Key Laboratory for Advanced Technology in Environmental Protection of Jiangsu Province, Yancheng Institute of Technology, Yancheng 224051, China
b.
School of Environmental Ecology and Biological Engineering, Key Laboratory of Green Chemical Engineering Process of Ministry of Education, Engineering Research Center of Phosphorus Resources Development and Utilization of Ministry of Education, Wuhan Institute of Technology, Wuhan 430205, China
Received Date:
28 May 2024 Accepted Date:
18 July 2024 Revised Date:
12 July 2024 Available Online:
15 January 2025
Abstract:
Photocatalytic overall pure water splitting is a promising method for generating green hydrogen energy under mild conditions. However, this process is often hindered by sluggish electron-hole separation and transport. To address this, a step-scheme (S-scheme) B-doped N-deficient C3N4/O-doped C3N5 (BN-C3N4/O-C3N5) heterojunction with interfacial B-O bonds has been constructed. Utilizing Pt and Co(OH)2 as co-catalysts, BN-C3N4/O-C3N5 S-scheme heterojunction demonstrates significantly enhanced photocatalytic activity for overall pure water splitting under visible light, achieving H2 and O2 evolution rates of 40.12 and 19.62 µmol/h, respectively. Systematic characterizations and experiments revealed the performance-enhancing effects of the enhanced built-in electric field and the interfacial B-O bonding. Firstly, the strengthened built-in electric field provides sufficient force for rapid interfacial electron transport. Secondly, by reducing the transport energy barrier and transfer distance, the interfacial B-O bonds facilitate rapid recombination of electrons and holes with relatively low redox potential via the S-scheme charge-transfer route, leaving the high-potential electrons and holes available for H+ reduction and OH− oxidation reactions. Overall, the photocatalytic efficiency of BN-C3N4/O-C3N5 S-scheme heterojunction was significantly improved, making it a promising approach for green hydrogen production through overall pure water splitting.
Light-driven water splitting into H2 and O2 offers a promising pathway to sustainable energy production [1-3]. Despite the design and synthesis of various photocatalysts, their photocatalytic activities remain relatively low [4,5]. It is widely accepted that single-component photocatalytic systems face inherent challenges for overall water splitting due to the thermodynamic contradiction between optical absorption and redox potentials [6-8]. In contrast, heterojunction photocatalytic systems, which utilize two components, are particularly appealing for enhancing photocatalytic activity [9-13].
An efficient heterojunction photocatalytic system typically requires well-matched band structures and an unobstructed pathway for electron transfer. The former ensures that the overall water splitting reaction can proceed, while the latter ensures that it can proceed rapidly [14,15]. Among the various heterojunction photocatalysts designed, C3N4-based photocatalysts show great potential due to their strong reduction and oxidation abilities for water splitting [16-19]. For instance, with Pt and RuO2 as cocatalysts, a α-Fe2O3/C3N4 heterojunction has achieved visible-light-driven overall water splitting [20]. Similarly, WO3·H2O/C3N4 [21], Fe2O3/RGO/C3N4 [22], g-C3N4/MnTiO3 [23], Bi4O5Br2/g-C3N4 [24], CoTiO3/g-C3N4 [25] and BiVO4/C3N4 [26] have all demonstrated certain levels of photocatalytic overall water splitting performance. However, the photocatalytic activities of C3N4-based photocatalysts remain low due to unsatisfactory electron transfer efficiency [27-30]. Typically, inorganic semiconductor components are attached to the surface of C3N4via van der Waals forces, which are not strong enough to facilitate rapid interfacial transport of photoinduced electrons. To address this issue, all-polymer heterojunctions have been developed [31,32]. For example, the C3N4/RGO/perylene diimide polymer heterojunction has shown high-flux interfacial charge transfer with lower charge-transfer resistance [33]. Furthermore, a BN-C3N4/C3N4 heterojunction achieved stoichiometric H2 and O2 evolution in the presence of Pt and Co(OH)2 cocatalysts, with a solar-to-hydrogen efficiency of 1.16% under one-sun illumination [34]. As a member of the polymeric carbon nitride family, g-C3N5 is an efficient H2-evolving photocatalyst under visible light due to its narrow bandgap (1.7 eV) and sufficiently negative conduction band (CB) potential (−0.79 eV) [35]. Compared with C3N4, C3N5 has a higher electronic density and a more extended π conjugated network, which endows it with extremely high charge transfer efficiency [36,37]. Furthermore, owing to the nitrogen-rich property, C3N5 has an asymmetric structure with a 2.80 D dipole moment in one unit, which can harness photogenerated charge separation kinetics [38]. By modulating the band and electronic structure of C3N5 and C3N4, it is possible to construct an efficient polymeric photocatalyst heterojunction for overall water splitting.
Following the above hypothesis, we constructed an S-scheme BN-C3N4/O-C3N5 heterojunction for photocatalytic overall water splitting. In this system, BN-C3N4 and O-C3N5 are tightly connected via interfacial B-O bonds. With the addition of Pt and Co(OH)2 co-catalysts, BN-C3N4/O-C3N5 can efficiently split water into stoichiometric H2 and O2 under visible light, achieving evolution rates of 40.12 and 19.62 µmol/h, respectively. This superior performance is attributed to the interfacial B-O bonds and the built-in electronic field. The interfacial B-O bonds provide a direct S-scheme electron-transfer pathway between BN-C3N4 and O-C3N5, while the built-in electronic field supplies sufficient force to facilitate the transfer of photoinduced electrons.
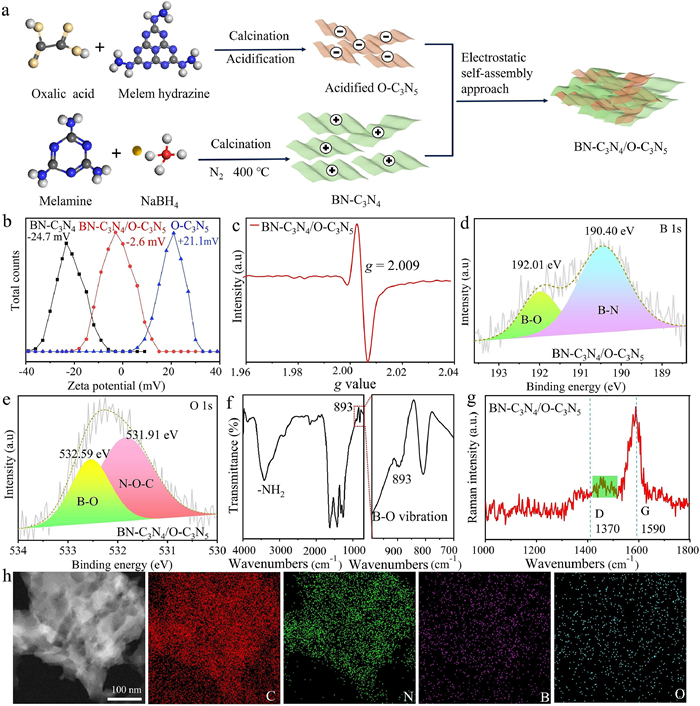
Fig. 1a presents the basic synthetic procedure for the S-scheme BN-C3N4/O-C3N5 heterojunction. Through rapid calcination of a mixture of NaBH4 and C3N4 nanosheets in an Ar atmosphere, B atoms were introduced into the C3N4 framework without compromising its structural integrity (Figs. S1 and S2 in Supporting information) [39]. The B atoms substitute carbon atoms at corner sites (inset in Fig. S3 in Supporting information) and form B-N bonds with adjacent N atoms (Fig. S4 in Supporting information) [40]. In BN-C3N4, B atoms are uniformly distributed (Fig. S5 in Supporting information). The C: N atom ratio in BN-C3N4 is 0.81 (Table S1 in Supporting information), slightly higher than that of pristine C3N4 (0.77). This increased C: N ratio indicates the successful introduction of nitrogen vacancies [41,42], as confirmed by 13C NMR and EPR spectra. Compared with C3N4, C3N4 shows two new peaks at 123.6 and 171.5 ppm in the 13C NMR spectrum (Fig. S6 in Supporting information) [43], which correspond to the C atoms of N≡C- groups and neighboring C atoms, respectively [34]. This results from the nitrogen defects in the C3N4 framework [44]. Furthermore, BN-C3N4 exhibits a significantly stronger EPR signal (Fig. S7 in Supporting information) [45] and a more pronounced ID Raman peak at 1370 cm−1 (Fig. S8 in Supporting information) compared to pristine C3N4, both indicating the formation of nitrogen vacancies [46-49].
Figure 1
Figure 1.
(a) Schematic of the synthesis of BN-C3N4/O-C3N5. (b) Zeta potential of the BN-C3N4 and O-C3N5 dispersed in deionized water at pH 7. (c) EPR spectrum of BN-C3N4/O-C3N5. (d) B 1s XPS spectrum of BN-C3N4. (e) O 1s XPS spectrum of O-C3N5. (f) FTIR spectrum of BN-C3N4/O-C3N5. (g) Raman spectrum of BN-C3N4/O-C3N5. (h) Mapping images of BN-C3N4/O-C3N5.
The as-synthesized O-C3N5 nanosheets, which obtained by exfoliation with sulfuric acid (Fig. S9 in Supporting information), exhibit a smaller interplanar distance compared to BN-C3N4, indicated by the slightly higher 2θ of (002) peaks (Fig. S10 in Supporting information). This difference is due to the enhanced π-conjugation degree of the C3N5 layers [36]. The absence of a (100) peak, a specific feature of in-plane packing, suggests the distortion of C3N5 framework and the broadening of nanochannel distance between heptazine units due to azo (-N=N-) bridging linkage [36]. By partially substituting N with O in the s-triazine units, O atoms are successfully incorporated into the C3N5 framework (Fig. S11 in Supporting information). Additionally, the increased ID/IG ratio of O-C3N5 compared to C3N5 suggests successful O atom doping (Fig. S12 in Supporting information) [50]. The doped O atoms form C-O-C or N-O-C bonds with neighboring C or N atoms, as evidenced by the XPS peak at 531.91 eV [51]. These O atoms are uniformly distributed within the material (Fig. S13 in Supporting information).
The S-scheme BN-C3N4/O-C3N5 heterojunction was formed through a spontaneous self-assembly process due to the completely opposite zeta potentials of BN-C3N4 (−24.7 mV) and O-C3N5 (21.1 mV) in pure water (Fig. 1b). The nitrogen vacancies remained stable during the self-assembly process (Fig. 1c), and the components BN-C3N4 and O-C3N5 were tightly connected (Fig. S14 in Supporting information). In the B 1s XPS spectrum of BN-C3N4/O-C3N5, the peak at 192.01 eV, attributed to the B-O bond [52], was not detected in BN-C3N4 alone (Fig. 1d). For comparison, the sample BN-C3N4/O-C3N5-MIX, obtained by physically mixing BN-C3N4 and O-C3N5, showed a much smaller shift in the B 1s binding energy and lacked B-O bonds (Fig. S15 in Supporting information). Furthermore, the interfacial B-N bonds were homogeneously distribution in BN-C3N4/O-C3N5 for the peaks of B-N bond were detected in different etching depths (EDs, Fig S16 in Supporting information). The additional peak at 532.60 eV in the O 1s spectrum of BN-C3N4/O-C3N5 further confirmed the formation of interfacial B-O bonds (Fig. 1e) [53]. In the FTIR spectrum, the band in the range of 891-939 cm−1 was ascribed to the B-O out-of-plane bending vibration (Fig. 1f) [54]. Moreover, the Raman spectrum of BN-C3N4/O-C3N5 exhibited a distinct additional peak at approximately 1442 cm−1, which is related to the B-O vibration (Fig. 1g) [55]. Thus, the interfacial B-O bonds were successfully introduced into the BN-C3N4/O-C3N5 heterojunction. The doped B and O atoms were highly dispersed in the as-developed BN-C3N4/O-C3N5 heterojunctions (Fig. 1h). The nitrogen vacancies remained stable throughout the self-assembly process.
The optical properties and band structures of BN-C3N4 and O-C3N5 were determined using ultraviolet-visible diffuse reflectance spectroscopy (UV-vis DRS) and VB XPS spectroscopy. As shown in Fig. S17a (Supporting information), BN-C3N4 exhibits a maximum absorption edge at 525 nm, with an absorption tail extending to 800 nm, further confirming the generation of dopant and defect-related midgap states [38,56]. O-C3N5, on the other hand, shows a maximum absorption edge at 708 nm. The calculated intrinsic bandgaps of BN-C3N4 and O-C3N5 are 2.31 and 1.75 eV, respectively (Fig. S17b in Supporting information). According to the VB XPS spectra (Fig. S18 in Supporting information), the VB maximum of BN-C3N4 and O-C3N5 are 2.29 and 0.92 eV, respectively. Consequently, the CB positions of BN-C3N4 and O-C3N5 are −0.02 and −0.83 eV, respectively. The band positions of BN-C3N4 and O-C3N5 are illustrated in Fig. S19 (Supporting information). Notably, the CB of O-C3N5 is sufficiently negative for the water reduction reaction, while the VB position of BN-C3N4 is sufficiently positive for the water oxidation reaction. Therefore, the constructed S-scheme BN-C3N4/O-C3N5 heterojunction is well-suited for photocatalytic overall water splitting [57].
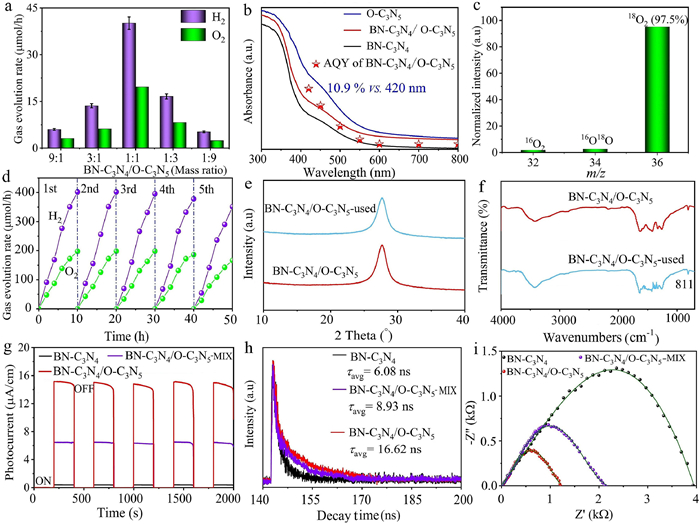
The photocatalytic overall water-splitting experiments were conducted in pure water without the addition of sacrificial reagents under visible light illumination (λ ≥ 420 nm). Pt and Co(OH)2 were loaded onto O-C3N5 and BN-C3N4 as co-catalysts for water reduction and oxidation, respectively (Figs. S20 and S21 in Supporting information). The resulting photocatalyst was denoted as Co(OH)2-BN-C3N4/Pt-O-C3N5. Almost no gas was detected in the photocatalytic systems of Co(OH)2-BN-C3N4 and Pt-O-C3N5 under the same conditions (Fig. S22 in Supporting information). The trace amount of hydrogen gas detected over Pt-O-C3N5 was due to the O dopant which can act as a recombination center for electrons and holes (Fig. S23 in Supporting information) [58,59]. However, when combined, Co(OH)2-BN-C3N4/Pt-O-C3N5 exhibited significantly enhanced photocatalytic activity, with the molar ratio of Co(OH)2-BN-C3N4 to Pt-O-C3N5 playing a crucial role in the visible-light-driven photocatalytic activity (Fig. 2a). Among the tested ratios, Co(OH)2-BN-C3N4/Pt-O-C3N5 with a molar ratio of 1:1 demonstrated the highest photocatalytic activity for overall pure water splitting. The corresponding H2 and O2 evolution rates reached approximately 40.12 and 19.62 µmol/h, respectively, with a molar ratio of detected H2 to O2 being nearly 2:1. In contrast, Co(OH)2-BN-C3N4/Pt-O-C3N5-MIX exhibited H2 and O2 evolution rates of 18.1 and 8.9 µmol/h, respectively, much lower than those of Co(OH)2-BN-C3N4/Pt-O-C3N5 heterojunction. Clearly, the strengthened interface interaction between O-C3N5 and BN-C3N4 is crucial for the well-managed charge-transfer process and the improved performance of photocatalytic overall water splitting.
Figure 2
Figure 2.
(a) Effect of the mass ratio of BN-C3N4 to O-C3N5 on the photocatalytic activity. (b) AQY of BN-C3N4/O-C3N5. (c) 18O isotope-labelled water experiment results of BN-C3N4/O-C3N5. (d) The photostability of BN-C3N4/O-C3N5. (e) XRD patterns of BN-C3N4/O-C3N5 before and after the reaction. (f) FTIR spectra of BN-C3N4/O-C3N5 before and after the reaction. (g) I-t curve of BN-C3N4/O-C3N5. (h) Time-resolved PL of BN-C3N4/O-C3N5. (i) EIS plot of BN-C3N4/O-C3N5.
The apparent quantum yield (AQY) of Co(OH)2-BN-C3N4/Pt-O-C3N5 was measured under the same photocatalytic reaction conditions with different monochromatic light wavelengths of 420, 450, 500, 550, and 600 nm. The AQY was calculated according to the two-step excitation process. A maximum AQY of 10.9% was achieved at 420 nm, gradually decreasing to 7.6% at 450 nm, 3.5% at 500 nm, 0.9% at 550 nm, and 0.0% at 600 nm. As depicted in Fig. 2b, the consistency between the optical absorption of Co(OH)2-BN-C3N4/Pt-O-C3N5 and the AQYs at different wavelengths further suggests that the water-splitting reaction is indeed driven by the light absorption of Co(OH)2-BN-C3N4/Pt-O-C3N5. In the 18O isotope-labelled water experiments (Fig. 2c), H218O (98%) replaced H216O in the photocatalytic performance assessment. The detected content of the evolved 18O2 was 97.4%, which closely matched the theoretical value (98.0%), confirming that the O2 is indeed from the water splitting reaction. The photocatalytic activity after five cycles of Co(OH)2-BN-C3N4/Pt-O-C3N5 maintained 96.1% of its initial activity (Fig. 2d), implying good stability in the photocatalytic performance of Co(OH)2-BN-C3N4/Pt-O-C3N5. The almost unchanged FTIR, TEM, and XRD spectra before and after the long-term photocatalytic reaction (Figs. 2e and f, and Fig. S24 in Supporting information) further demonstrate the excellent structural stability of the self-assembled Co(OH)2-BN-C3N4/Pt-O-C3N5 heterojunction.
Thanks to the enhanced interface interaction, the photo- and photoelectro- performances of BN-C3N4/O-C3N5 were significantly improved. The photo-induced carrier separation efficiency is enhanced, as evidenced by the enlarged photocurrent, decreased photoluminescence (PL), and prolonged electron lifetime. As depicted in Fig. 2g, BN-C3N4/O-C3N5 exhibits a much higher photocurrent density compared to BN-C3N4/O-C3N5-MIX, suggesting that the successive photo-generated electrons are transferred through the interface between BN-C3N4 and O-C3N5 [60-62]. Consequently, the recombination between photoinduced electrons and holes is suppressed, as indicated by the significantly decreased PL intensity (Fig. S25 in Supporting information) and prolonged electron lifetime (Fig. 2h) of BN-C3N4/O-C3N5. The electron lifetime of BN-C3N4/O-C3N5 is 16.62 ns, which is remarkably longer than those in BN-C3N4 (6.08 ns) and BN-C3N4/O-C3N5-MIX (8.93 ns) (Table S2 in Supporting information), which echoed with the significantly reduced radius of BN-C3N4/O-C3N5 (Fig. 2i) [63,64].
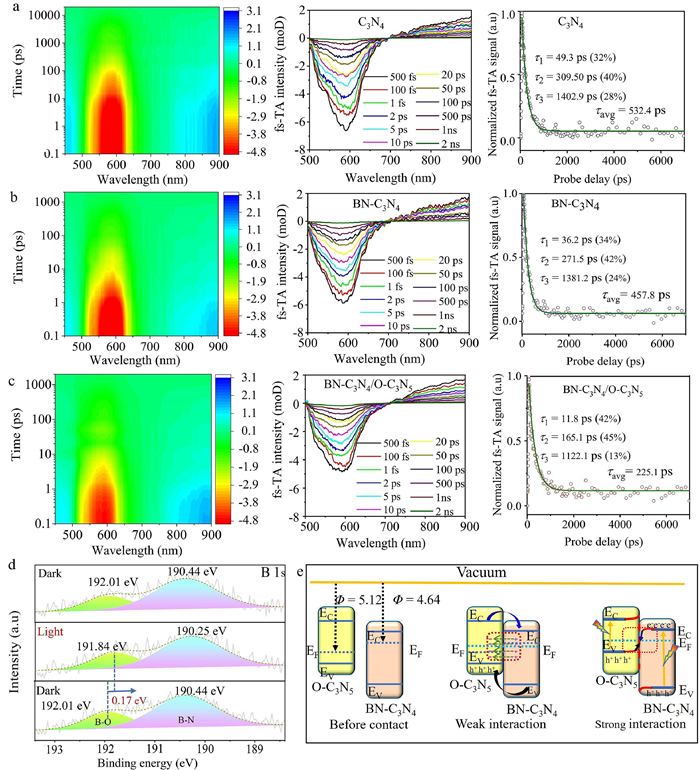
To further explore the charge transfer dynamics behavior in BN-C3N4/O-C3N5, femtosecond transient absorption (fs-TA) characterization was performed. Under excitation, the prominent peaks at approximately 580 nm are attributed to the ground state bleach (GSB) signals of C3N4, BN-C3N4, and BN-C3N4/O-C3N5, each exhibiting different time delays (Figs. 3a-c) [65,66]. Generally, GSB signals are related to the state-filling effect and are proportional to the number of photo-excited electrons in the CB [67]. Additionally, peaks with wavelengths greater than 710 nm are assigned to the photoinduced absorption (PA) bands of BN-C3N4. The fitted results of GSB signals in C3N4, BN-C3N4, and BN-C3N4/O-C3N5 are summarized in Table S3 (Supporting information). The lifetime τ1 is related to the electron transfer to the surface trap level of BN-C3N4, while τ2 represents interband charge recombination. Compared to τ1 of C3N4 (49.3 ps), the decreased τ1 of BN-C3N4 at 36.2 ps indicates promoted exciton energy transfer from C3N4 to B atom doping levels due to the hybridization of the BN-C3N4 electronic states and the S state of B3+ [68]. The shortened τ2 of BN-C3N4 suggests significantly improved recombination with B doping for faster electron transfer. With increasing delay time, GSB signals shift due to the Stark effect-induced exciton [69]. When BN-C3N4 is assembled with O-C3N5 (Fig. 3b), the built-in field facilitates charge transfer [70,71]. Therefore, τ1 of GSB decay in BN-C3N4/O-C3N5 is reduced to 11.8 ps. Affected by the interfacial Ni-O electron transferring channel and stronger built-in field, τave is shortened to 225.1 ps, suggesting faster electron transfer to B atoms for the production of H2 [72,73]. This further verifies that the built-in electric field efficiently facilitates the separation of photoinduced electron-hole pairs and rapid interfacial electron transfer by the B-O bonds [74,75]. According to previously reported literature, PA peaks are mainly contributed by photogenerated holes [76]. The decreased fitted τave of BN-C3N4/O-C3N5 (295.09 ns) compared to BN-C3N4 (685.69 ns) suggests improved hole consumption due to electron transfer from O-C3N5 to BN-C3N4. This result aligns with the S-scheme charge transferring process [77,78].
Figure 3
Figure 3.
2D mapping TA spectra signals, and normalized decay kinetic curves of C3N4 (a), BN-C3N4 (b), BN-C3N4/O-C3N5 (c). (d) In-situ B 1s XPS spectra of BN-C3N4/O-C3N5. (e) Simulation of band bending and electron transfer of BN-C3N4/O-C3N5.
The accelerated interface electron transport behavior is attributed to the presence of interfacial B-O bonds, which persist throughout the entire photocatalytic process, as evidenced by in-situ X-ray photoelectron spectroscopy of boron element. As depicted in Fig. 3d, after 30 min of light illumination, the peak corresponding to B-O bonds undergoes a significant blue shift of 0.17 eV, indicating the flow of photo-generated electrons through these interfacial bonds. Upon cessation of light, the binding energy of B-O bonds reverts to its original position, signifying the absence of photo-induced electrons through the interfacial B-O bonds. While O 1s exhibits an opposite change trend to that of B 1s XPS spectra. Under the light illumination, the peak of B-O bonds undergoes a significant red shift of 0.1 eV (Fig. S26 in Supporting information), indicating the photo-generated electrons was injected into the O atoms [79,80]. Therefore, in the BN-C3N4/O-C3N5 system, photogenerated electrons can be transferred from B atoms to O atoms more effectively due to the presence of interfacial B-O bonds.
Usually, for heterogeneous photocatalysts, the interfacial charge-transfer process is closely related to the work function (Φ) of the components [81]. The diffusion direction of electrons and holes from one semiconductor to another also greatly influences carrier transport and transfer efficiency. To address this, UPS characterization was performed to measure the work function of BN-C3N4 and O-C3N5 (Fig. S27 in Supporting information). The work function was calculated using the following equation (Eq. 1):
(1)
where Φ is the work function, hν is 21.22 eV, and E and E are the cutoff and Fermi edges, respectively.
The calculated work functions of BN-C3N4 and O-C3N5 are 4.64 and 5.42 eV, respectively. Additionally, BN-C3N4 and O-C3N5 display SPV values of 29.9 and 21.7 mV (Fig. S28 in Supporting information), respectively. For BN-C3N4/O-C3N5, the SPV value changes to 155.0 mV, significantly higher than that of BN-C3N4/O-C3N5-MIX. The significantly enhanced SPV value demonstrates that the strong interface interaction of BN-C3N4/O-C3N5 can promote the transfer of photogenerated charge carriers [82]. Based on these results, a schematic of Fermi level distributions is presented in Fig. 3e. As illustrated, the Fermi level of O-C3N5 is more negative than that of BN-C3N4 under light illumination. When BN-C3N4 and O-C3N5 come into contact, electrons migrate from the CB of BN-C3N4 to the VB of O-C3N5via the interface B-O bonds until their levels reach equilibrium. As a result, the positively charged BN-C3N4, along with the negatively charged O-C3N5, creates an internal electric field at the interface with a direction from BN-C3N4 to O-C3N5. Affected by the built-in electric field, the electrons in O-C3N5 are repulsed, leading to an increase in potential energy and an upward band bend [83]. Conversely, in the BN-C3N4 side, the potential energy of electrons decreases, and the bands bend downwards [84]. Affected by the built-in electric field and the interfacial band bending, the photoinduced electrons at the CB of BN-C3N4 are transferred to the VB of O-C3N5 through the interfacial B-O bonds and recombine with the photoinduced holes. This suggests the S-scheme charge-transfer process in the BN-C3N4/O-C3N5 heterojunction [85]. As a result, the stronger reductive photoinduced electrons in O-C3N5 and stronger oxidative photoinduced holes in BN-C3N4 are preserved, and thus BN-C3N4/O-C3N5 could realize efficient H2 and O2 evolution simultaneously.
To investigate the influence of boron dopants and nitrogen defects on the formation of BN-C3N4, DFT calculations were conducted. Fig. S29 (Supporting information) illustrates the optimized configurations of C3N4, B-doped C3N4, N-deficient C3N4 and BN-C3N4. The calculated formation energies of B-doped C3N4, N-deficient C3N4, and BN-C3N4 were 1.78, 2.12, and 1.56 eV, respectively. The relatively lower formation energy of BN-C3N4 may result from the synergistic effect of nitrogen defects and boron dopant on relaxing the BN-C3N4 structure [86,87].
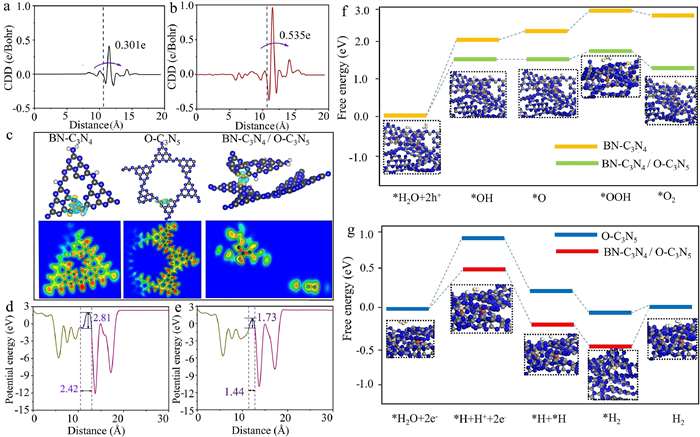
In theory, there were three types of interfacial bonding, named B-C bonding, B-N bonding, and B-O bonding, as shown in Fig. S30 (Supporting information). From an energetic standpoint, the BN-C3N4/O-C3N5 heterojunction with interfacial B-O bonding was the most stable, consistent with our experimental results. To unveil the effect of interfacial B-O bonds on the built-in electric field, the differential charge density and electronic location functions over BN-C3N4/O-C3N5 and BN-C3N4/O-C3N5-MIX were simulated. There are significant differences in the charge density distributions (DCD) between BN-C3N4/O-C3N5 and BN-C3N4/O-C3N5-MIX (Figs. 4a and b), suggesting that owing to the interfacial B-O bonding, the charge is transferred from O-C3N5 and accumulated on the BN-C3N4 side. Specifically, by introducing the interfacial B-O bonds, the interfacial charge transfer is enhanced by about 77%, verifying the formation of the built-in electric field and its lifting effect on the interfacial charge transport. Furthermore, as indicated by the results of the electronic location functions (ELFs), electrons are evenly distributed between B and O atoms (ELF value ~0.5) (Fig. 4c) [88,89]. Therefore, in the BN-C3N4/O-C3N5 system, the interfacial B-O bonds act as the highway for charge transport, profoundly improving the separation and transfer efficiency of photo-generated charges [90]. Additionally, Figs. 4d and e present the calculation results of the calculation of interfacial electrostatic potential. The energy barrier for electron transport from O-C3N5 to BN-C3N4 components in BN-C3N4/O-C3N5-MIX is 2.81 eV and the corresponding transmission distance is 2.42 Å, while in BN-C3N4/O-C3N5 system, the energy barrier and transfer distance reduce to 1.73 eV and 1.44 Å, respectively, significantly boosting the interfacial transport of photoinduced electrons.
Figure 4
Figure 4.
Energy barrier of interfacial charge transfer (a) without and (b) with B-O bond. (c) 2D mapping of electronic location function of BN-C3N4, BN-C3N4/O-C3N5, and O-C3N5. Charge density difference distribution (unit in e/Bohr) of BN-C3N4/O-C3N5-MIX (d) and BN-C3N4/O-C3N5 (e). (f) Gibbs free energy of HER on Co(OH)2-BN-C3N4/Pt-O-C3N5 and Pt-O-C3N5. (g) Gibbs free energy of OER on Co(OH)2-BN-C3N4/Pt-O-C3N5 and Co(OH)2-BN-C3N4.
To gain insight into the reaction mechanism of the water splitting reaction over Co(OH)2-BN-C3N4/Pt-O-C3N5 systems, the elementary reactions of water on the water reduction and oxidation were studied theoretically from an energy perspective (Fig. 4f). The energy barrier for the first H atom (H*) formation on Co(OH)2-BN-C3N4/Pt-O-C3N5 was 0.52 eV, lower than that on the O-C3N5 model (0.91 eV). This suggests that the band potential of photoinduced electrons on the CB of O-C3N5 in Co(OH)2-BN-C3N4/Pt-O-C3N5 was more negative, possibly originating from the band bending effect. This positive influence also appeared at both the second H atom formation and the H2* formation step (H*+H*→H2). Therefore, the S-scheme Co(OH)2-BN-C3N4/Pt-O-C3N5 was determined to decrease the thermodynamic energy barrier of H2 formation. The Gibbs free energy of multiple reaction intermediates (*OH, *O, and *OOH) was also examined (Fig. 4g). It can be observed that on Co(OH)2-BN-C3N4, the *OH radical formation energy was approximately 2.40 eV, while on Co(OH)2-BN-C3N4/Pt-O-C3N5, it dramatically reduces to 1.81 eV. Moreover, the energy of *O formation on Co(OH)2-BN-C3N4/Pt-O-C3N5 was also lower than that on Co(OH)2-BN-C3N4, indicating that the O2 formation from water oxidation can proceed more smoothly over the S-scheme Co(OH)2-BN-C3N4/Pt-O-C3N5. It can be concluded that the S-scheme Co(OH)2-BN-C3N4/Pt-O-C3N5 can significantly reduce the energy barriers of H2 and O2 formation.
In conclusion, based on the extended homogeneity of carbon nitride materials, an S-scheme BN-C3N4/O-C3N5 heterojunction was successfully constructed for photocatalytic overall water splitting, wherein the O2-evolving component BN-C3N4 and the H2-evolving component O-C3N5 are connected by interfacial B-O bonds. Co(OH)2-BN-C3N4/Pt-O-C3N5 exhibits satisfying visible-light driven photocatalytic activity, with H2 and O2 evolution rates reaching 40.12 and 19.62 µmol/h, respectively. During the photocatalytic process, the interfacial B-O bonds act as a highway for rapid electron transport. Under the synergistic effect of the built-in electric field and interfacial B-O bonds, the electron transfer rates are effectively improved following a S-scheme charge-transfer rout, by which electrons and holes with low redox potential recombine, leaving high potential ones for H+ reduction and OH− oxidation reactions. Our study provides a new strategy to construct highly efficient S-scheme heterojunctions for photocatalytic overall water splitting.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work is supported by the National Natural Science Foundation of China (No. 62004143), the Key R & D Program of Hubei Province (No. 2022BAA084), and the Major Project of Natural Science Foundation of Jiangsu Universities, China (No. 23KJA150010).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.110288.
[1]
Y. Bai, C. Li, L. Liu, et al., Angew. Chem. Int. Ed. 61 (2022) e202201299. doi: 10.1002/anie.202201299
Z. Zhu, H. Huang, L. Liu, et al., Angew. Chem. Int. Ed. 61 (2022) e202203519. doi: 10.1002/anie.202203519
[90]
X. Sun, M. Song, F. Liu, et al., Appl. Catal. B: Environ. Energy 342 (2024) 123436. doi: 10.1016/j.apcatb.2023.123436
Figure 1
(a) Schematic of the synthesis of BN-C3N4/O-C3N5. (b) Zeta potential of the BN-C3N4 and O-C3N5 dispersed in deionized water at pH 7. (c) EPR spectrum of BN-C3N4/O-C3N5. (d) B 1s XPS spectrum of BN-C3N4. (e) O 1s XPS spectrum of O-C3N5. (f) FTIR spectrum of BN-C3N4/O-C3N5. (g) Raman spectrum of BN-C3N4/O-C3N5. (h) Mapping images of BN-C3N4/O-C3N5.
Figure 2
(a) Effect of the mass ratio of BN-C3N4 to O-C3N5 on the photocatalytic activity. (b) AQY of BN-C3N4/O-C3N5. (c) 18O isotope-labelled water experiment results of BN-C3N4/O-C3N5. (d) The photostability of BN-C3N4/O-C3N5. (e) XRD patterns of BN-C3N4/O-C3N5 before and after the reaction. (f) FTIR spectra of BN-C3N4/O-C3N5 before and after the reaction. (g) I-t curve of BN-C3N4/O-C3N5. (h) Time-resolved PL of BN-C3N4/O-C3N5. (i) EIS plot of BN-C3N4/O-C3N5.
Figure 3
2D mapping TA spectra signals, and normalized decay kinetic curves of C3N4 (a), BN-C3N4 (b), BN-C3N4/O-C3N5 (c). (d) In-situ B 1s XPS spectra of BN-C3N4/O-C3N5. (e) Simulation of band bending and electron transfer of BN-C3N4/O-C3N5.
Figure 4
Energy barrier of interfacial charge transfer (a) without and (b) with B-O bond. (c) 2D mapping of electronic location function of BN-C3N4, BN-C3N4/O-C3N5, and O-C3N5. Charge density difference distribution (unit in e/Bohr) of BN-C3N4/O-C3N5-MIX (d) and BN-C3N4/O-C3N5 (e). (f) Gibbs free energy of HER on Co(OH)2-BN-C3N4/Pt-O-C3N5 and Pt-O-C3N5. (g) Gibbs free energy of OER on Co(OH)2-BN-C3N4/Pt-O-C3N5 and Co(OH)2-BN-C3N4.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
