Henan Key Laboratory of Crystalline Molecular Functional Materials and College of Chemistry, Zhengzhou University, Zhengzhou 450001, China
* Corresponding author. E-mail address: yingjieliu@zzu.edu.cn (Y.-J. Liu). 1 These authors contributed equally to this work.
Received Date:
28 June 2024 Accepted Date:
08 August 2024 Revised Date:
18 July 2024 Available Online:
15 September 2025
Abstract:
Non-covalent interactions-driven host-guest assembly based on metallo-tweezers has been used to construct varied optical functional materials with attractive structures and properties. We reported here two pairs of chiral gold(Ⅰ)-based metallo-tweezers as hosts to clip AgⅠ or CuⅠ cations for circularly polarized phosphorescence (CPP), driven by the integration of two-fold coordination and heterometallophilic interactions. The AuⅠ-based hosts and metal ions-guests formed sandwich structures in 1:1 ratio with high binding affinity. The achieved tweezer/cation adducts exhibited red-shifted absorption bands and circular dichroism signals, which were attributed to the newly formed ligand to metal-metal charge transfer process. Remarkably, the host-guest supramolecular adducts showed turn-on phosphorescence and CPP, which benefited from rigidifying effect of multiple intermolecular interactions and shorter excited-state lifetime. Overall, our findings bring new insights into the feasibility to achieve and modulate CPP performance by fabricating metallo-tweezer-based host-guest complexes.
Optical functional materials displaying circularly polarized luminescence (CPL) have attracted attentions due to their promising applications in the fields of 3D displays [1], anti-counterfeiting [2,3], asymmetric catalysis [4,5]. CPL materials are required to meet two prerequisites of chirality and luminescence properties [6-8]. Recent advancements have expanded into circularly polarized phosphorescence (CPP) materials, which were characterized by large Stokes shifts and long excited lifetime via boosting triplet excitons utilization [9-11]. Due to their intrinsic heavy atom effect, d10 transition metal complexes, mainly AuⅠ, AgⅠ and CuⅠ complexes, have been harnessed for developing phosphorescence materials [12-14]. Thereinto, the relativistic effects of Au endow AuⅠ complexes with strong intermolecular noncovalent Au···Au interaction referred to as “aurophilicity” [15-17]. The Au···Au contacts play vital roles in their self-assembly behaviors in the ground states and Au···Au chemical bonding in the excited states. So, it offers opportunities to achieve photofunctional materials with distinct photoluminescence properties through modulating the Au···Au separations by means of tuning the assembly conditions or exerting external stimuli [18-20]. Therefore, the past two decades have witnessed increasing interests in phosphorescence properties of mono-/poly-nuclear AuⅠ complexes [21-23]. A large number of AuⅠ complexes have been reported to show great potential applications in optoelectronic materials such as anti-counterfeiting, light-emitting diodes (LEDs) [24]. To date, although many chiral AuⅠ complexes have been reported, few of them showed CPL activity [25-28].
Self-assembly of chiral AuⅠ molecules has become an effective method for constructing CPL materials [29-31]. Generally, assembly driving forces mainly involve non-covalent interactions, including hydro-/halogen bonds, coordination bonds, Coulomb interactions and metallophilic interactions, which play key roles both in the aggregate structure and luminescent properties [32-36]. In our previous work, synergistic effect of aurophilicity and H-bonds were utilized to fabricate helical assemblies based on mono-nuclear carbene AuⅠ ionic complex to obtain decent CPL performance [37]. However, for weak or non-emissive AuⅠ complexes, it is still necessary to seek new strategies to improve their CPL properties [29].
Host-guest recognition has been proven to be an effective way to endow optical functional materials with enhanced performance [38-40]. As novel host-guest systems, metallo-tweezers integrating metal cations or complexes have been extensively investigated [41-43]. Compared with organic receptors, the predictable metallophilicity and coordination configuration of transition-metal segments allow the construction of predefined supramolecular architectures, through carefully selecting the ligands and metal centers [44-48]. Peris and co-workers have established a variety of AuⅠ-supramolecular tweezers as novel architectural motifs that form dimers in the presence of several M+ cations [49,50]. Wang and co-workers reported a case of host-guest recognition that AuⅢ molecular tweezers encapsulated chiral PtⅡ guest complex for amplified CPP properties [51]. The obtained rigid co-assembly structure could protect the emitter from external quenchers, such as O2 and solvent molecules, thus inhibit nonradiative decay of triplet excitons and exhibit turn-on CPP. On this basis, we considered that if host-guest assemblies based on chiral AuⅠ complexes and Ag+/Cu+ driven by hetero-metallophilic interactions could induce CPP properties, which have been rarely studied.
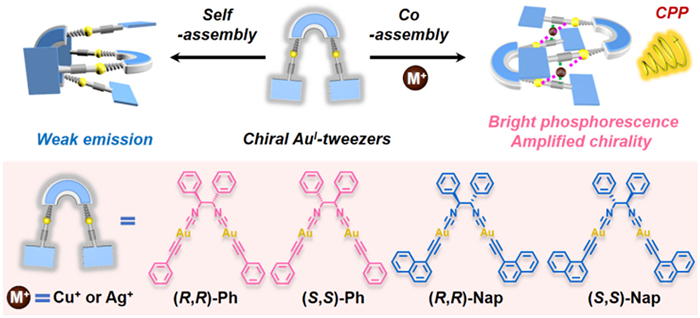
Therefore, we herein utilized rigid alkynyl-AuⅠ fragments decorated with the chiral isocyanide ligands to prepare two pairs of enantiomeric pure AuⅠ-tweezers (R,R)-/(S,S)-Ph and (R,R)-/(S,S)-Nap (Scheme 1). Such AuⅠ-based tweezers were selected as chiral inducers and hosts, which were non-emissive in solution-state. With the addition of M+ (Ag+ or Cu+) cations, each two chiral AuⅠ-based tweezers together encapsulated two M+ and formed sandwich-like structures driven by the integration of two-fold coordination and heterometallophilic interactions. Thanks to the rigidifying effects and formation of ligand to metal charge-transfer process, the achieved tweezer⊂guest complexes (abbreviated as M+ adducts) showed switch-on phosphorescent emission in solution state. Meanwhile, supramolecular chirality was induced for the obtained M+ adducts. Remarkably, the resulted non-covalent adducts of (R,R)-/(S,S)-Ph and Ag+ exhibited turn-on CPP both in O2 and nitrogen (N2) states. Overall, the present study offers a pre-organized approach to prepare CPP materials via AuⅠ-tweezer-based host-guest complexation.
Scheme 1
Scheme 1.
Schematic representation for the supramolecular assembly structures of AuⅠ-tweezers before and after host-guest recognition with the complementary Cu+ or Ag+ guests. The chemical structures of AuⅠ-tweezers receptors (R,R)-Ph, (S,S)-Ph, (R,R)-Nap and (S,S)-Nap, are shown in the frame.
The chiral AuⅠ-tweezers (R,R)-Ph and (S,S)-Ph (Scheme 1) were readily synthesized by reaction of (1R,2R)−1,2-diphenylethane-1,2-diisocyanide ((R,R)-NC) or (1S,2S)−1,2-diphenylethane-1,2-diisocyanide ((S,S)-NC) with [AuⅠ-(C≡C)Ph]n, respectively (Schemes S1 and S2 in Supporting information). Besides, (R,R)-Nap and (S,S)-Nap were prepared by replacing [AuⅠ-(C≡C)Ph]n with [AuⅠ-(C≡C)Nap]n (Scheme S3 in Supporting information). The obtained complexes were well characterized by NMR spectroscopy (Figs. S1-S12 in Supporting information), electrospray ionization mass spectrometry (ESI-MS) (Fig. S13 in Supporting information), and elemental analysis. Similar to the traditional linearly two-coordinated AuⅠ complexes, the as-prepared chiral AuⅠ complexes were non-emissive in diluted dichloromethane (DCM) solutions.
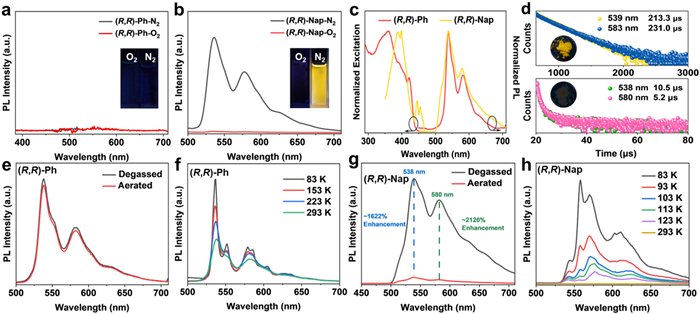
For the individual AuⅠ-tweezers (R,R)-/(S,S)-Ph (Fig. S14 in Supporting information), vibronic structured absorbance were present in the range of 250–325 nm with maximum at 290 nm in DCM. Meanwhile, the individual (R,R)-/(S,S)-Nap exhibited red-shifted absorbance peaked at 340 nm. The theoretical results (Fig. S16 in Supporting information) showed that the both lowest-energy electron absorption transition of (R,R)-Ph and (R,R)-Nap from the highest occupied molecular orbital (HOMO) to the lowest unoccupied molecular orbital (LUMO) were attributed to the ligand-ligand charge transfer (LLCT) process. So, the bathochromic shift of (R,R)-/(S,S)-Nap were attributed to larger π-conjugation and electron-donating capacity of naphthalene compared with benzene [52]. The photoluminescence (PL) spectra of the AuⅠ-tweezers in dilute DCM solution (1 × 10−4 mol/L) were further recorded under N2 and O2 conditions (Figs. 1a and b, Fig. S17 in Supporting information). Unaffected by O2, (R,R)-/(S,S)-Ph consistently showed negligible emission. However, (R,R)-/(S,S)-Nap were non-emissive under O2 conditions; while at N2 conditions, two O2-quenched peaks at 536 and 580 nm appeared. The relatively long emission lifetime (32.8–35.7 µs, Fig. S18 in Supporting information) suggests that (R,R)-/(S,S)-Nap are phosphorescent, similar to previously reported aryl gold isocyanide complexes [53]. The obvious O2 sensitivity for the phosphorescence signal indicates that the non-emissive nature of (R,R)-/(S,S)-Nap solution was mainly attributed to O2 quenching, which facilitate the triplet excitons to decay non-radiatively [54,55].
Figure 1
Figure 1.
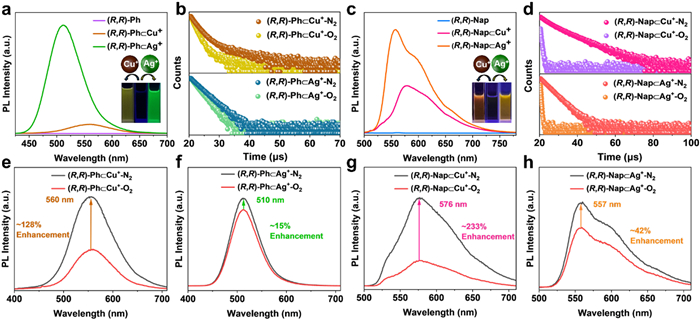
PL spectra of (a) (R,R)-Ph and (b) (R,R)-Nap in DCM solutions under N2 and O2 conditions. (c) Excitation and PL spectra of (R,R)-Ph and (R,R)-Nap crystals. (d) Luminescence decay curves and luminescent photos of (R,R)-Ph (up) and (R,R)-Nap (below) crystals. PL spectra of (e) (R,R)-Ph crystals and (g) (R,R)-Nap crystals under degassed and aerated conditions. Temperature-dependent PL spectra of (f) (R,R)-Ph and (h) (R,R)-Nap crystals.
Crystallization of (R,R)-/(S,S)-Ph from dichloromethane/acetonitrile (DCM/CH3CN) afforded yellow-emitting crystals (Fig. 1d, insets). However, the identical crystallization procedure for (R,R)-/(S,S)-Nap gave rise to negligible orange-emissive needle crystals (Fig. 1d, insets). The powder X-ray diffraction (PXRD) analyses results indicated their high crystallinities (Fig. S15 in Supporting information). The PL and excited-state lifetime (Figs. 1c and d) of crystalline solids were further studied to evaluate the aggregation effect on the luminescent process. Both crystals of (R,R)-Ph and (R,R)-Nap exhibited resolved emission bands, showing the maximum (λem) at ≈ 538 nm with shoulder peaks at ≈ 580 nm. (R,R)-Ph is yellow emissive with PL quantum yield (PLQY) of 15.2%. However, (R,R)-Nap is negligible orange-emissive with lifetime of 10.5 µs and 5.2 µs (Fig. 1d), which were significantly shorter than (R,R)-Ph crystals (213.3 µs and 231.0 µs). Notably, for (R,R)-/(S,S)-Nap, the dilute solution in N2 condition and crystals (Fig. S19 in Supporting information) exhibited similar phosphorescent emission bands, suggesting that the phosphorescence were attributed to the Au perturbed intramolecular charge transfer [18]. Removing O2 exerted no visible change in the PL intensity of (R,R)-Ph, while emission of (R,R)-Nap were greatly enhanced by nearly 103 times with slightly shorter lifetime at 538 nm (Figs. 1e and g, Fig. S20 and Table S1 in Supporting information).
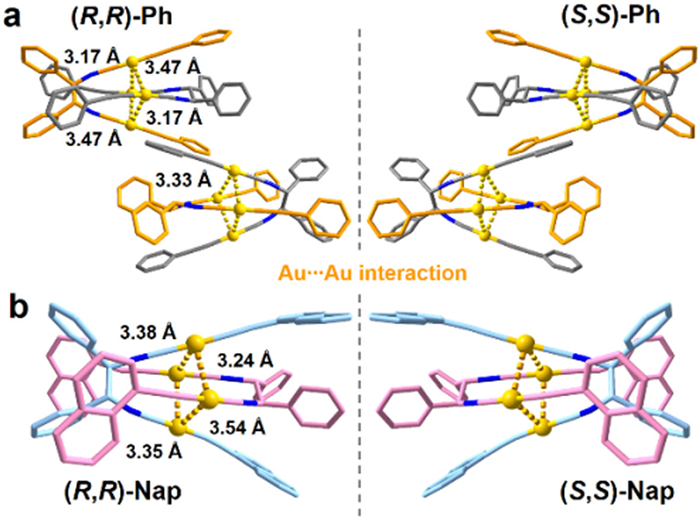
To investigate the self-assembly behaviors and structure-luminescent relationships of these AuⅠ-tweezers, X-ray crystallography analysis were conducted on (R,R)-/(S,S)-Ph and (R,R)-/(S,S)-Nap crystals (Tables S2 and S3 in Supporting information). As expected, they have analogous coordination mode and molecular structures (Fig. S21 in Supporting information), wherein two [AuⅠ-(C≡C)Ph] or [AuⅠ-(C≡C)Nap] units are connected by the chiral (R,R)-/(S,S)-NC ligands in (R,R)-/(S,S)-Ph and (R,R)-/(S,S)-Nap, respectively. However, different alkynyl ligands make them adopt distinct stacking modes. Specifically, (R,R)-/(S,S)-Ph crystallized in C2 chiral space group. In (R,R)-Ph crystal (Fig. S22a in Supporting information), the intramolecular dihedral angles between the planes of the [-(C≡C)Ph] ligands are 35.90° and 42.70°, indicating V-shaped AuⅠ-tweezers. Driven by aurophilic interactions, each two molecules aggregate into a dimer unit (Fig. 2a), in which the two phenylacetylene fragments sandwich the (R,R)-NC ligands together of the complementary molecule. The four Au atoms form into rectangles with Au···Au distances of 3.17–3.47 Å, which are clearly suggestive of moderate aurophilic interactions (Fig. 2a), thereby, leading to yellow phosphorescent emission from the crystals. The dimers are isolated by DCM molecules and further arrange into layered structures (Fig. S23 in Supporting information). Different from (R,R)-/(S,S)-Ph, (R,R)-/(S,S)-Nap crystallized in the P212121 space group. In (R,R)-Nap crystal (Fig. S22b in Supporting information), the intramolecular dihedral angles between the planes of the [-(C≡C)Nap] ligands are 40.62°, also indicating V-shaped AuⅠ-tweezers. Each two (R,R)-Nap also form a dimer unit, which are connected by very weaker aurophilic interactions with larger Au···Au separations of 3.24–3.54 Å (Fig. 2b). Additionally, in the molecular layered structure (Fig. S24 in Supporting information), the adjacent dimers are isolated by CH3CN and DCM molecules. Notably, (R,R)-Nap crystals (ρNap = 1.910 g/cm3) exhibit smaller density than (R,R)-Ph crystals (ρPh = 2.028 g/cm3), which might be attributed to the greater steric resistance of naphthalene compared to benzene ring. It suggests that the tighter packing in (R,R)-Ph could effectively insulate the O2, which allows brighter emission. Additionally, both the crystal structures of (R,R)-Nap and (R,R)-Ph are disordered (Fig. S25 in Supporting information). So, the loosely packing affords enough space for the vibrational motions of ligands, which will strengthen the non-radiative transition [56]. Therefore, it is conjectured that the combination of weaker aurophilicity and looser stacking lead to the non-luminescence of (R,R)-Nap crystals.
Figure 2
Figure 2.
Dimer structures and Au···Au interactions in (a) (R,R)-/(S,S)-Ph, and (b) (R,R)-/(S,S)-Nap crystals. All H atoms were omitted for clarity (orange, gray, pale blue and pink: C atoms; blue: N atoms; yellow: Au atoms).
To in-depth clarify the luminescent behaviors, temperature-variable PL spectra (Figs. 1f and h) and lifetime (Fig. S26 and Table S4 in Supporting information) of (R,R)-Ph/-Nap crystals at temperatures from 83 K to 293 K were systematically collected. Their luminescence peaks (Figs. 1f and h) remained unchanged during the whole cooling process, indicating that their exciton species were temperature-independent. For (R,R)-Ph from 293 K to 83 K, the short-wavelength band at 537 nm was enhanced by 2 times while the long-wavelength band nearly remains constant, accompanied with 1.1-fold lifetimes. For (R,R)-Nap, the decreased temperature to 83 K resulted in a 103-fold intensity in the maximum peak. Thus, its non-emissive nature under 293 K was resulted from strong thermally activated non-radiative transition process, due to the loosely packing mode and weaker intermolecular interactions in (R,R)-Nap crystal, which could be restricted under 83 K. It was further verified by the fact that the excited-state lifetime under 83 K (544 nm, 87.1 µs) is nearly an order of magnitude greater than that under 293 K (538 nm, 10.5 µs).
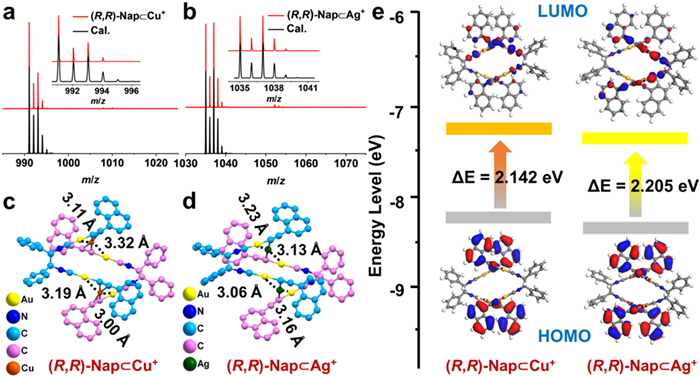
The above analyses demonstrate that the quasiorthogonal disposition of the diisocyanide and aromatic alkyne ligands, combining with the existence of the AuⅠ centers, provide the self-complementarity required to form such dimer structures in solid-state. In addition, the tight packing and aurophilicity in (R,R)-Ph crystal together inhibit the triplet exciton quenching caused by O2 and vibration, leading to the stronger phosphorescence. However, the existing intermolecular Au···Au interactions are not sufficient to drive the molecules to emit CPP both in solution and solid state. Thus, non-covalent complexation between AuⅠ-tweezers ((R,R)-/(S,S)-Ph and (R,R)-/(S,S)-Nap) and M+ cations (Cu+ or Ag+) were investigated to induce host-guest assembly to light CPP. ESI-MS was firstly employed to confirm the structures of M+ adducts, abbreviated as (R,R)-/(S,S)-Ph⊂Cu+, (R,R)-/(S,S)-Ph⊂Ag+, (R,R)-/(S,S)-Nap⊂Cu+ and (R,R)-/(S,S)-Nap⊂Ag+, which were prepared by mixing the AuⅠ-tweezers and M+ in 1:1 molar ratios. The analysis of the Cu+ and Ag+ adducts of (R,R)-/(S,S)-Ph by ESI-MS (Figs. S27 and S28 in Supporting information) revealed main peaks at m/z 891.07 and 935.01, which were assigned to the mass of one (R,R)-/(S,S)-Ph plus one Cu+ or Ag+ cation, respectively. In addition, signals at m/z 991.06 and 1035.15 were detected, indicating the formation of monovalent ions composed of one (R,R)-/(S,S)-Nap and one Cu+ or Ag+ cation, which were verified after matching with the calculated theoretical m/z values (Figs. 3a and b, Fig. S29 in Supporting information). Additionally, Job’s plots established a 1:1 binding stoichiometry of (R,R)-/(S,S)-Ph or (R,R)-/(S,S)-Nap with Ag+, as well as (R,R)-/(S,S)-Nap with Cu+ (Fig. S30 in Supporting information), which were coincided with the above ESI-MS analysis results [57]. Moreover, the binding constants (Ks) of AuⅠ-tweezers with Cu+ or Ag+ ions were calculated ranging from 4.10 × 104 L/mol to 3.94 × 107 L/mol (Table S5 in Supporting information) by the nonlinear curve-fitting of the absorbance titration data for the 1:1 model (Fig. S31 and Eq. S1 in Supporting information) [58]. The above results illustrated high binding affinity for AuⅠ-tweezers with Ag+/Cu+ guests.
Figure 3
Figure 3.
Positive mode ESI-MS of (a) (R,R)-Nap⊂Cu+ and (b) (R,R)-Nap⊂Ag+. Insets: Enlarged portion of the ESI-MS exhibiting the measured and simulated isotopic distribution patterns. The optimized structures of (c) (R,R)-Nap⊂Cu+ and (d) (R,R)-Nap⊂Ag+. (e) Calculated FOMs diagrams of (R,R)-Nap⊂Cu+ (left) and (R,R)-Nap⊂Ag+ (right).
Since the high-quality single crystals of M+ adducts suitable for X-ray crystallography were not successfully obtained, the energy-minimized structures of the M+ adducts were simulated and optimized via DMol3 module in Material Studio (MS) modeling 4.0 software package. In the optimized geometry of (R,R)-Ph⊂M+ and (R,R)-Nap⊂M+ (Figs. 3c and d, Fig. S32 in Supporting information), each M+ (Cu+ or Ag+) cations are fixed by two AuC≡C–fragments by M+−bis(µ2-alkyne) coordination bonds and heterometallophilicity to form 1:1 sandwich structures. Therefore, two AuⅠ-tweezers are connected by two M+ ions. In both Cu+ adducts, all the eight Cu-C bond distances (Figs. S33a and S34a, Tables S6 and S8 in Supporting information) are between 2.00 Å and 2.13 Å, and thus alkyne ligands display one type of coordinated modes: µ2-η1, η2 mode [59]. While in both Ag+ adducts (Figs. S33b and S34b, Tables S7 and S9 in Supporting information), four of the Ag-C bond distances range from 2.20 Å to 2.24 Å; and the other four Ag···C distances are in the range of 2.45–2.63 Å. The later indicate negligible coordination interactions. It suggested the alkyne ligands in Ag+ adducts display one type of coordinated modes: µ2-η2 mode. The M+−alkyne coordination bonds were further verified by Fourier transform infrared (FT-IR) spectroscopy, due to the blue-shifts of C≡C stretching vibration from 2121 cm-1 ((R,R)-Ph) and 2113 cm-1 ((R,R)-Nap) to the higher wavenumber region (2194 cm−1 for (R,R)-Ph⊂M+ and 2196 cm−1 for (R,R)-Nap⊂M+, Fig. S35 in Supporting information) [60,61]. More importantly, in both Cu+ adducts, the distances between the each Cu+ and two adjacent Au centers range between 3.00 Å and 3.32 Å, while for each Ag+ adduct, the Ag···Au separations are ranging from 3.06 Å to 3.23 Å, thus illustrating that the introduction of cations partially contributed to occurrence of strong metallophilic interactions. Hence, strong coordination and metallophilic bonds are prerequisite for the construction of M+ adducts.
Frontier molecular orbits (FMOs) calculations based on the optimized structures of M+ adducts (Fig. 3e and Fig. S36 in Supporting information) indicate that the introduction of M+ induces a mixture of ligand to metal-metal charge transfer (LMMCT) and ligand to ligand charge transfer (LLCT) as the transition to the lowest excited state, i.e., the transition from the HOMO (mainly distributed at the [-(C≡C)Ph] or [-(C≡C)Nap] moiety) to the LUMO (mainly distributed at the M+ and (R,R)-NC-Au moiety). Thus, the Ag+ and Cu+ could modulate the FMOs of AuⅠ tweezers via noncovalent clipping. In addition, the addition of Ag+ and Cu+ could cause internal fixation of (R,R)-Ph and (R,R)-Nap through multiple coordination and metallophilic interactions, which confine rotational and vibrational freedom. It contributes to restrain structural rearrangement at the excited state, and further allow luminescent variations between AuⅠ-tweezers and the obtained M+ adducts. Therefore, high binding affinity and rigid structures are benefit to the phosphorescence and CPP lighting of AuⅠ emitters.
To our delight, the introduction of metal ions (Cu+ or Ag+) can significantly light up the luminescence of the solution of AuⅠ-based tweezers ((R,R)-/(S,S)-Ph and (R,R)-/(S,S)-Nap). Firstly, non-covalent complexation between metal ions (Cu+ or Ag+) and AuⅠ- tweezers generated obvious changes in UV–vis absorption (Fig. S37 in Supporting information). Upon mixing Cu+ with tweezers in the molar ratio of 1:1, lower-energy absorption bands red-shifted to 320 nm for (R,R)-/(S,S)-Ph⊂Cu+ and 360 nm for (R,R)-/(S,S)-Nap⊂Cu+, respectively. Analogously, the addition of 1 equivalent Ag+ caused the band-edge redshift of the absorption band for (R,R)-/(S,S)-Ph⊂Ag+ and (R,R)-/(S,S)-Nap⊂Ag+. Emission changes upon guest complexation were then investigated (Figs. 4a and c, Fig. S38 in Supporting information). With the addition of guest metal ions, bright green emission or yellow emission centered at 560 nm ((R,R)-Ph⊂Cu+, τ = 1.4 µs), 510 nm ((R,R)-Ph⊂Ag+, τ = 1.8 µs), 576 nm ((R,R)-Nap⊂Cu+, τ = 1.6 µs) and 551 nm ((R,R)-Nap⊂Ag+, τ = 1.1 µs) emerged for the mixture DCM solution, respectively (Fig. S38). Intriguingly, the emission intensity of (R,R)-Ph⊂Ag+ (or (R,R)-Nap⊂Ag+) was much higher than that of (R,R)-Ph⊂Cu+ (or (R,R)-Nap⊂Cu+) under the same circumstances. The M+ adducts of (S,S)-enantiomers exhibited similar properties (Fig. S39 in Supporting information). Moreover, it should be noted that the lifetimes of M+ adducts are drastically shorter than that of individual AuⅠ-tweezers, which are beneficial to hinder the influence of O2 quenching on the luminescence process. Indeed, upon removing O2 from the DCM solution of M+ adducts, the triplet emission enhanced in small degrees (Figs. 4e-h and Fig. S40 in Supporting information), accompanied by prolonging of the emission lifetime (Figs. 4b and d, Tables S10 and S11 in Supporting information). Notably, emission intensity of Ag+ adducts displayed a much lower dependence on O2. As a consequence, we rationalized that the key reason of phosphorescence lighting is the shortened lifetime of excited-state caused by heterometallic doping. Moreover, the phosphorescent behaviors of M+ adducts in solid state were investigated. Upon grinding the mixed powders of AuⅠ-tweezers and M+, different emission from green to red emerged (Figs. S41 and S42 in Supporting information), with slight longer lifetime than those in solution state (Fig. S43 and Table S12 in Supporting information). The red emission color of (R,R)-Nap⊂Cu+ and (R,R)-Nap⊂Ag+ were easily visualized under irradiation of 365 nm UV lamp, distinct from the non-emission profile of (R,R)-Nap crystal (Fig. S42). And the EDS-mapping of different ground powder of M+ adducts proved the Cu+/Ag+ have uniformly interacted with the AuⅠ-tweezers (Fig. S44 in Supporting information). Overall, it is demonstrated that phosphorescent emission of AuⅠ-tweezers were enhanced upon non-covalent complexation with M+.
Figure 4
Figure 4.
PL spectra (λex = 365 nm) from different M+ adducts of (a) (R,R)-Ph and (c) (R,R)-Nap. Insets: luminescent images of solutions under irradiation of UV lamp. Comparisons of luminescence decay curves from different M+ adducts of (b) (R,R)-Ph and (d) (R,R)-Nap under N2 and O2 conditions. (e-h) Comparisons of PL spectra from different M+ adducts under N2 and O2 conditions.
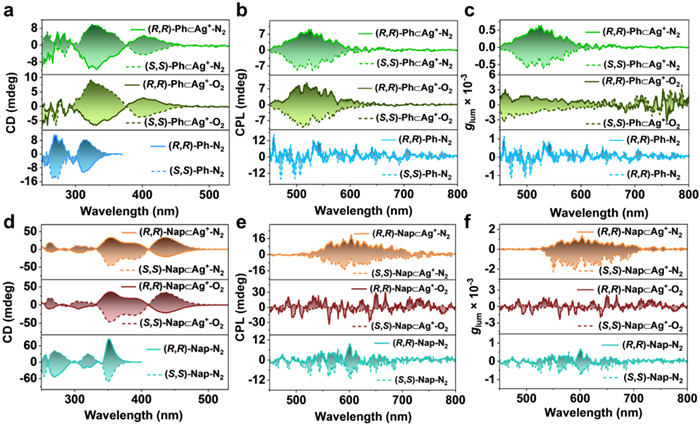
Considering the optically active (R,R)-/(S,S)-NC units in AuⅠ-tweezers, chiroptical properties were further performed. As expected, mirror-imaged circular dichroism (CD) signals were observed for the individual (R,R)-/(S,S)-Ph and (R,R)-/(S,S)-Nap (Fig. S45 in Supporting information). However, no CPL signals were detected for these AuⅠ-tweezers in DCM under N2/O2 conditions (Fig. S46 in Supporting information). Upon adding one equimolar amount of M+ into AuⅠ-tweezers solutions, new CD signals appeared in the long-wavelength regions with maxima at 400 nm (Ph⊂Ag+), 420 nm (Nap⊂Cu+) and 440 nm (Nap⊂Ag+), respectively (Figs. S47-S49 in Supporting information, Figs. 5a and d). Accordingly, the red-shifted CD signals demonstrate the occurrence of supramolecular chirality from M+ adducts. Unfortunately, no CPL signals were detected for the (R,R)-/(S,S)-Ph⊂Cu+ and (R,R)-/(S,S)-Nap⊂Cu+ under the N2 conditions (Figs. S50 and S51 in Supporting information). On the contrary, (R,R)-/(S,S)-Ph⊂Ag+ exhibited obvious mirror-imaged CPL signals even under the O2 conditions (Fig. 5b) with luminescent dissymmetry factor |glum| of |1 × 10–3| (Fig. 5c). In comparison, (R,R)-/(S,S)-Nap⊂Ag+ exhibited O2 quenched CPP. Concretely, no signals were detected for the (R,R)-/(S,S)-Nap⊂Ag+ in O2 conditions (Fig. 5e). After excluding O2, (R,R)-/(S,S)-Nap⊂Ag+ exhibited turn-on CPP with |glum| of |1 × 10–3| (Fig. 5f). These results indicated that (R,R)-/(S,S)-Ph⊂Ag+ display the most robust CPL towards O2 quenching, and similar phenomenon has been observed in its PL spectra. Hence, CPP signals arisen for chiral AuⅠ-tweezers emitters upon co-assembly with M+, which is promising for recognition and anti-counterfeiting applications.
Figure 5
Figure 5.
(a) CD, (b) CPL and (c) corresponding glum spectra of (R,R)-/(S,S)-Ph in DCM before and after the addition of Ag+ under N2/O2 condition. (d) CD, (e) CPL and (f) corresponding glum spectra of (R,R)-/(S,S)-Nap in DCM before and after the addition of Ag+ under N2/O2 condition.
In summary, a non-covalent host-guest strategy has been employed as an effective strategy toward lighting-up CPP of triplet alkyne-AuⅠ tweezers. It benefits from the design of dual-arms receptors (R,R)-/(S,S)-Ph and (R,R)-/(S,S)-Nap, which displayed high non-covalent binding affinity with Cu+ and Ag+ due to two-fold driving interactions. The synergistic alkyne-Ag+ (or Cu+) coordination and M+···Au heterometallic interactions have afforded the driving force to induce the host-guest co-assembly for sandwich supramolecular architectures. Benefiting from their shorten lifetimes, which could prevent O2 from quenching the phosphorescence, the obtained M+ adducts displayed switch-on and tunable phosphoresce. Furthermore, co-assembly-induced rigid structures by multiple intermolecular interactions played a crucial role in CPP amplification from Ag+ adducts. These interesting results would open up new opportunities for the chiral co-assembly of alkyne-AuⅠ complexes from simple coordination and metal-metal bonding subunits toward high-performance CPP materials.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was financially supported by the National Natural Science Foundation of China (Nos. 92356304, 92061201, and 22105177), the National Key R & D Program of China (No. 2021YFA1200301), the China Postdoctoral Science Foundation (No. 2021TQ0294), and the Zhongyuan Thousand Talents (Zhongyuan Scholars) Program of Henan Province (No. 234000510007).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.110326.
[1]
Y. Yang, R.C.d. Costa, M.J. Fuchter, et al., Nat. Photonics 7 (2013) 634–638. doi: 10.1038/nphoton.2013.176
[2]
F. Zhang, Q. Li, C. Wang, et al., Adv. Funct. Mater. 32 (2022) 2204487. doi: 10.1002/adfm.202204487
F. Rey-Tarrío, S. Simón-Fuente, J.M. Cuerva, et al., Angew. Chem. Int. Ed. 63 (2024) e202318454. doi: 10.1002/anie.202318454
Scheme 1
Schematic representation for the supramolecular assembly structures of AuⅠ-tweezers before and after host-guest recognition with the complementary Cu+ or Ag+ guests. The chemical structures of AuⅠ-tweezers receptors (R,R)-Ph, (S,S)-Ph, (R,R)-Nap and (S,S)-Nap, are shown in the frame.
Figure 1
PL spectra of (a) (R,R)-Ph and (b) (R,R)-Nap in DCM solutions under N2 and O2 conditions. (c) Excitation and PL spectra of (R,R)-Ph and (R,R)-Nap crystals. (d) Luminescence decay curves and luminescent photos of (R,R)-Ph (up) and (R,R)-Nap (below) crystals. PL spectra of (e) (R,R)-Ph crystals and (g) (R,R)-Nap crystals under degassed and aerated conditions. Temperature-dependent PL spectra of (f) (R,R)-Ph and (h) (R,R)-Nap crystals.
Figure 2
Dimer structures and Au···Au interactions in (a) (R,R)-/(S,S)-Ph, and (b) (R,R)-/(S,S)-Nap crystals. All H atoms were omitted for clarity (orange, gray, pale blue and pink: C atoms; blue: N atoms; yellow: Au atoms).
Figure 3
Positive mode ESI-MS of (a) (R,R)-Nap⊂Cu+ and (b) (R,R)-Nap⊂Ag+. Insets: Enlarged portion of the ESI-MS exhibiting the measured and simulated isotopic distribution patterns. The optimized structures of (c) (R,R)-Nap⊂Cu+ and (d) (R,R)-Nap⊂Ag+. (e) Calculated FOMs diagrams of (R,R)-Nap⊂Cu+ (left) and (R,R)-Nap⊂Ag+ (right).
Figure 4
PL spectra (λex = 365 nm) from different M+ adducts of (a) (R,R)-Ph and (c) (R,R)-Nap. Insets: luminescent images of solutions under irradiation of UV lamp. Comparisons of luminescence decay curves from different M+ adducts of (b) (R,R)-Ph and (d) (R,R)-Nap under N2 and O2 conditions. (e-h) Comparisons of PL spectra from different M+ adducts under N2 and O2 conditions.
Figure 5
(a) CD, (b) CPL and (c) corresponding glum spectra of (R,R)-/(S,S)-Ph in DCM before and after the addition of Ag+ under N2/O2 condition. (d) CD, (e) CPL and (f) corresponding glum spectra of (R,R)-/(S,S)-Nap in DCM before and after the addition of Ag+ under N2/O2 condition.

 DownLoad:
DownLoad:

 下载:
下载:







 下载:
下载:
