict their catalytic performance in ammonia oxidation. This study utilized adsorption descriptors of *N to establish correlations with catalytic activity, allowing for the efficient screening of potential catalysts based on their predicted performance metrics [91]. The Pt3Ru1/2Co1/2 catalyst, which matched the prediction, exhibited a mass activity of 174.0 A/g and an onset potential of 0.42 V under practical testing conditions of 1.0 mol/L KOH with 0.1 mol/L NH3 at 900 rpm and 5 mV/s. The synergistic application of theoretical predictions and experimental methodologies has substantially enhanced the performance of catalysts, thereby significantly reducing the cycle time required for traditional catalyst design. This acceleration in development is pivotal for rapid innovation in catalytic technologies. As computational capacities continue to advance and as research into high-entropy alloy materials progresses, we are poised to witness the emergence of breakthrough catalysts.
In the field of ammonia oxidation on Pt-based catalysts, a variety of studies have assessed the catalytic efficiency of commercially available Pt/C and PtIr/C catalysts. Notably, these investigations have yielded divergent activity levels. Such discrepancies in the results highlight the critical need for standardized methodologies in the evaluation of these catalysts. Establishing uniform testing protocols is essential to accurately discern the underlying variables that influence catalytic performance, thus facilitating more consistent and reliable research outcomes. Here, we discuss the testing paradigm in the hope that it will be useful in the future for different reference comparisons of electrocatalytic ammonia oxidation (Fig. 6).
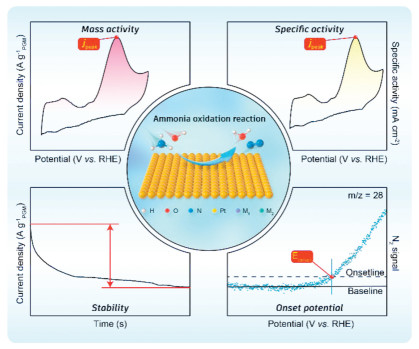
The electrode and electrolyte represent the two critical components in the testing conditions for the electrocatalytic AOR. Electrodes are categorized into glassy carbon electrodes (GCEs) and self-supported electrodes. Self-supported electrodes allow for a higher loading of active material, which is beneficial for scalability and practical applications. Nevertheless, they often suffer from less controlled surface properties, which can lead to variability in catalytic activity and complicate the interpretation of electrochemical data. On the other hand, GCEs are widely favored for their chemical inertness and high conductivity, which make them excellent substrates for a broad range of electrochemical studies. The smooth and homogeneous surface of GCEs ensures the uniform deposition of catalyst materials, leading to reproducible results. Additionally, GCEs are suitable for rotating disk electrode (RDE) tests, which are critical for evaluating the kinetics and mechanism of electrocatalytic reactions under controlled hydrodynamic conditions.
For electrolytes, it is important to note that Pt-based catalysts are active only in alkaline environments, under these conditions, ammonia usually undergoes ionization equilibrium. Consequently, the concentration of KOH in the electrolyte not only influences the shift in the RHE but also affects the concentration of ammonia molecules within the solution. Additionally, variability in ammonia sources is a common observation in the scientific literature, as the ammonia concentration itself can change during prolonged storage. To achieve more accurate electrolyte compositions, we recommend the use of NH4Cl or (NH4)2SO4 as ammonia sources in conjunction with KOH. Additionally, the ammonia oxidation reaction is notably temperature sensitive, with its activity increasing as the temperature increases. To minimize errors associated with temperature fluctuations, it is advisable to conduct experiments over short intervals. To standardize the testing procedures, these conditions are explicitly detailed in the specific test parameters provided below.
Determining the onset potential is a pivotal step in evaluating the activity of catalysts for AOR. The onset potential is the voltage at which a noticeable current begins to flow, marking the initiation of electrochemical activity. The onset potential is an important index of catalyst activity and can objectively describe the direct activity of a catalyst. Most of the articles chose to read the value of the onset potential from the CV curve [54, 55, 78, 89]. To enhance the accuracy of these measurements and to capture the intrinsic catalytic activity more reliably, we propose the use of DEMS to ensure that the intrinsic catalytic activity of the catalysts can be obtained. For the purpose of ensuring consistency and enabling meaningful comparisons, it is recommended that 1 mol/L KOH with 0.1 mol/L NH4Cl (or 0.05 mol/L (NH4)2SO4) was used as the reaction electrolyte, and the scanning rate was 5 mV/s for such comparison.
Mass activity and specific activity are important measures of catalyst performance. Mass activity measures the catalytic activity per unit mass of the catalyst and is particularly useful in assessing the overall efficiency of the catalyst from an economic perspective. Comparing the effectiveness of different catalysts is beneficial when cost reduction is a priority. However, mass activity can sometimes be misleading if the catalysts under comparison have significantly different densities or if the distribution of active sites within the catalyst material is uneven. On the other hand, specific activity refers to the activity per unit surface area of the catalyst, with a focus on the intrinsic efficiency of the active sites themselves. This metric is advantageous for understanding fundamental catalytic processes at the molecular level and for identifying how modifications at the atomic scale affect catalytic performance. In Pt-based catalysts, mass activity is obtained by the ratio of current to mass, and the mass is obtained by inductively coupled plasma mass spectrometry (ICP-MS), while the specific activity of the catalyst is obtained by normalizing the electrochemical surface area (ECSA). CV is a widely used electrochemical technique to determine the ECSA of catalysts, However, these adsorption peaks are not always specific to the active sites of interest. Thus, we advocate that the electrochemical surface area of Pt-based catalysts be measured by CO stripping or Hupd. The ECSA was calculated by the following equation [92, 93].
|
|
(1) |
|
|
(1) |
where S represents the measured Hupd charge, and mPt is the mass of the as-loaded Pt. These two test methods have different degrees of accuracy when different Pt-based catalysts are tested. Specifically, taking PtM alloy as an example, when using Hupd method, the empirical value of the charge density on the surface of M will be greater than 2.1 C/cm2 when M is partially precious metal, and less than 2.1 C/cm2 when M is mostly non-precious metal. In addition, the PtM alloy catalyst blurs the absorption H desorption peaks, which leads to inaccuracies in the calculation of the area [94]. Pt alloying also alters the empirical constant of the charge density on the catalyst surface in CO-stripping tests. However, the greater adsorption capacity of Pt for CO has a minimal impact on the peak area, making the measurement of the ECSA using CO stripping more accurate for platinum-alloyed catalysts [95].
For both metrics, a CV scan needs to be included, taking the peak current to mPt or the SECSA ratio. The peak current density reflects the highest ammonia oxidation conversion rate and is directly related to the device power. In the powder system, we suggest using the test method of Wu et al. with 1 mol/L KOH and 0.1 mol/L NH4Cl (or 0.05 mol/L (NH4)2SO4) as the reaction electrolyte, a scanning rate of 5 mV/s, and a rotational speed of 900 rpm [41]. The rotational speed not only eliminates the bubbles in time away from the surface of the catalyst but also accelerates mass transfer. Therefore, in self-supported electrodes, we suggest a higher NH3 concentration to accelerate the mass transfer process of NH3 molecules; for example, Yang et al. utilized 5 mol/L KOH + 1 mol/L NH3 as the electrolyte with 5 mV/s as the scanning rate [50].
Stability plays a crucial role in the practical application of catalysts. The dissolution of the catalyst and the poisoning effect can severely limit the stability of the catalyst. For electrocatalytic ammonia oxidation, the test of stability is mainly divided into chronopotentiometry and CV scanning of multiple loops. However, the use of multiple CV scans to compare changes in peak current density does not accurately reflect the stability of Pt-based catalysts. This is because, during the CV cycles, the poisoned species adsorbed on the Pt-based catalyst at high potentials are reduced in the low potential range [37]. However, under operational conditions, the potentials are consistently maintained at high levels, and such a detoxification process at low potentials is absent. Therefore, we recommend the use of chronopotentiometry as a more appropriate metric for assessing stability.
Common errors and mistakes in electrochemical performance testing often arise during the calculation of the onset potential and specific activity. For the onset potential, the use of ammonia as the reaction electrolyte can lead to inaccuracies because prolonged storage results in ammonia volatilization, which reduces its concentration. Additionally, the pH of the electrolyte is prone to errors due to the ionization equilibrium of ammonia in solution, necessitating real-time pH measurements during each test. When calculating specific activity, the determination of the ECSA for Pt-based catalysts is typically performed via Hupd or CO stripping. However, for Pt-alloy catalysts, CO stripping is preferred for ECSA measurement because the empirical constant in Hupd is influenced by alloying, which blurs the adsorption and desorption peaks. In contrast, CO stripping offers a more accurate measurement of the ECSA for Pt-alloy catalysts.
In this review, we provide a comprehensive summary of the advancements in the mechanisms of AORs and discuss various methods for detecting reaction intermediates. We explore the evolution of Pt-based catalysts, tracing their development from monometallic forms to complex multimetallic alloys, and examine the influences of facets, morphology, and support structures. Despite years of development, Pt-based catalysts still face significant challenges that hinder their commercial application, particularly concerning overpotential and stability. To address these issues, we propose several avenues for improvement aimed at accelerating the advancement of AOR technologies. These suggestions are intended to refine catalyst design and enhance operational efficacy, thereby bridging the gap between laboratory research and industrial application.
For the electrocatalysis mechanism of ammonia oxidation, although most studies follow the G-M theory, very few studies have detected the presence of intermediates by direct characterization methods. As discussed in the Chapter Mechanisms of AOR, developing more sensitive detection methods is crucial for capturing the transient signals of these intermediates. Such advancements would not only refine our understanding of the reaction mechanism but also enhance the reproducibility and validity of the experimental results. Furthermore, the issue of catalyst poisoning in Pt-based systems represents a critical bottleneck in the application of these catalysts. Although theoretical calculations point to *N as the primary poisoning species, the lack of direct detection methods impedes our ability to confirm these findings empirically. This gap suggests a potential misalignment between theoretical predictions and practical outcomes, which could be addressed by integrating more sophisticated analytical techniques in experimental setups. Advancements in this area could provide invaluable insights into the dynamics of catalyst poisoning, thereby informing the design of more durable and efficient catalysts.
After years of development, Pt-based catalysts have undergone extensive design modifications, including adjustments in facets, structure, support, and alloying. Despite these advancements, they still fall short in overcoming the challenges of catalyst poisoning and high overpotential. It is imperative that future design strategies incorporate machine learning techniques to enhance the efficiency of catalyst screening. Presently, a significant gap exists, as few reported catalysts leverage in situ characterization to fundamentally optimize N—N coupling. This disconnect between performance and mechanistic understanding has led to an unclear direction in the design of AOR catalysts. To address these deficiencies, future catalyst designs should integrate in situ characterization with theoretical calculations. This combined approach will not only elucidate the underlying mechanisms at play but also guide the development of more effective and resilient catalyst systems.
The advancement of AORs is fundamentally linked to the performance of catalysts, making their characterization a pivotal aspect of research in this field. Currently, the lack of specific evaluation standards and uniform benchmarking protocols impedes effective peer comparisons and hinders the field's progress. Establishing rigorous, universally accepted metrics and testing procedures is crucial for enabling accurate assessments of catalyst performance. Such standards would not only facilitate meaningful comparisons across different studies but also drive the development of more efficient and robust catalysts. The implementation of these benchmarks is also essential for advancing our understanding and optimization of ammonia oxidation processes.
The three-electrode system is quite helpful in elucidating both the mechanism and the intrinsic activity of catalysts in electrocatalytic studies. However, the ultimate application of these catalysts in ammonia oxidation fuel cells necessitates their integration as anodes in a full-cell configuration. This transition from half-cell to full-cell setups reveals a significant research gap that must be addressed through the development of innovative device assemblies. To overcome these challenges and optimize ammonia fuel cells for commercial viability, further research is needed to develop robust catalysts that can operate effectively at higher scales and in more complex systems. Like methanol fuel cells, ammonia fuel cells integrate the catalyst onto the membrane through a membrane electrode assembly to reduce mass and charge transfer resistance. However, ammonia fuel cells operate in more alkaline environments, placing greater demands on the stability of the electrode materials. Beyond developing high-activity catalysts, effective electrolyte flow channel design and efficient hydrothermal management are also crucial for improving ammonia transport and reaction efficiency, thereby enhancing cell performance. For the stability of the devices, the design of excellent anti-poisoning catalysts is the key to the long operation of the cell. Furthermore, indirect ammonia oxidation fuel cells can also be considered as an option, where ammonia is first converted to hydrogen through an electrolytic cell, and the resulting hydrogen is then used to assemble a hydrogen-oxygen fuel cell. An et al. pioneered a series of assembly and characterization methods for direct ammonia fuel cells (DAFCs), providing a valuable framework for future efforts to construct full-cell systems using subsequent catalysts [96]. From a broader perspective, the integration of such ammonia fuel cells into the energy system could significantly contribute to decarbonization efforts.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Youpeng Wang: Writing – review & editing, Writing – original draft, Conceptualization. Yuan Ji: Writing – review & editing, Visualization, Conceptualization. Chengbo Li: Writing – review & editing, Visualization, Validation. Zhaoyang Chen: Writing – review & editing, Visualization. Xu Li: Writing – review & editing. Tingting Zheng: Writing – review & editing. Qiu Jiang: Writing – review & editing, Supervision, Conceptualization. Chuan Xia: Writing – review & editing, Writing – original draft, Supervision, Conceptualization.
Xia acknowledges the National Key Research and Development Program of China (No. 2022YFB4102000), the National Natural Science Foundation of China (Nos. 22102018 and 52171201) and the Huzhou Science and Technology Bureau (No. 2022GZ45). Q. Jiang acknowledges the China Postdoctoral Science Foundation-Funded Project (No. 2022M710601), the Huzhou Science and Technology Bureau (No. 2023GZ02), and the Natural Science Foundation of Sichuan Province (No. 24NSFSC5779). T. Zheng acknowledges the National Natural Science Foundation of China (Nos. 22322201 and 22278067) and the Natural Science Foundation of Sichuan Province (No. 2023NSFSC0094).
X. Ding, Y. Ji, H. Huang, et al., Chem. Catal. 4 (2024) 100932.
S.A. Lee, M.G. Lee, H.W. Jang, Sci. China Mater. 65 (2022) 3334–3352. doi: 10.1007/s40843-022-2111-2
Z.H. Lyu, J. Fu, T. Tang, J. Zhang, J.S. Hu, EnergyChem 5 (2023) 100093. doi: 10.1016/j.enchem.2022.100093
Y. Zhao, B.P. Setzler, J. Wang, et al., Joule 3 (2019) 2472–2484. doi: 10.1016/j.joule.2019.07.005
H. Kim, S. Hong, H. Kim, et al., Appl. Mater. Today 29 (2022) 101640. doi: 10.1016/j.apmt.2022.101640
A.I. Osman, N. Mehta, A.M. Elgarahy, et al., Environ. Chem. Lett. 20 (2022) 153–188. doi: 10.1007/s10311-021-01322-8
S. Sun, Q. Jiang, D. Zhao, et al., Renew. Sustain. Energy Rev. 169 (2022) 112918. doi: 10.1016/j.rser.2022.112918
Z. Xia, X. Zhang, H. Sun, S. Wang, G. Sun, Nano Energy 65 (2019) 104048. doi: 10.1016/j.nanoen.2019.104048
T.Y. Burshtein, Y. Yasman, L. Muñoz-Moene, J.H. Zagal, D. Eisenberg, ACS Catal. 14 (2024) 2264–2283. doi: 10.1021/acscatal.3c05657
W.S. Chai, Y. Bao, P. Jin, G. Tang, L. Zhou, Renew. Sustain. Energy Rev. 147 (2021) 111254. doi: 10.1016/j.rser.2021.111254
F. Valentini, A. Marrocchi, L. Vaccaro, Adv. Energy Mater. 12 (2022) 2103362. doi: 10.1002/aenm.202103362
N.M. Adli, H. Zhang, S. Mukherjee, G. Wu, J. Electrochem. Soc. 165 (2018) J3130–J3147. doi: 10.1149/2.0191815jes
B. Lee, L.R. Winter, H. Lee, et al., ACS Energy Lett. 7 (2022) 3032–3038. doi: 10.1021/acsenergylett.2c01615
C.H. Christensen, T. Johannessen, R.Z. Sørensen, J.K. Nørskov, Catal. Today 111 (2006) 140–144. doi: 10.1016/j.cattod.2005.10.011
F. Jiao, B. Xu, Adv. Mater. 31 (2019) 1805173. doi: 10.1002/adma.201805173
A. Yapicioglu, I. Dincer, Renew. Sustain. Energy Rev. 103 (2019) 96–108. doi: 10.1016/j.rser.2018.12.023
X. Fu, V.A. Niemann, Y. Zhou, et al., Nat. Mater. 23 (2024) 101–107. doi: 10.1038/s41563-023-01702-1
N. Salmon, R. Bañares-Alcántara, Nat. Synth. 2 (2023) 604–611. doi: 10.1038/s44160-023-00309-3
J. Huang, Z. Chen, J. Cai, et al., Nano Res. 15 (2022) 5987–5994. doi: 10.1007/s12274-022-4279-5
G. Jeerh, M. Zhang, S. Tao, J. Mater. Chem. A 9 (2021) 727–752. doi: 10.1039/d0ta08810b
O. Siddiqui, I. Dincer, Fuel Cells 18 (2018) 379–388. doi: 10.1002/fuce.201800052
Y. Liu, Z. Pan, O.C. Esan, et al., J. Power Sources 570 (2023) 233057. doi: 10.1016/j.jpowsour.2023.233057
E. Muller, F. Spitzer, Z. Elektrochem. Angew. Phys. Chem. 11 (1905) 917–931. doi: 10.1002/bbpc.19050115002
H.G. Oswin, M. Salomon, Can. J. Chem. 41 (1963) 1686–1694. doi: 10.1139/v63-243
H. Gerischer, A. Mauerer, J. Electroanal. Chem. 25 (1970) 421–433. doi: 10.1016/S0022-0728(70)80103-6
F.J. Vidal-Iglesias, J. Solla-Gullón, J.M. Pérez, A. Aldaz, Electrochem. Commun. 8 (2006) 102–106. doi: 10.1016/j.elecom.2005.11.002
T. Matsui, S. Suzuki, Y. Katayama, et al., Langmuir 31 (2015) 11717–11723. doi: 10.1021/acs.langmuir.5b02330
Y. Katayama, T. Okanishi, H. Muroyama, T. Matsui, K. Eguchi, J. Catal. 344 (2016) 496–506. doi: 10.1016/j.jcat.2016.10.020
J.F.E. Gootzen, A.H. Wonders, W. Visscher, R.A. Van Santen, J.A.R. Van Veen, Electrochim. Acta 43 (1998) 1851–1861. doi: 10.1016/S0013-4686(97)00285-5
K. Nakajima, H. Toda, K. Sakata, Y. Nishibayashi, Nat. Chem. 11 (2019) 702–709. doi: 10.1038/s41557-019-0293-y
D.W. McKee, A.J. Scarpellino, I.F. Danzig, M.S. Pak, J. Electrochem. Soc. 116 (1969) 562. doi: 10.1149/1.2411963
S. Wasmus, E.J. Vasini, M. Krausa, H.T. Mishima, W. Vielstich, Electrochim. Acta 39 (1994) 23–31. doi: 10.1016/0013-4686(94)85006-2
E. Moran, C. Cattaneo, H. Mishima, et al., J. Solid State Electrochem. 12 (2008) 583–589. doi: 10.1007/s10008-007-0407-0
W. Peng, L. Xiao, B. Huang, L. Zhuang, J. Lu, J. Phys. Chem. C 115 (2011) 23050–23056. doi: 10.1021/jp2081148
D.A. Finkelstein, E. Bertin, S. Garbarino, D. Guay, J. Phys. Chem. C 119 (2015) 9860–9878. doi: 10.1021/acs.jpcc.5b00949
H. Kim, M.W. Chung, C.H. Choi, Electrochem. Commun. 94 (2018) 31–35. doi: 10.4068/cmj.2018.54.1.31
H. Kim, W. Yang, W.H. Lee, et al., ACS Catal. 10 (2020) 11674–11684. doi: 10.1021/acscatal.0c02413
I. Katsounaros, M.C. Figueiredo, F. Calle-Vallejo, et al., J. Catal. 359 (2018) 82–91. doi: 10.1016/j.jcat.2017.12.028
A.C.A. de Vooys, M.F. Mrozek, M.T.M. Koper, et al., Electrochem. Commun. 3 (2001) 293–298. doi: 10.1016/S1388-2481(01)00156-4
Y. Katayama, T. Okanishi, H. Muroyama, T. Matsui, K. Eguchi, ACS Catal. 6 (2016) 2026–2034. doi: 10.1021/acscatal.6b00108
K. Endo, Y. Katayama, T. Miura, Electrochim. Acta 50 (2005) 2181–2185. doi: 10.1016/j.electacta.2004.09.024
K. Siddharth, P. Alam, M.D. Hossain, et al., J. Am. Chem. Soc. 143 (2021) 2433–2440. doi: 10.1021/jacs.0c13178
F.J. Vidal-Iglesias, N. Garcı́a-Aráez, V. Montiel, J.M. Feliu, A. Aldaz, Electrochem. Commun. 5 (2003) 22–26. doi: 10.1016/S1388-2481(02)00521-0
G. Fu, C. Liu, R. Wu, et al., J. Mater. Chem. A 2 (2014) 17883–17888. doi: 10.1039/C4TA03601H
S. He, Z. Wu, S. Li, J. Lee, Int. J. Hydrog. Energy 41 (2016) 1990–1996. doi: 10.1016/j.ijhydene.2015.10.069
R.A. Martı́nez-Rodrı́guez, F.J. Vidal-Iglesias, J. Solla-Gullón, C.R. Cabrera, J.M. Feliu, J. Am. Chem. Soc. 136 (2014) 1280–1283. doi: 10.1021/ja411939d
J. Liu, C. Zhong, Y. Yang, et al., Int. J. Hydrog. Energy 37 (2012) 8981–8987. doi: 10.1016/j.ijhydene.2012.03.015
Y. Zhou, G. Zhang, M. Yu, et al., ACS Sustain. Chem. Eng. 6 (2018) 8437–8446. doi: 10.1021/acssuschemeng.8b00586
J. Liu, B. Liu, Y. Wu, et al., Catalysts 9 (2019) 4.
Y. Yang, J. Kim, H. Jo, et al., J. Mater. Chem. A 9 (2021) 11571–11579. doi: 10.1039/d1ta00363a
J. Gwak, M. Choun, J. Lee, ChemSusChem 9 (2016) 403–408. doi: 10.1002/cssc.201501046
K.R. Lee, D. Song, S.B. Park, J. Han, RSC Adv. 4 (2014) 5638–5641. doi: 10.1039/c3ra44057e
N. Hanada, Y. Kohase, K. Hori, H. Sugime, S. Noda, Electrochim. Acta 341 (2020) 136027. doi: 10.1016/j.electacta.2020.136027
Z. Li, Y. Wang, G.G. Botte, Electrochim. Acta 228 (2017) 351–360. doi: 10.1016/j.electacta.2017.01.020
H. Jin, S. Lee, Y. Sohn, et al., Int. J. Energy Res. 45 (2021) 18281–18291. doi: 10.1002/er.6988
S. Johnston, B.H.R. Suryanto, D.R. MacFarlane, Electrochim. Acta 297 (2019) 778–783. doi: 10.1016/j.electacta.2018.12.014
J. Solla-Gullón, F.J. Vidal-Iglesias, P. Rodríguez, et al., J. Phys. Chem. B 108 (2004) 13573–13575. doi: 10.1021/jp0471453
L. Cunci, C.A. Velez, I. Perez, et al., ACS Appl. Mater. Interfaces 6 (2014) 2137–2145. doi: 10.1021/am4052552
J. Liu, B. Chen, Y. Kou, et al., J. Mater. Chem. A 4 (2016) 116–1168.
J. Liu, X. Du, Y. Yang, et al., Electrochem. Commun. 58 (2015) 6–10. doi: 10.1016/j.elecom.2015.05.015
J. Galipaud, C. Roy, M.H. Martin, et al., ChemElectroChem 2 (2015) 1187–1198. doi: 10.1002/celc.201500045
J. Ye, J. Lin, Z. Zhou, et al., J. Electroanal. Chem. 819 (2018) 495–501. doi: 10.1016/j.jelechem.2017.12.062
Y. Zhou, G. Zhang, M. Yu, et al., ACS Sustain. Chem. Eng. 6 (2018) 8437–8446. doi: 10.1021/acssuschemeng.8b00586
S. Li, H. Chen, J. Liu, et al., ACS Appl. Mater. Interfaces 9 (2017) 27765–27772. doi: 10.1021/acsami.7b08604
A.C.A. de Vooys, M.T.M. Koper, R.A. van Santen, J.A.R. van Veen, J. Electroanal. Chem. 506 (2001) 127–137. doi: 10.1016/S0022-0728(01)00491-0
A.O. Elnabawy, J.A. Herron, S. Karraker, M. Mavrikakis, J. Catal. 397 (2021) 137–147. doi: 10.1016/j.jcat.2021.03.010
K. Endo, K. Nakamura, Y. Katayama, T. Miura, Electrochim. Acta 49 (2004) 2503–2509. doi: 10.1016/j.electacta.2004.01.025
N. Sacré, M. Duca, S. Garbarino, et al., ACS Catal. 8 (2018) 2508–2518. doi: 10.1021/acscatal.7b02942
M.H.M.T. Assumpção, R.M. Piasentin, P. Hammer, et al., Appl. Catal. B 174-175 (2015) 136–144. doi: 10.1016/j.apcatb.2015.02.021
J.C.M. Silva, S.G. Silva, R.F.B. De Souza, et al., Appl. Catal. A 490 (2015) 133–138. doi: 10.1016/j.apcata.2014.11.015
Y.T. Chan, K. Siddharth, M. Shao, Nano Res. 13 (2020) 1920–1927. doi: 10.1007/s12274-020-2712-1
B. Achrai, Y. Zhao, T. Wang, et al., J. Electrochem. Soc. 167 (2020) 134518. doi: 10.1149/1945-7111/abbdd1
S. Morita, E. Kudo, R. Shirasaka, et al., J. Electroanal. Chem. 762 (2016) 29–36. doi: 10.1016/j.jelechem.2015.12.017
J.C.M. Silva, S. Ntais, E. Teixeira-Neto, et al., Int. J. Hydrog. Energy 42 (2017) 193–201. doi: 10.1016/j.ijhydene.2016.09.135
T.L. Lomocso, E. Baranova, Electrochim. Acta 56 (2011) 8551–8558. doi: 10.1016/j.electacta.2011.07.041
B.K. Boggs, G.G. Botte, Electrochim. Acta 55 (2010) 5287–5293. doi: 10.1016/j.electacta.2010.04.040
L. Cunci, C.A. Rao, C. Velez, Y. Ishikawa, C.R. Cabrera, Electrocatalysis 4 (2013) 61–69. doi: 10.1007/s12678-012-0120-3
S. Suzuki, H. Muroyama, T. Matsui, K. Eguchi, J. Power Sources 208 (2012) 257–262. doi: 10.1016/j.jpowsour.2012.02.043
S. Le Vot, L. Roué, D. Bélanger, J. Power Sources 223 (2013) 221–231. doi: 10.1016/j.jpowsour.2012.08.048
T. Okanishi, Y. Katayama, H. Muroyama, T. Matsui, K. Eguchi, Electrochim. Acta 173 (2015) 364–369. doi: 10.1016/j.electacta.2015.05.066
K. Yao, Y.F. Cheng, Int. J. Hydrog. Energy 33 (2008) 6681–6686. doi: 10.1016/j.ijhydene.2008.07.023
K. Yao, Y.F. Cheng, J. Power Sources 173 (2007) 96–101. doi: 10.1016/j.jpowsour.2007.04.081
K. Endo, Y. Katayama, T. Miura, Electrochim. Acta 49 (2004) 1635–1638. doi: 10.1016/S0013-4686(03)00993-9
Q. Xue, Y. Zhao, J. Zhu, et al., J. Mater. Chem. A 9 (2021) 8444–8451. doi: 10.1039/d1ta00426c
Y.T. Chan, K. Siddharth, M. Shao, Nano Res. 13 (2020) 1920–1927. doi: 10.1007/s12274-020-2712-1
N. Sacr´e, M. Duca, S. Garbarino, et al., ACS Catal. 8 (2018) 2508–2518. doi: 10.1021/acscatal.7b02942
Y. Katayama, T. Okanishi, H. Muroyama, T. Matsui, K. Eguchi. J. Phys. Chem. C 119 (2015) 9134–9141. doi: 10.1021/acs.jpcc.5b01710
X. Lin, X. Zhang, Z. Wang, et al., J. Colloid Interface Sci. 601 (2021) 1–11. doi: 10.4236/oalib.1107953
Y. Li, X. Li, H.S. Pillai, et al., ACS Catal. 10 (2020) 3945–3957. doi: 10.1021/acscatal.9b04670
Y. Li, H.S. Pillai, T. Wang, et al., , Energy Environ. Sci. 14 (2021) 1449–1460. doi: 10.1039/D0EE03351K
H.S. Pillai, Y. Li, S. Wang, et al., Nat. Commun. 14 (2023) 792. doi: 10.1038/s41467-023-36322-5
S. Anantharaj, S. Noda, Mater. Today Energy 32 (2023) 101234. doi: 10.1016/j.mtener.2022.101234
E.G. Ciapina, S.F. Santos, E.R. Gonzalez, J. Electroanal. Chem. 815 (2018) 47–60. doi: 10.1016/j.jelechem.2018.02.047
M. Escudero-Escribano, A. Verdaguer-Casadevall, P. Malacrida, et al., J. Am. Chem. Soc. 134 (2012) 16476–16479. doi: 10.1021/ja306348d
S. Moniri, T. Van Cleve, S. Linic, J. Catal. 345 (2017) 1–10. doi: 10.1016/j.jcat.2016.11.018
Y. Liu, Z. Pan, O.C. Esan, X. Xu, L. An, Energy Fuels 36 (2022) 13203–13211. doi: 10.1021/acs.energyfuels.2c02951

Table 2. Different detection methods and different adsorption of intermediate species for AOR.
 下载: 导出CSV
下载: 导出CSV
Table 3. Comparison of the AOR performances of single Pt catalysts.
| Catalyst | Electrolyte | Scan rate (mV/s) | Onset potential | Peak current density (mA/cm2) | Ref. |
| Pt | 0.05 mol/L KOH + 0.1 mol/L NH3 | 20 | – | 4 | [51] |
| Pt/C | 0.2 mol/L NH4OH + 1.0 mol/L KOH | 15 | – | 0.920 | [52] |
| Pt nanoparticle | 0.1 mol/L NH3 + 1.0 mol/L KOH | 10 | −0.4 VHg/HgO | 58 | [53] |
| Pt/C | 1.0 mol/L NH3 + 1.0 mol/L KOH | 20 | −0.46 VSHE | 6.150 | [54] |
| Pt cubesH-S | 0.5 mol/L NH4OH + 0.5 mol/L KOH | 100 | 0.450 VRHE | 5.240 | [55] |
| PtR | 1 mol/L NH3 + 1 mol/L KOH | 10 | – | 0.400 | [56] |
| Colloidal Pt | 0.1 mol/L NH3 + 0.2 mol/L NaOH | 10 | – | 1.35 | [57] |
| er-GOx/Pt hydrid materials | 1.0 mol/L NH4OH | 10 | −0.3 VAg/AgCl | 0.255 | [58] |
| Pt decorated Ni | 0.1 mol/L NH3 + 1.0 mol/L KOH | 10 | – | 0.75 | [59] |
| Pt nanoparticles | 0.1 mol/L NH3 + 1.0 mol/L KOH | 10 | – | 0.8 | [60] |
| Ptseed | 0.1 mol/L NaOH +0.1 mol/L NH3 | 20 | – | 0.53 | [61] |
| Dendritic Pt | 0.1 mol/L NaOH +0.03 mol/L NH3 | 50 | −0.55 VSCE | – | [62] |
| Pt/NGA-800 | 1 mol/L NH3 + 1 mol/L KOH | – | – | 27.5 | [63] |
| Pt/ITO | 0.1 mol/L NH3 + 1.0 mol/L KOH | 10 | – | – | [64] |
下载: 导出CSV
Table 4. Comparison of the AOR performances of binary Pt alloy catalysts.
| Catalyst | Electrolyte | Scan rate (mV/s) | Onset potential | Peak current density (mA/cm2) | Ref. |
| PtIr/C | 1 mol/L NH3 | 10 | 0.470 VRHE | – | [72] |
| Pt/Ir/MWCNT nanoparticle | 0.1 mol/L NH3 + 1.0 mol/L KOH | 10 | – | – | [73] |
| Pt90Ru10/C | 1.0 mol/L NH4OH + 1.0 mol/L KOH | 20 | – | 0.92 | [74] |
| Pt7Pd3 | 0.5 mol/L NH4OH + 1.0 mol/L KOH | 30 | −0.490 VRHE | 7 | [75] |
| PtIr | – | 5 | – | 110 | [76] |
| RGO/PtIr | 1.0 mol/L NH4OH | 10 | − 0.4 VAg/AgCl | 20 | [77] |
| PtRu | 0.1 mol/L NH3 + 1.0 mol/L KOH | 20 | 0.3 VRHE | 1.8 | [78] |
| Pt90Ir10 | 0.08 mol/L NH3 + 1.0 mol/L KOH | 20 | – | 0.4 | [79] |
| SnO2-Pt/C | 0.1 mol/L NH3 + 0.1 mol/L KOH | 20 | 0.45 VRHE | 1.62 | [80] |
| PtNi | 0.1 mol/L NH3 + 1.0 mol/L KOH | 10 | – | 1.1 | [81] |
| PtNi | 0.1 mol/L NH3 + 1.0 mol/L KOH | 5 | – | 0.8 | [82] |
| Pt0.8Ir0.2 | 0.1 mol/L NH3 + 1.0 mol/L KOH | 1 | – | 0.038 | [83] |
| Pt6Ru-NCs | 1.0 mol/L NH3 + 1.0 mol/L KOH | 50 | 0.5 VRHE | – | [84] |
| Pt-Zn/C nanocubes | 0.1 mol/L NH3 + 1.0 mol/L KOH | 50 | – | – | [85] |
| PtxIr100-x/MgO | 0.1 mol/L NH3 + 0.2 mol/L NaOH | 20 | 0.53 VRHE | 1 | [86] |
| Pt/PBI/MWNT-CeO2 | 0.1 mol/L NH3 + 1.0 mol/L KOH | 20 | – | – | [87] |
下载: 导出CSV

 扫一扫看文章
扫一扫看文章

扫一扫关注我们





 下载:
下载: