State Key Laboratory for Modification of Chemical Fibers and Polymer Materials, College of Materials Science and Engineering, Donghua University, Shanghai 201620, China
b.
Beijing Key Laboratory of Ionic Liquids Clean Process, Institute of Process Engineering, Chinese Academy of Sciences, Beijing 100190, China
c.
School of Chemical Sciences, University of Chinese Academy of Sciences, Beijing 100049, China
d.
National Energy R & D Center for Coal to Liquid Fuels, Synfuels China Technology Co., Ltd., Beijing 101407, China
e.
Shimadzu (China) Co., Ltd., Shanghai 200233, China
f.
School of Chemistry and Physics & Centre for Materials Science, Queensland University of Technology, Brisbane, QLD 4001, Australia
wluo@dhu.edu.cn (W. Luo). 1 These authors contributed equally to this work.
Received Date:
07 May 2024 Accepted Date:
26 July 2024 Revised Date:
20 July 2024 Available Online:
15 January 2025
Abstract:
In this work, we developed plasmonic photocatalyst composed of CuPd alloy nanoparticles supported on TiN, the optimized Cu3Pd2/TiN catalyst shows excellent conversion (> 96%) and selectivity (> 99%) for Heck reaction at 50 ℃ under visible light irradiation. By in-situ spectroscopic investigations, we find that visible light excitation could achieve stable metallic Cu species on the surface of CuPd alloy nanoparticles, thereby eliminating the inevitable surface oxides of Cu based catalyst. The in-situ formed metallic Cu species under irradiation take advantage of the strong interactions of Cu with visible light, and manifest in the localized surface plasmon resonances (LSPR) photoexcitation. Visible light excitation could further promote the charge transfer between catalytic Pd component and the support TiN, resulting in electron-rich Pd sites on CuPd/TiN. Moreover, light excitation on CuPd/TiN generates strong chemisorption of iodobenzene and styrene, favoring the activation of reactants for Heck reaction. DFT calculations suggest that electron-rich CuPd sites ideally lower the activation energy barrier for the coupling reaction. This work provides valuable insights for mechanistic understanding of plasmonic photocatalysis.
Photocatalytic synthesis of organic compounds has garnered considerable interest among researchers due to its advantages, including mild reaction conditions, high substrate functional group compatibility, enhanced product selectivity, and reduced energy consumption [1-3]. Traditional semiconductor photocatalysts have been limited in their application to photocatalytic organic synthesis due to a narrow light-response range and the availability of catalytic active sites [4,5]. Consequently, developing new photocatalysts that can absorb a broad spectrum of visible light and exhibit high activity and selectivity is crucial for photocatalytic organic synthesis. Recent developments have shown that supported metal nanoparticles exhibit exceptional advantages in photocatalytic reactions, possessing an extensive range of light absorption and superior electron transport properties. These metal nanoparticle photocatalysts are regarded as having significant potential in advancing application for organic synthesis [6-8].
Notably, plasmonic metal nanoparticles (Au, Ag, Cu or Al) have emerged as a new class of photocatalysts that can efficiently absorb visible light and generate high energetic electrons, rendering them highly suitable for catalyzing organic synthesis reactions [9,10]. However, plasmonic monometallic nanoparticles still exhibit certain limitations in their applications, for example, the number of chemical reactions that can be catalyzed by monometallic Au catalysts are relatively few comparing with the range that can be catalyzed by conventional transition metals [11]. Inherent catalytic behavior of the transition metals should be considered. In this regard, bimetallic plasmonic photocatalysts could utilize one metal as the light-absorber and another as the catalytic center, creating a synergistic effect that enhances their catalytic performance [11-13]. These catalysts are distinguished by their tunable geometric and electronic structures, enabling superior performance over monometallic counterparts. Previous research has revealed that the properties of bimetallic nanoparticle catalysts not only optimize catalytic efficiency but also expand the range of applications in organic synthesis [14]. Despite significant advancements in bimetallic photocatalytic systems, there remains a substantial gap in understanding of their mechanisms, including energy transfer processes and the surface charge states of catalysts under illumination [15,16]. Crucially, the adsorption and activation of reactants on the catalyst surface are key to enhance catalytic performance [17]. The Heck cross-coupling reaction, as a fundamental strategy for constructing C—C bonds, plays a pivotal role in the synthesis of fine chemicals, polymers, pharmaceuticals, and natural products [18]. Recently, we utilized a zirconia supported AuPd bimetallic catalyst for photocatalytic Heck reaction. We found that under irradiation, significant charge transfer between plasmonic Au and catalytic Pd component delivering electron-deficient Pd sites on the surface of nanoparticles, this in-situ formed active Pd sites plays a pivotal role in the photocatalytic process [19]. Noteworthily, the charge flow within the bimetallic catalyst under irradiation apparently influence the surface states of the catalyst and dominate the overall photocatalytic performance, while the intrinsic mechanism also largely depends on the components of the bimetallic plasmonic photocatalysts.
Cu as a low-cost plasmonic metal, not only rivals Au in optical performance but also possesses a broader range of LSPR peak [20,21]. Previous studies have demonstrated that the LSPR effect in Cu can induce photoreduction of its oxidized surfaces, thereby enhancing stability [22]. The alloying of plasmonic Cu with catalytically active Pd not only markedly improves this stability but also modulates the plasmonic properties of the material. Consequently, CuPd alloy nanoparticles exhibit considerable potential in photocatalytic systems [23]. Selecting suitable supporting materials represents another critical factor in the development of effective plasmonic nanoparticle photocatalytic systems. Titanium nitride (TiN), a non-metallic plasmonic material, features a band structure analogous to noble metals and is distinguished by its excellent conductivity and charge carrier properties. TiN not only absorbs visible and near-infrared (NIR) light effectively but also demonstrates its unique chemical stability and catalytic activities [24,25]. In recent studies, researchers have identified electron transfer between TiN and Cu nanoparticles, which not only preserves the stability of the Cu nanoparticles but also contributes to their efficacy in the photocatalytic epoxidation of olefins [25]. However, there remains a significant gap in understanding the mechanisms of energy transfer and charge transfer between TiN supports and bimetallic nanoparticles, necessitating further investigation.
In our work, bimetallic CuPd nanoparticles supported by TiN were synthesized to serve as heterogeneous catalysts for photocatalytic Heck reaction. We employed the conventional impregnation-reduction method to prepare monometallic catalysts comprising Cu and Pd, and bimetallic CuPd alloy nanoparticles with a fixed metal loading of 3% on TiN supports. The detailed preparation methods are shown in the Experimental section of Supporting information. These catalysts were respectively designated as Cu/TiN, Pd/TiN and CuxPdy/TiN, where x and y are the molar ratio of Cu/Pd.
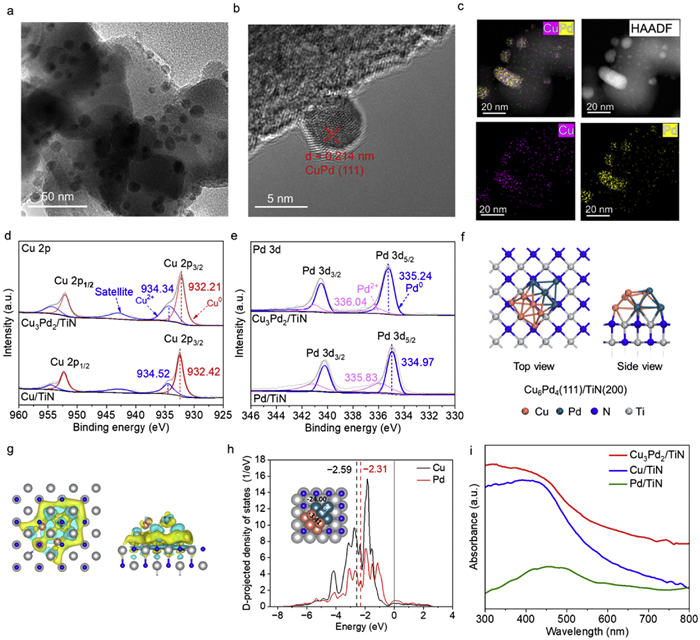
TEM images of a representative Cu3Pd2/TiN bimetallic photocatalysts shows uniform distribution of metal nanoparticles with a mean diameter of 5–6 nm on the TiN support (Fig. 1a, Figs. S1 and S2 in Supporting information). In the high-resolution TEM images of Cu3Pd2/TiN (Fig. 1b), a lattice spacing of 0.214 nm characteristic of CuPd alloy (111) crystal planes was observed [26]. The EDS elemental mapping images of Cu3Pd2/TiN (Fig. 1c) revealed the uniform distribution of Pd and Cu species, with nearly overlapping signals confirming the alloy state of CuPd photocatalyst. The actual Cu/Pd loadings in the catalysts were determined using inductively coupled plasma optical emission spectroscopy (ICP-OES), with results closely aligning with the nominal amounts (Table S1 in Supporting information). For instance, in the Cu3Pd2/TiN sample, the total metal loading was targeted at 3%, with a Cu to Pd molar ratio of 3:2. The actual loading amont of Cu and Pd is measured to be 1.4 wt% and 1.3 wt%, respectively. Powder X-ray diffraction (P-XRD) was employed to measure the crystal structure of Cu3Pd2/TiN. The XRD patterns as shown in Fig. S3 (Supporting information) distinctly display the diffraction peaks of TiN nanoparticles. The characteristic peaks of Cu or Pd were not observed due to the low metal loading.
Figure 1
Figure 1.
(a–c) HRTEM images, and corresponding EDS elemental mapping of Cu3Pd2/TiN. (d) High-resolution Cu 2p XPS spectra of Cu3Pd2/TiN and Cu/TiN. (e) High-resolution Pd 3d XPS spectra of Cu3Pd2/TiN and Pd/TiN. (f) Top and side views of the Cu6Pd4(111)/TiN(200). (g) The corresponding charge density difference of Cu6Pd4(111)/TiN(200) structure. (h) The projected density of states (pDOS) plots of the d-orbitals for Cu and Pd with Bader charge analysis inserted. (i) UV–vis DRS spectra of Cu3Pd2/TiN, Cu/TiN and Pd/TiN measured with TiN as the reference.
The surface chemical states of the Cu3Pd2/TiN photocatalyst were studies using XPS. As shown in Fig. 1d, the high-resolution Cu spectra demonstrate the co-presence of Cu0 and Cu2+ species. For monometallic Cu/TiN, the prominent peaks at 932.42 eV (Cu 2p3/2) and 952.05 eV (Cu 2p1/2) belong to metallic Cu, while peak at 934.52 eV (Cu 2p3/2) is assigned to the oxidized Cu2+ species. The peaks of Cu 2p in Cu3Pd2/TiN shift towards to lower binding energy (about ~0.2 eV) relative to that of Cu/TiN. The Cu2+ species were observed at 934.34 eV and 934.52 eV for both Cu3Pd2/TiN and Cu/TiN samples, suggesting that the Cu species are unstable in air and tends to oxidize to form Cu2+ species on the surface [20]. A more prominent Cu2+ peak can be seen in the bimetallic Cu3Pd2/TiN spectrum, which indicates a partial surface oxidation on the catalysts due to the exposure to oxygen in air [25]. As depicted in Fig. 1e, Pd/TiN exhibits two principal peaks for Pd 3d centered at 334.97 and 340.15 eV, corresponding to the Pd0 states of Pd 3d5/2 and Pd 3d3/2, respectively [27]. The binding energy for Pd0 is observed at 335.24 eV, which is about 0.27 eV higher as compared to that of monometallic Pd/TiN. The elevated binding energy of Pd and reduced binding energy of Cu in Cu3Pd2/TiN as compared to Pd/TiN, signifies an electron transfer between Cu and Pd, a consequence of the alloying process.
The DFT calculations were also performed by taking in account the loading of CuPd on TiN and the morphology demonstrated by TEM. The model structure of Cu6Pd4(111)/TiN(200) (Fig. 1f) was successfully constructed to represent the Cu3Pd2/TiN catalyst with accurate stoichiometric ratios of Cu, Pd and TiN (Fig. S4 in Supporting information). The charge density difference (Fig. 1g) combined with the Bader charge analysis (Fig. 1h inserted) indicated that the TiN surface (−24.00, negatively charged) transfer electrons to CuPd alloy that enriched the electron density on CuPd surface. The negatively charged Cu (−3.41) and positively charged Pd (3.92) implied the electrons transfer inside the CuPd alloy nanoparticles. In addition, the d-band center of the present alloy catalyst has also been calculated. The projected density of states (pDOS) showed that the d-band center of Pd cluster on TiN surface is closer to the Fermi level than Cu (Fig. 1h, −2.31 vs. −2.59), suggesting that Pd is more active than Cu and more likely to serve as the dominant catalytic active site.
The light absorption properties of Pd/TiN, Cu/TiN, and Cu3Pd2/TiN were analyzed using UV–vis DRS with TiN as the reference background (Fig. 1i). Cu3Pd2/TiN exhibits broad light absorption in the visible range, and the spectrum of CuPd alloy nanoparticles has significant deviations from those of the monometallic nanoparticles. It is known that monometallic Cu nanoparticles feature a significant absorption peak near 580 nm (Fig. S5 in Supporting information) [25], which is attributed to the LSPR absorption characteristics of Cu nanoparticles. However, the LSPR absorption of Cu nanoparticles closely overlaps with that of TiN [28,29], delivering no characteristic LSPR absorption in the Cu/TiN sample. Moreover, there is a significant metal-support interaction between the metal nanoparticles and the TiN substrate, resulting in the substantial shift and broadening of light absorption peaks for the TiN supported photocatalysts. The UV–vis DRS data indicates that the Cu3Pd2/TiN photocatalyst is able to utilize most of the irradiation energy delivered in the solar spectrum.
A series of photocatalysts composed of nanoparticles with different ratios of Cu and Pd supported on TiN were utilized for the Heck cross-coupling reaction under visible light irradiation. Through the optimization of reaction conditions (Tables S2 and S3 in Supporting information), the photocatalytic Heck reaction had been enabled to proceed under exceptionally mild conditions (50 ℃) using iodobenzene and styrene as reactants. As shown in Fig. 2a, monometallic Cu/TiN could hardly drive Heck reaction, and Pd/TiN delivers poor photocatalytic activity (10%). By contrast, formation of CuPd alloy nanoparticles could significantly enhance the activities. The Cu3Pd2/TiN photocatalyst (with molar ratio of Cu: Pd = 3:2) exhibited the optimal performance, achieving 96% reaction conversion and a high selectivity of 99% for the desired product stilbene. Further raising the reaction temperature to 100 ℃ could achieve a decent catalytic activity (73.2%), but this thermal-driven activity is still lower compared with that obtained under visible light irradiation conducted at 50 ℃. The above results clearly suggest that Cu3Pd2/TiN is an efficient photocatalyst to drive Heck reaction using visible light under very mild conditions instead of conventional thermal heating.
Figure 2
Figure 2.
(a) Photocatalytic performance of Heck reaction using catalysts with different Cu/Pd ratios under visible light irradiation at 50 ℃. (b) Dependence of the photocatalytic activity on the light intensity using Cu3Pd2/TiN catalysts for Heck reaction. (c) The action spectra for Heck reaction. UV–vis DRS spectra of Cu3Pd2/TiN and Cu/TiN were included for comparison. Reaction activities presented as apparent quantum efficiencies (AQE) are plotted against the wavelength of irradiation at 400, 450, 500, 550, 600 and 650 nm (light intensity 0.5 W/cm2). (d) Catalytic activity of Cu3Pd2/TiN catalysts for the Heck reaction at different temperatures under visible light irradiation (red dot) and thermal dark conditions (blue dot). (e) The kinetic study of the photocatalytic reaction and thermal reaction.
To gain deeper insights into the photocatalytic Heck reaction, we explored the impact of light intensity. Reaction conversion increased with the increment in irradiation intensity from 0.1 W/cm² to 0.2, 0.3, 0.4, and 0.5 W/cm² (Fig. 2b). A linear relationship between irradiation intensity and photocatalytic conversion was evident: higher intensity corresponded to higher conversion, suggesting a significant contribution of light irradiation to the photocatalytic reaction. Notably, when an intensity of 0.1 W/cm² was applied, the contribution of light irradiation was only 10%, with a subsequent increase in light intensity resulting in higher conversion rates following an almost linear trend. Higher light intensity induces an elevation in the number of excited energetic electrons and a stronger electromagnetic field near the surface of the plasmonic nanoparticles [30], thereby facilitating the photocatalytic reaction.
In gaining insight into the photocatalytic mechanism, the irradiation wavelength significantly influences the photocatalytic reaction, as indicated by the action spectra analysis, revealing variations in the photocatalytic activity (apparent quantum efficiencies) with changing irradiation wavelengths (Fig. 2c). It was observed that the AQEs align with the trend of light absorption depicted in the UV–vis DRS spectra of Cu3Pd2/TiN. The peak AQE is observed at the shorter wavelength (400 nm), while longer wavelength photons demonstrate reduced activity. The noticeable variance in AQE values across different wavelengths implies that the reaction under shorter wavelength irradiation may be propelled by the excitation electron transfer pathway. Initially, the reliance on wavelength implies that the role of photothermal effects in the reaction rate is negligible. The light absorption of nanoparticles can induce a temporary elevation in lattice temperature, known as the photothermal effect [31]. The elevated lattice temperature at the nanoparticle surface promotes the occurrence of catalytic reaction; furthermore, the longer the wavelength of the light, the more pronounced this temperature elevation becomes. Nevertheless, light irradiation with wavelengths > 600 nm failed to effectively catalyze the reaction. For instance, Cu NPs demonstrate significant absorption at 580 nm; however, its catalytic activity is diminished under irradiation at this wavelength. Conversely, the photocatalytic reaction employing a shorter wavelength with higher energy demonstrates a highest activity, implying that the photothermal effect is not the main cause for the reaction. In this case, the photons with shorter wavelength could excite energetic electrons to higher energy levels, making the charge transfer process more readily to initiate the catalytic reaction. Consequently, we conclude that the Heck reaction is propelled not by photothermal effects but rather by direct photon energy. Secondly, the pronounced decline in catalytic performance observed at wavelengths > 550 nm suggests the presence of a threshold level of photon energy necessary for the reaction to occur [32].
To further explore the impact of reaction temperature on the catalytic process, we performed both photocatalytic and thermal Heck reactions at varied temperatures, and the reaction temperatures were maintained ranging from 10 ℃ to 100 ℃ controlled through external heating or cooling. We observed that a moderate increase of reaction temperature to 50 ℃ significantly accelerated the photocatalytic Heck reaction (Fig. 2d). In contrast, the thermal reaction conducted in the dark achieved very low activity (5.6%), further increasing temperature to 100 ℃, the thermal reaction could achieve a decent catalytic activity (73.2%). This acceleration associated with temperature is attributed to the enhanced vibrational states of the adsorbed reactant molecules, which decrease the energy required for photo-excited electrons to overcome activation barriers [33]. Kinetic analysis of both photocatalytic and thermal catalytic processes was conducted (Fig. 2e), it is shown that under traditional thermal conditions in the dark, the apparent activation energy for the Heck reaction was approximately 119.09 kJ/mol, while the photocatalytic process exhibited a much lower apparent activation energy of approximately 69.95 kJ/mol. These findings conclusively demonstrate the effective utilization of visible light in driving Heck reaction, and light irradiation could lower reaction activation energy compared with conventional thermal catalysis.
Photocatalytic Heck cross-coupling reactions were investigated on a small scope utilizing various substituted aryl iodides with styrene (Table S4 in Supporting information). Visible light irradiation was capable of driving the reaction using aryl iodides featuring either electron-donating or electron-withdrawing groups, resulting in reasonable conversions and excellent selectivity. Control experiments conducted in the dark exhibited no activity. Unfortunately, no detectable reaction was observed when bromobenzene or chlorobenzene was employed, this can be attributed to the substantially higher bonding activation energies associated with the respective C—Br and C—Cl bonds, rendering the photocatalytic process considerably challenging under ambient conditions.
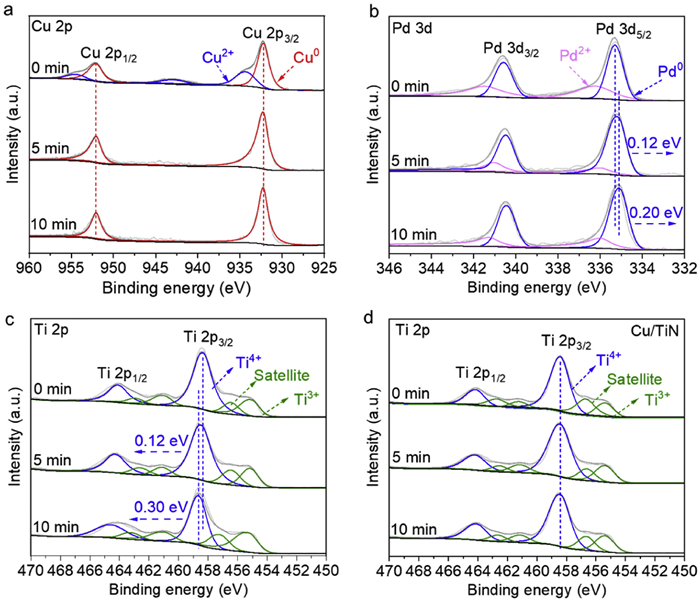
To clearly elucidate the surface electronic properties of photocatalyst under photocatalytic reaction conditions, we conducted in-situ XPS measurements on Cu3Pd2/TiN under visible light irradiation. As shown in Fig. 3a, before irradiation, the Cu2+ peak is clearly observed in Cu 2p spectrum, which is due to the slight oxidation of the surface Cu species in the alloy sample, and this result is in line with that shown in Fig. 1d. The Cu2+ peak disappeared under irradiation, and Cu0 exists as the major component in the CuPd alloy nanoparticles. This result suggests that the surface oxidized Cu species could be reduced under irradiation, the in-situ formed metallic Cu species in the alloy nanoparticles could take advantage of the strong interactions of Cu with visible light manifested in the LSPR photoexcitation. Cu-based nanoparticles often suffer from inevitable partial oxidation of their surface atoms upon exposure to air, which negatively impacts their catalytic activity. It has been shown that LSPR photoexcitation could switch the oxidation states of surface atoms of Cu nanoparticles, thereby controlling the photocatalytic performance with unique product selectivity [22]. Under light irradiation, the LSPR photoexcited transient electrons could transfer to the antibonding of Cu—O orbitals of the surface oxidized Cu species, leading to the facile reduction of the oxidized Cu species to metallic Cu. As evidence by the XPS data, the in-situ formed metallic Cu is rather stable under continuous irradiation, therefore the LSPR of Cu could be fully excited and maintained under irradiation.
Figure 3
Figure 3.In-situ high-resolution XPS spectra under visible light irradiation (0–10 min): (a) Cu 2p, (b) Pd 3d, (c) Ti 2p XPS spectra of Cu3Pd2/TiN and (d) Ti 2p XPS spectra of Cu/TiN.
As observed in Fig. 3b, the binding energy of the Pd 3d3/2 core level (Pd0) decreased by approximately 0.2 eV after 10 min of irradiation. Considering the concurrent changes in the Pd2+ composition, we infer that this reduction is attributed to electron transfer to Pd from other components, resulting in electron-enriched Pd sites. Furthermore, Fig. 3c demonstrates an increase of about 0.3 eV in the binding energy of Ti 2p after 10 min of irradiation, confirming the occurrence of electron transfer between the Pd species of CuPd alloy and the Ti species of TiN support. To corroborate these findings, we also conducted in-situ XPS analysis on the monometallic Cu/TiN (Fig. 3d and Fig. S6 in Supporting information). The results from Fig. 3d indicate that the binding energy of Ti 2p on Cu/TiN did not change under illumination, suggesting that Cu, serving as the photonic capture site, is excited via its LSPR excitation without significant electron gain or loss. The electron transfer between TiN and Pd components is also promoted due to the plasmonic enhancement. Moreover, the transformation of Pd into an electron-rich site facilitates its role as the active sites for catalytic reactions [19].
In Heck reactions, the adsorption of reactants (iodobenzene and styrene) onto the active sites of catalysts is crucial for the activation process. Accordingly, this study utilized in-situ DFTIR to meticulously examine the dynamic surface reactions of catalysts under illumination. As depicted in Fig. 4, in the absence of light, the Cu3Pd2/TiN catalyst exhibited pronounced characteristic peaks for aromatic C═C stretching vibrations between 1450 cm−1 (Fig. 4a), whereas the corresponding peaks for iodobenzene on the TiN substrate were notably weaker (arrow in Fig. 4b) [34]. This contrast underscores the superior chemisorption capabilities of CuPd alloy nanoparticles for iodobenzene. Under illuminated conditions, both catalysts demonstrated enhanced adsorption of iodobenzene, with an incremental increase in peak intensity correlating with the duration of light exposure. Concurrently, testing on styrene under identical conditions revealed no detectable peaks for styrene in the dark for either Cu3Pd2/TiN or TiN substrates (Figs. 4c and d). However, under illumination, peaks corresponding to styrene were observed for both, corroborating the positive effect of light on enhancing the chemisorption of styrene on the catalysts. These findings highlight the distinct advantage of CuPd alloy nanoparticles over TiN in the chemisorption of reactants and reveal the significant role of light in augmenting the adsorptive capacities on catalysts.
Figure 4
Figure 4.In-situ DRIFTS spectra of iodobenzene and styrene adsorbed on catalysts. (a, b) Infrared spectra of iodobenzene on Cu3Pd2/TiN catalysts and on TiN support under continuous visible light irradiation. (c, d) Infrared spectra of styrene on Cu3Pd2/TiN catalysts and on TiN support under continuous visible light irradiation.
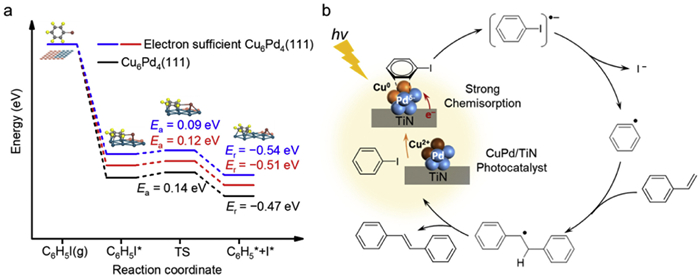
Since the Heck reaction takes place on the CuPd surface, it is rational to simulate the reaction coordinate on the alloy surface without considering the effect of support (Fig. S7 in Supporting information). Previous in-situ XPS results and Bader charge analysis have indicated that the CuPd surface is electron enriched under light irradiation, especially for the electron transfer between TiN surface and Pd site. Therefore, further comparing the activation of reactant on both CuPd and electron-rich CuPd sites would be imperative for the Heck reaction mechanism in photocatalysis. As the activation of iodobenzene (C—I bond) has been reported as the elemental step for cross-coupling reactions [19], in this work, we calculated the iodobenzene dissociation on both Cu6Pd4(111) and electron-rich Cu6Pd4(111) surfaces. In advance, the adsorption of iodobenzene was evaluated to find the most stable adsorption site (Fig. S8 in Supporting information). According to our calculated adsorption energies, the surface Pd NPs are the thermodynamically ideal sites (Fig. S8a, −3.43 eV) for iodobenzene activation rather than Cu sites, which further confirmed that Cu mainly play the role of light harvesting, while Pd are the dominant active sites for catalytic reaction. With the above data in hand, we successfully depicted the reaction pathway for iodobenzene dissociation. As shown in Fig. 5a, the dissociation barrier on pure Cu6Pd4(111) is 0.14 eV and it is exothermic by −0.47 eV, which is favored kinetically and thermodynamically. The corresponding structures during dissociation are listed in Fig. S9 (Supporting information). In comparison, the dissociation of iodobenzene is much easier on electron-rich Cu6Pd4(111) as it only requires overcoming a barrier of 0.12 eV (Fig. 5a, red line) which is lower than that on pure Cu6Pd4(111). Meanwhile, it is also an exothermic process on electron-rich surface and it is even more favored thermodynamically than that on pure surface (−0.51 eV vs. −0.47 eV). To further verify our speculation, we further enhanced the electron amount on Cu6Pd4(111). It has been found that the energy barrier (Ea) and reaction energy (Er) subsequently decreased to 0.09 and −0.51 eV, respectively (Fig. 5a, blue line). Based on our theoretical calculation, the function of electron-rich CuPd sites has been confirmed in the promotion of this catalytic process.
Figure 5
Figure 5.
(a) Potential energy surface of iodobenzene dissociation over Cu6Pd4(111) and electron-rich Cu6Pd4(111) surfaces. (b) Proposed photocatalytic Heck reaction mechanism.
Based on our experimental findings and the DFT calculation, we propose a plausible mechanism (Fig. 5b). Initially, a fraction of Cu atoms on the alloy nanoparticles' surface underwent oxidation to CuO via O2 in the atmosphere. This Cu2+ species was subsequently reduced to Cu0 upon visible light irradiation. Concurrently, Cu3Pd2/TiN facilitated the excitation of electron-hole pairs under irradiation, with Pd on the alloy nanoparticles' surface gaining electrons. Previous studies have demonstrated that, in non-homogeneous phase catalytic systems, the Heck reaction predominantly occurs on the surface of Pd nanoparticles [19]. The electron-rich Pd sites promote the strong chemisorption and activation of iodobenzene, facilitating the breaking of the C—I bond to generate phenyl radicals (Table S5 in Supporting information). Subsequently, the olefins undergo migratory insertion and β-H elimination, yielding the desired products. The synergistic effect of bimetallic CuPd NPs and TiN effectively suppresses electron-hole pair recombination, thereby enhancing the overall photocatalytic performance [23].
In summary, we developed plasmonic photocatalyst composed of CuPd alloy nanoparticles supported on TiN for visible-light-driven Heck reaction. The Cu3Pd2/TiN photocatalyst exhibited high activity (> 96%) and selectivity (> 99%) in catalyzing Heck reaction under visible light irradiation at 50 ℃. The surface charge state of CuPd alloy nanoparticles was characterized through in-situ XPS measurements. Visible light excitation could achieve stable metallic Cu species on the surface of CuPd alloy nanoparticles, thereby eliminating the inevitable surface oxides of Cu based catalyst. The in-situ formed metallic Cu species under irradiation take advantage of the strong interactions of Cu with visible light, and demonstrate LSPR excitation. Photogenerated electrons were efficiently transferred to the Pd sites on the CuPd alloy nanoparticles, resulting in electron enrichment on the Pd surface and facilitating the activation of the C—I bond. Furthermore, it was demonstrated for the first time that effective visible light promoted chemisorption of reactants on Cu3Pd2/TiN, as confirmed by in-situ DRIFTS, thereby promoting the reaction. This study offers insights that may inspire future research in the construction of bimetallic nanoparticles for organic synthesis under light irradiation. Additionally, it serves as a reference for further exploration of the mechanism underlying photocatalytic organic synthesis.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by Shanghai Pujiang Program (No. 21PJ1400400), the Research Start-up Fund at Donghua University, the Foundation of State Key Laboratory of Coal Combustion (No. FSKLCCA2309), the National Natural Science Foundation of China (No. 22302109), and the Australian Research Council (No. DP230102740).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.110299.
Z. Xi, J. Li, D. Su, et al., J. Am. Chem. Soc. 139 (2017) 15191–15196. doi: 10.1021/jacs.7b08643
[27]
Y. Xu, C. Zhang, L. Zhang, et al., Energy Environ. Sci. 9 (2016) 2410–2417. doi: 10.1039/C6EE00830E
[28]
W. Li, U. Guler, N. Kinsey, et al., Adv. Mater. 26 (2014) 7959–7965. doi: 10.1002/adma.201401874
[29]
A.A. Hussain, B. Sharma, T. Barman, et al., ACS Appl. Mater. Interfaces 8 (2016) 4258–4265. doi: 10.1021/acsami.6b00249
[30]
Q. Xiao, S. Sarina, E. Jaatinen, et al., Green Chem. 16 (2014) 4272–4285. doi: 10.1039/C4GC00588K
[31]
S. Mukherjee, F. Libisch, N. Large, et al., Nano Lett. 13 (2013) 240–247. doi: 10.1021/nl303940z
[32]
S. Sarina, E. Jaatinen, Q. Xiao, et al., J. Phys. Chem. Lett. 8 (2017) 2526–2534. doi: 10.1021/acs.jpclett.7b00941
[33]
Q. Xiao, Z. Liu, A. Bo, et al., J. Am. Chem. Soc. 137 (2015) 1956–1966. doi: 10.1021/ja511619c
[34]
J. Tan, H. Zhu, S. Cao, et al., RSC Adv. 10 (2020) 43175–43186. doi: 10.1039/D0RA08653C
Figure 1
(a–c) HRTEM images, and corresponding EDS elemental mapping of Cu3Pd2/TiN. (d) High-resolution Cu 2p XPS spectra of Cu3Pd2/TiN and Cu/TiN. (e) High-resolution Pd 3d XPS spectra of Cu3Pd2/TiN and Pd/TiN. (f) Top and side views of the Cu6Pd4(111)/TiN(200). (g) The corresponding charge density difference of Cu6Pd4(111)/TiN(200) structure. (h) The projected density of states (pDOS) plots of the d-orbitals for Cu and Pd with Bader charge analysis inserted. (i) UV–vis DRS spectra of Cu3Pd2/TiN, Cu/TiN and Pd/TiN measured with TiN as the reference.
Figure 2
(a) Photocatalytic performance of Heck reaction using catalysts with different Cu/Pd ratios under visible light irradiation at 50 ℃. (b) Dependence of the photocatalytic activity on the light intensity using Cu3Pd2/TiN catalysts for Heck reaction. (c) The action spectra for Heck reaction. UV–vis DRS spectra of Cu3Pd2/TiN and Cu/TiN were included for comparison. Reaction activities presented as apparent quantum efficiencies (AQE) are plotted against the wavelength of irradiation at 400, 450, 500, 550, 600 and 650 nm (light intensity 0.5 W/cm2). (d) Catalytic activity of Cu3Pd2/TiN catalysts for the Heck reaction at different temperatures under visible light irradiation (red dot) and thermal dark conditions (blue dot). (e) The kinetic study of the photocatalytic reaction and thermal reaction.
Figure 3In-situ high-resolution XPS spectra under visible light irradiation (0–10 min): (a) Cu 2p, (b) Pd 3d, (c) Ti 2p XPS spectra of Cu3Pd2/TiN and (d) Ti 2p XPS spectra of Cu/TiN.
Figure 4In-situ DRIFTS spectra of iodobenzene and styrene adsorbed on catalysts. (a, b) Infrared spectra of iodobenzene on Cu3Pd2/TiN catalysts and on TiN support under continuous visible light irradiation. (c, d) Infrared spectra of styrene on Cu3Pd2/TiN catalysts and on TiN support under continuous visible light irradiation.

 DownLoad:
DownLoad:



 下载:
下载:






 下载:
下载:
