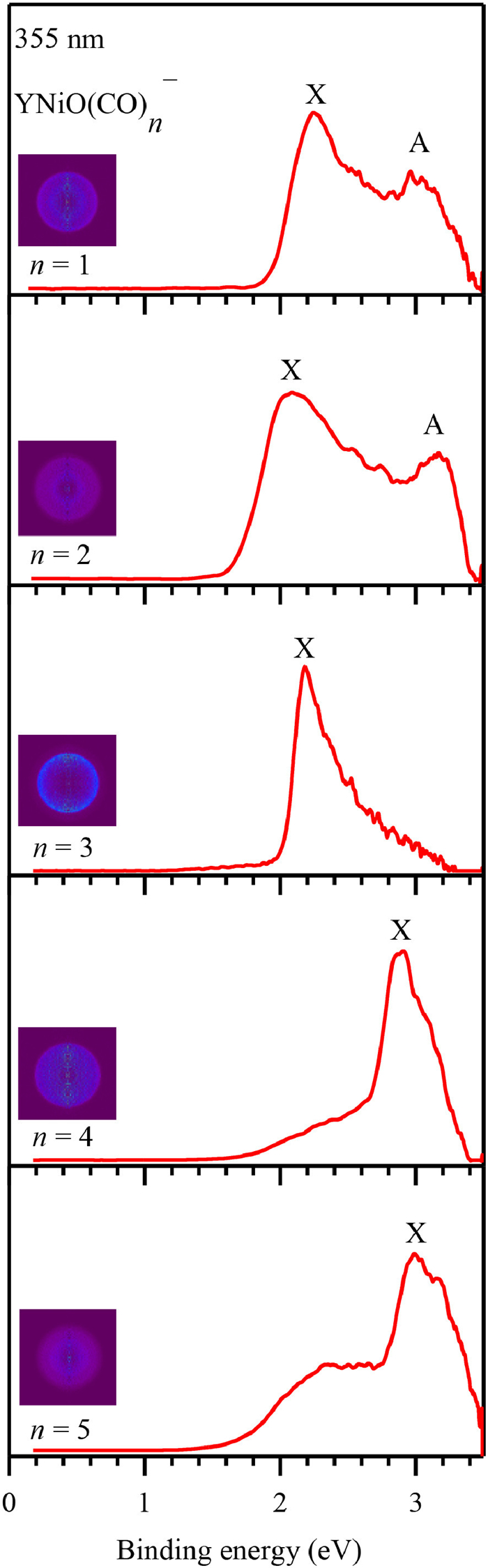
Figure 1.
Photoelectron spectra of YNiO(CO)n− (n = 1–5) recorded at 355 nm (3.496 eV). Photoelectron images after inverse Abel transformation are embedded in the photoelectron spectra.
Ligand-mediated reactivity in CO oxidation of yttrium-nickel monoxide carbonyl complexes
Jumei Zhang , Ziheng Zhang , Gang Li , Hongjin Qiao , Hua Xie , Ling Jiang

The oxidation of carbon monoxide (CO) is not only of practical importance for its removal in fuel gas cleanup, but also an important prototypical reaction that has drawn significant attention in heterogeneous catalysis [1–3]. Identifying the active sites and reaction mechanism of catalysts is a central subject of heterogeneous catalysis. Gas phase studies under isolated and controlled condition can provide a clear structure−reactivity understanding in heterogeneous catalysis at the molecular level [4–10]. Reactions of CO with noble metal-containing clusters have been extensively studied under well-defined conditions to understand single-atom catalysis by isolated noble metal atoms dispersed on oxide supports [11–14]. Recently, transition metal oxide clusters as substitutes for noble metal have attracted much attention due to their high catalytic activity, stability, and controllability [9,15–17]. Reactions of CO with heteronuclear transition metal oxide clusters have been widely studied to explore the effects of factors such as cluster composition [18], oxidation state [19], electric charge [20], and support [9]. However, the role of CO ligands adsorbed on heteronuclear transition metal oxide clusters has rarely been mentioned during the CO oxidation [8,17,21].
Recently, it was found that while CuAl4O7–9− and Ni2VO4− are less favorable for the oxidation of CO to CO2, the adsorption of an additional CO ligand to form CuAl4O7–9CO− and Ni2VO4CO− could promote CO oxidation. In the NbNiO(CO)n− (n = 5−8) series, the NbNiO(CO)n− (n = 5−6) anions were characterized as O-bridged complexes, while the NbNiO(CO)n− (n = 7−8) anions were characterized as η2-CO2-tagged complexes, which indicated that multiple adsorbed CO molecules could facilitate CO oxidation. In the TaNiO(CO)n− (n = 5−8) series, CO oxidation can occur at n = 8 through both Langmuir–Hinshelwood-like and Eley–Rideal-like mechanisms. These results indicate that CO ligands may play an important role in the process of CO oxidation, and this finding may be crucial in the design of efficient catalysts based on transition metals.
Group 3 metal oxide clusters are promising candidates for catalytic applications in the activation of CO2, which is the reverse reaction of CO oxidation [22–25]. Whereas the relevant studies have indicated that metal oxides were able to activate the CO2 molecule, the reverse reaction was predicted to be thermodynamically unfavorable. Herein we present a combined photoelectron velocity-map imaging spectroscopic and theoretical study of CO oxidation in the heterodinuclear yttrium-nickel monoxide anion during the progressive CO adsorption.
YNiO(CO)n− (n = 1−5) complexes were generated via pulsed laser vaporization of yttrium-nickel alloy with 5% CO seeded in helium and subsequently characterized by coupled photoelectron velocity−map imaging spectrometry (see Supporting information for experimental details). Photoelectron imaging and the corresponding photoelectron spectra of YNiO(CO)n− (n = 1−5) recorded at 355 nm were shown in Fig. 1. Vertical detachment energies (VDE) were directly measured from the well-defined band maxima. The spectrum of YNiO(CO)− featured two apparent peaks (X and A). The ground-state peak X was stronger than the excited-state band A. The ground-state VDE and the first excited-state VDE, which obtained from the maximum of band X and band A, were 2.25 ± 0.06 eV, and 3.00 ± 0.02 eV, respectively. The spectrum of YNiO(CO)2− was similar to that of YNiO(CO)−, showing one intense band X and a weak bands A, corresponding to VDEs of 2.08 ± 0.07, and 3.18 ± 0.02 eV, respectively. While for YNiO(CO)3−, there was only one sharp peak, with the band centered at 2.17 ± 0.07 eV. Spectral similarity was observed between YNiO(CO)4− and YNiO(CO)5−, in which the ground state VDEs were blue-shifted compared to those of the smaller clusters and were preceded by long tails. The ground-state VDEs, obtained from the maximum of band X, were 2.88 ± 0.03 and 3.00 ± 0.02 eV, respectively. Since no vibrational features for band X, the ground-state adiabatic detachment energies (ADEs) were roughly estimated by drawing a straight line along its leading edge and then adding the instrumental resolution to the intersection with the binding energy axis. The ADE values of the ground states for YNiO(CO)n− (n = 1−5) were evaluated to be 2.10 ± 0.07, 1.87 ± 0.07, 2.07 ± 0.07, 2.65 ± 0.04, and 2.76 ± 0.04 eV, respectively. The experimental observations of spectral similarities and differences as well as the abrupt band shift, implied the occurrence of structural evolution during the progressive adsorption of CO.
Density functional theory calculation at the B3LYP level were performed to explore the structures and reactivities of YNiO(CO)n− (n = 1−5) (see theoretical details in Supporting information). The optimized ground states and selected low-lying structures were presented in Fig. 2. Three types of structures were predicted for all of these complexes. The first type consisted of a linear YONi− anion core terminally bonded only by multiple CO ligands. We defined this type as a μ2-O-linear structure, labeled as nA. The second type, labeled as nB, involved a terminal O atom bonded to the Y atom. The third type had a CO2-taged structure, which was defined as nC. For n = 1, 2, the most stable structure was nA. The nB and nC structures were 1.49/2.00 eV, and 0.49/0.84 eV higher in energy than nA. For n = 3, the most stable structure was 3B, with 3A and 3C 0.41/0.61 eV higher in energy than 3B. While for n = 4, 5, the most stable structure was nC. The nA and nB structures were 1.12/0.40 eV and 0.80/0.28 eV higher in energy than nC, respectively. The energy differences between nA, nB, and nC decreasd in the beginning of the series, then first reversed at n = 3 with 3B becoming dominant. The second reverse occurred at n = 4 with 4C becoming favored. The theoretical prediction of relative energetics of the three structure types was confirmed by the spectroscopic experiments.
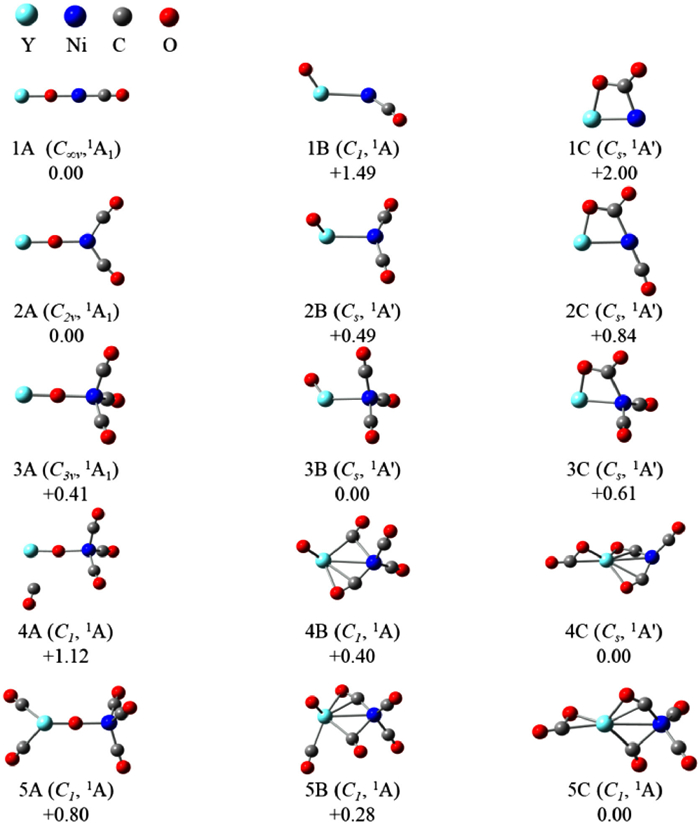
To clarify the possible structures that may exist under the experimental conditions, the theoretical VDE and ADE values for each structure were compared with the experimental date in Table S1 (Supporting information). To further facilitate comparison with experimental data, simulated spectra of each structure were shown in Fig. S1 (Supporting information). For n = 1, the VDE1, VDE2 and ADE values of the most stable 1A were calculated as 2.15, 3.03, and 2.04 eV, respectively, consistent with the experimental values. The simulated spectrum of 1A matched well with the experimental spectrum. The calculated VDE1, VDE2, and ADE of 1B (2.08/2.90/1.75 eV, respectively) were smaller compared with the experimental values. The calculated VDE1, and ADE of 1C (1.34/1.28 eV, respectively) were smaller compared with the experimental values. The simulated spectra of 1B and 1C were disagreement with the experimental spectra. Apart from the aforementioned calculated VDEs and simulated spectra, 1B and 1C were too high in energy to exist under the experimental conditions. Similarly, in the cases n = 2–5, the most stable 2A, 3B, 4C and 5C were identified by their VDE and simulated spectrum matching best to the strongest peak of the experimental spectrum. Therefore, 1A, 2A, 3B, 4C and 5C structures were considered as the dominant structures for the experimental observations.
The potential energy profiles for the consecutive CO adsorption on the YNiO(CO)n-1− (n = 1−4) and YNiO(CO)n-1− (n=5) complexes were presented in Fig. 3 and Fig. S2 (Supporting information), respectively, to provide insights into the dynamic reacivity of YNiO(CO)n-1− (n = 1−4) toward CO oxidation. Overall, the reaction channels of YNiO(CO)n-1− (n = 1−5) toward CO were predicted to be thermodynamically favorable for YNiO−, YNiO(CO)−, YNiO(CO)2−, YNiO(CO)3−, and YNiO(CO)4−, accompanied by energy releases of 2.98, 0.68, 0.60, 0.33, and 0.59 eV, respectively. For n = 1 − 3, the isomerization process from nA to nC can be viewed as gradual cleavage of Y-O bond as CO approached the O atom to form CO2-tagged structure nC via a twisted four-membered ring transition state (TS2, TS4, TS6). The isomerization processes undergone a nearly uphill pathway to generate structure nC. The energy barrier of 1A to 1C and from 2A to 2C was 2.63, 1.20 eV, respectively, disfavoring CO oxidation. While for n = 3, an energy barrier of 0.44 eV had to be surmounted to convert 3A into 3C. While the reverse process of 3A to 3C was thermodynamically exothermic and kinetically more facile. The isomerization from nA to nB was viewed as a process of Ni-O bond breaking and separation. Energy barrier of 1.60, 0.59, and 0.26 eV, respectively, needed to be surmounted in the conversion of nA to nB (n = 1−3), which was lower than that for the conversion of nA into nC. For n = 1, 2, the reverse process was thermodynamically exothermic and kinetically facile. Therefore, 1A and 2A prevailed over 2A and 2B. While for n = 3, the isomerization process of 3A to 3B was thermodynamically exothermic and kinetically facile, and therefore 3B was dominant for n = 3.
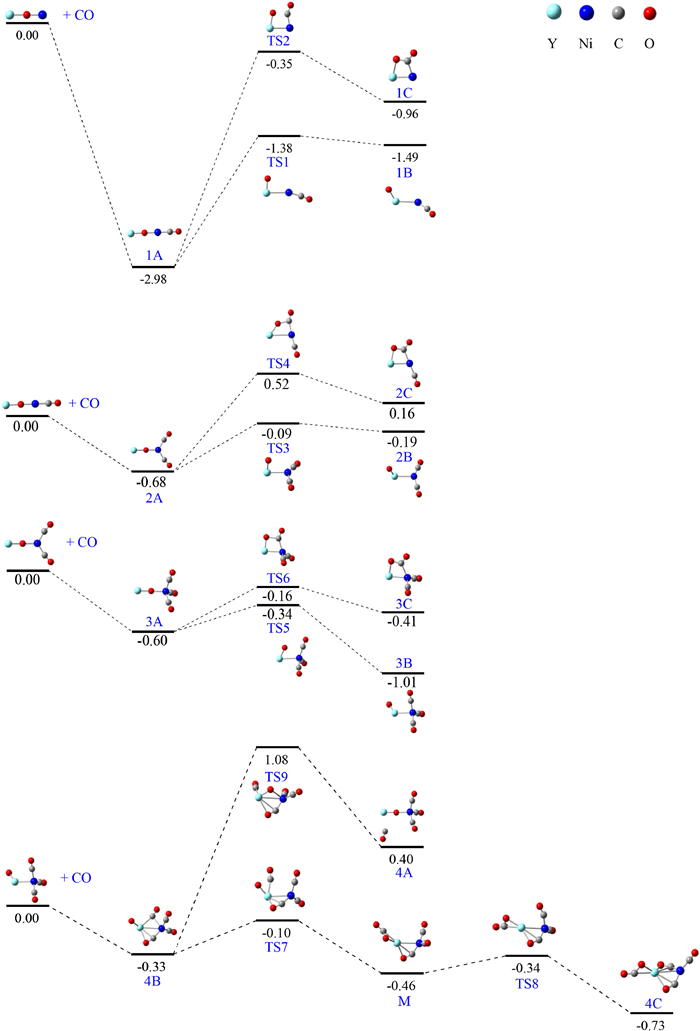
Attachment of one CO molecule to the most stable 3B generated 4B with an energy release of 0.33 eV. The isomerization from 4B to 4A had to overcome an energy of 1.41 eV. The process was disfavored by a positive barrier of 1.08 eV with respect to the ground-state reactants. The isomerization from 4B to 4C was viewed as the bridging CO approaching the terminal O atom as separated from the Ni atom to form a CO2 moiety structure (M) via transition state TS7, followed by the terminal CO bonded to Ni atom approaching to Y atom to form 4C via transition states TS8. The isomerization of 4B to 4C involved energy barrier of 0.23 eV and 0.12 eV to pass through TS7 and TS8, respectively, and the process followed a thermodynamically exothermic and kinetically facile pathway. Therefore, CO oxidation was easily achieved at n = 4. Attachment of one CO ligand to 4C gave rise to the ground state of 5C. The isomerization from 5C to 5B and 5A followed an endothermic pathway. Therefore, 5C was the dominant for n = 5. Overall, the potential energy profiles indicated that nA was favorable for n = 1, 2, nB was dominant product for n = 3, and nC was preferred for n = 4, 5, and CO oxidation became thermodynamically exothermic and kinetically facile for n ≥ 4.
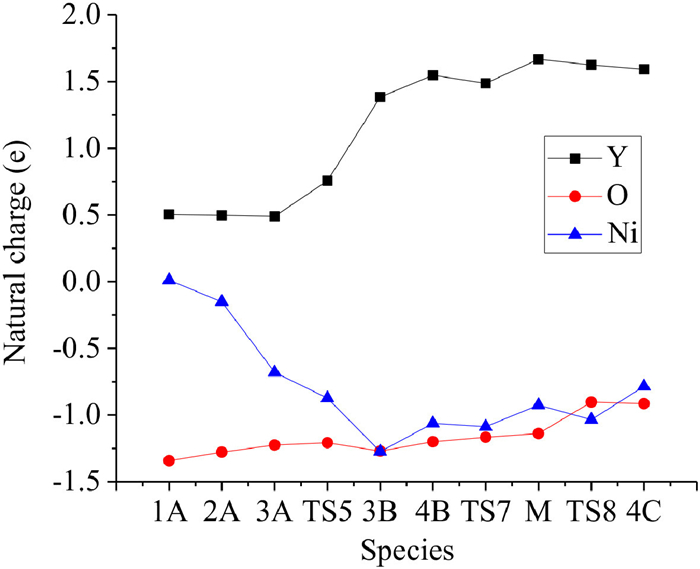
To gain further insight into the roles of Y and Ni in CO oxidation, natural population analysis was performed (see theoretical details in Supporting information). Natural charges of each isomer were shown in Table S2 (Supporting information) and the charge flows within the intermediates along the reaction pathway of YNiO(CO)n− (n = 1–5) were presented in Fig. 4. CO served as the major electron donor upon its adsorptions at n ≤ 3 while the Ni atom functioned as the electron acceptor to accumulate electrons [1A → 3A, ∆Q(Ni) = −0.70 e]. The Y atom served as the major electron donor and the Ni atom functioned as the electron acceptor during the structural rearrangement [3A → 3B, ∆Q(Ni) = −0.58 e, ∆Q(Y) = +0.89 e]. In the following steps of CO adsorption an oxidation, the Ni and the Y atoms both served as electron donors, and the contribution of Ni in donating electrons exceeded that of Y atom [3B → 4B → 4C, ∆Q(Ni)= +0.48 e, ∆Q(Y) = +0.21 e]. Therefore, the Y atom played an important role as an electron donor, while the Ni atom served as an electron reservoir to assist the adsorption and oxidation of CO.
In summary, the CO oxidation in yttrium-nickel monoxide carbonyl complexes was identified and well-illustrated by the combination of mass-selected photoelectron velocity imaging and density functional theory calculations. The YNiO(CO)n− (n = 1, 2) anions consisted of a linear Y-O-Ni anion core with CO terminally bonded to the Ni atom. In YNiO(CO)3−, the Y atom was bonded to the terminal O atom, and three carbonyls were terminally bonded to Ni atom. While YNiO(CO)n− (n = 4, 5) complexes had a CO2-moiety. The Ni atom acted as an electron acceptor to accumulate electrons at n ≤ 3, and served as electron donor along with the Y atom to contribute to electron density during CO oxidation at n > 3. The Ni atom played the role of an electron reservoir in assisting the adsorption and oxidation of CO. These results provided important insight for the rational design of highly active species for CO adsorption and oxidation.
The paper “Ligand-Mediated Reactivity in CO Oxidation of Yttrium-Nickel Monoxide Carbonyl Complexes” is not under consideration for publication and has not been published elsewhere in any medium including electronic journals and computer databases of a public nature. The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jumei Zhang: Writing – original draft, Visualization, Data curation. Ziheng Zhang: Investigation, Data curation. Gang Li: Investigation, Data curation. Hongjin Qiao: Writing – review & editing, Writing – original draft, Validation, Data curation. Hua Xie: Writing – review & editing, Writing – original draft, Validation, Supervision, Data curation. Ling Jiang: Writing – original draft, Validation, Supervision.
The authors gratefully acknowledge the Dalian Coherent Light Source (DCLS) for support and assistance. This work was supported by the Natural Science Foundation of Shandong Province (No. ZR2021QB215), the National Natural Science Foundation of China (Nos. 22273101, 22125303, 92061203, 21327901, and 22288201), Dalian Institute of Chemical Physics (No. DICP I202437), the Talent Induction Program for Youth Innovation Teams in Colleges and Universities of Shandong Province (No. 2022-2024), the Talent Introduction Research Start-up Funds of Ludong University (No. 2021-2026).
Supplementary material associated with this article can be found, in the online version, at doi:
M.M. Haruta, T. Kobayashi, H. Sano, N. Yamada, Chem. Lett. 16 (1987) 405–408. doi: 10.1246/cl.1987.405
X.W. Xie, Y. Li, Z.Q. Liu, M.M. Haruta, W. Shen, Nature 458 (2009) 746–749. doi: 10.1038/nature07877
H.J. Freund, G. Meijer, M. Scheffler, R. Schlögl, M. Wolf, Angew. Chem. Int. Ed. 50 (2011) 10064–10094. doi: 10.1002/anie.201101378
H. Schwarz, Angew. Chem. Int. Ed. 54 (2015) 10090–10100. doi: 10.1002/anie.201500649
S.M. Lang, T.M. Bernhardt, Phys. Chem. Chem. Phys. 14 (2012) 9255–9269. doi: 10.1039/c2cp40660h
A.W. Castleman, Catal. Lett. 141 (2011) 1243–1253. doi: 10.1007/s10562-011-0670-7
X.N. Li, X.P. Zou, S.G. He, Chin. J. Catal. 38 (2017) 1515–1527. doi: 10.1016/S1872-2067(17)62782-7
J.M. Zhang, Y. Li, Z.L. Liu, et al., J. Phys. Chem. Lett. 10 (2019) 1566–1573. doi: 10.1021/acs.jpclett.9b00205
C.X. Chi, H. Qu, L.Y. Meng, et al., Angew. Chem. Int. Ed. 56 (2017) 14096–14101. doi: 10.1002/anie.201707898
L.N. Wang, Z.Y. Li, Q.Y. Liu, et al., Angew. Chem. Int. Ed. 54 (2015) 11720–11724. doi: 10.1002/anie.201505476
B.T. Qiao, A.Q. Wang, X.F. Yang, et al., Nat. Chem. 3 (2011) 634–641. doi: 10.1038/nchem.1095
Z.Y. Li, Z. Yuan, X.N. Li, Y.X. Zhao, S.G. He, J. Am. Chem. Soc. 136 (2014) 14307–14313. doi: 10.1021/ja508547z
X.N. Li, Z. Yuan, J.H. Meng, Z.Y. Li, S.G. He, J. Phys. Chem. C 119 (2015) 15414–15420. doi: 10.1021/acs.jpcc.5b04218
J.J. Chen, X.N. Li, Q. Chen, et al., J. Am. Chem. Soc. 141 (2019) 2027–2034. doi: 10.1021/jacs.8b11118
L.N. Wang, X.N. Li, L.X. Jiang, et al., Angew. Chem. Int. Ed. 57 (2018) 3349–3353. doi: 10.1002/anie.201712129
X.P. Zou, L.N. Wang, X.N. Li, et al., Angew. Chem. Int. Ed. 57 (2018) 10989–10993. doi: 10.1002/anie.201807056
L.N. Wang, X.N. Li, S.G. He, J. Phy. Chem. Lett. 10 (2019) 1133–1138. doi: 10.1021/acs.jpclett.9b00047
J.B. Ma, Z.C. Wang, M. Schlangen, S.G. He, H. Schwarz, Angew. Chem. Int. Ed. 52 (2013) 1226–1230. doi: 10.1002/anie.201208559
H.J. Zhai, L.S. Wang, Chem. Phys. Lett. 500 (2010) 185–195. doi: 10.1016/j.cplett.2010.10.001
G.E. Johnson, J.U. Reveles, N.M. Reilly, et al., J. Phys. Chem. A 112 (2008) 11330–11340. doi: 10.1021/jp805186r
J.M. Zhang, Y. Li, Y. Bai, et al., Chin. Chem. Lett. 32 (2021) 854–860. doi: 10.1016/j.cclet.2020.05.029
Q.N. Zhang, H. Qu, M.H. Chen, M.F. Zhou, J. Phys. Chem. A 120 (2016) 425–432. doi: 10.1021/acs.jpca.5b11809
M.F. Zhou, L. Andrews, J. Am. Chem. Soc. 120 (1998) 13230–13239. doi: 10.1021/ja982900+
M.R. Sievers, P.B. Armentrout, Inorg. Chem. 38 (1999) 397–402. doi: 10.1021/ic981117f
D.Y. Hwang, A.M. Mebel, Chem. Phys. Lett. 357 (2002) 51–58. doi: 10.1016/S0009-2614(02)00438-4

Figure 1 Photoelectron spectra of YNiO(CO)n− (n = 1–5) recorded at 355 nm (3.496 eV). Photoelectron images after inverse Abel transformation are embedded in the photoelectron spectra.

Figure 2 Ground-state structures and selected low-lying isomers of YNiO(CO)n− (n = 1−5) anions calculated at the B3LYP/aug-cc-pVTZ/SDD level of theory. Relative energies are given in eV after zero-point energy correction.

Figure 3 Potential energy surface for the consecutive CO adsorption on YNiO(CO)n-1− (n = 1−4) anions calculated at the B3LYP/aug-cc-pVTZ/SDD level of theory. Energies are given in eV after zero-point energy correction.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:

 下载:
下载: