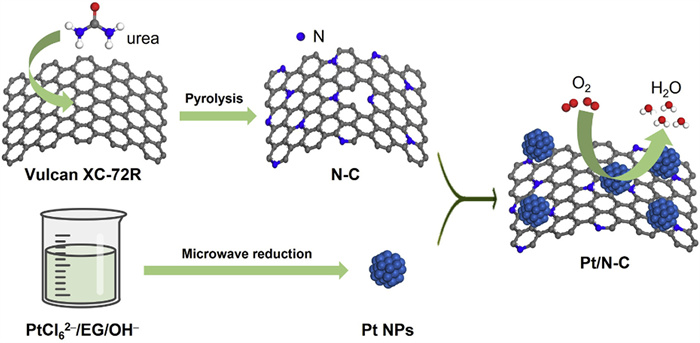
Scheme 1.
Schematic illustration for the preparation of Pt/N-C catalyst.
Interfacial Pt-N coordination for promoting oxygen reduction reaction
Jialin Cai , Yizhe Chen , Ruiwen Zhang , Cheng Yuan , Zeyu Jin , Yongting Chen , Shiming Zhang , Jiujun Zhang

Hydrogen fuel-based proton exchange membrane fuel cells (PEMFCs) with high energy/power densities, high conversion efficiency and zero-emission have been proved to be practicable clean and sustainable energy conversion and power systems for many large-scale applications including electric automobiles and stationary power plants [1–3]. In the effort to further improve the performance of PEMFCs, the sluggish kinetics of their cathodic oxygen reduction reaction (ORR) is identified as the major limiting factor [4,5]. Currently, carbon supported platinum (Pt/C) materials are used as the cathode electrocatalysts to speed up the ORR [6]. However, the weak interaction of Pt nanoparticles (NPs) with carbon support is prone to result in the severe degradation of catalytic activity and electrochemical stability [7–11]. These Pt NPs are susceptible to dissolution and aggregation on the surface of conventional carbon black, while the corrosion of the carbon support can cause the detachment of Pt NPs, leading to the degradation of PEMFC performance. In response to these challenges, the researchers have dedicated great endeavors to modifying commercial carbon black or developing novel support materials [12–14].
Studies have demonstrated that the single/co-doping of heteroatoms (such as B, N, P, S) into carbon supports can significantly enhance the electrocatalytic properties through enhancing the interaction between Pt NPs and carbon support [15–26]. In particular, the N-doping of carbon supports have attracted the most attention for the conspicuous improvement in ORR activity and electrochemical stability of Pt-based catalysts. This enhancement arises from the beneficial changes in both the electronic and structural properties of carbon support, induced by the high N-incorporation content in carbon support, the well dispersion of Pt NPs through N-doping, and the synergistic interaction of Pt with N-species [27–33]. For example, Zhang et al. [32] reported an oxygen spillover effect that the oxygenated intermediates formed on Pt NPs could transport to surface of the N-doped carbon black, promoting the ORR activity. Cai et al. [33] reported that the interfacial Pt-N-C site could provide a new active route, enhancing the electrocatalytic capacity in the catalyst system of N-doped carbon semi-tube (N-CST) supported Pt NPs. Despite some progress has been made, the fundamental understanding of the enhancement mechanisms of electrocatalytic ORR performance, especially the interaction of Pt NPs and N-doped carbon supports is still insufficient.
To identify the Pt-support effect on the enhancement of electrocatalytic ORR performance, this work has confirmed the presence of an interfacial Pt-N bond in a N-doped carbon supported Pt (Pt/N-C) catalyst and deeply studied the promotion mechanisms of ORR electrocatalysis. The N-doped carbon (N-C) support is successfully synthesized by high-temperature pyrolysis of a mixture comprising urea and commercial Vulcan XC-72R carbon black. This N-C material is used as the functional support, on which the microwave-reduced Pt NPs are decorated to form Pt/N-C catalysts. Several sophisticated instruments are used to characterize the morphologies, structures and compositions of such catalysts, electrochemical techniques are employed to validate their catalytic ORR performance, and theoretical calculations are also carried out for fundamental understanding of their catalytic enhancement mechanisms. The strongly interacting Pt-N bonds are found to form at the interface of Pt NPs and N-C support through electrons transferred from N-C support to Pt NPs. The interfacial Pt-N bonds can not only induce the weak adsorption of oxygen species on the Pt surface to trigger the excellent catalytic ORR activity, but also make the robust integration between Pt NPs and N-C support to stimulate the strong stability.
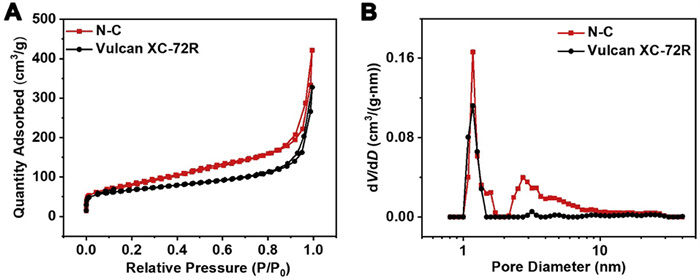
The synthesis procedure of one typical Pt/N-C catalyst is illustrated in Scheme 1. N-doped carbon (N-C) is prepared by implementing a high-temperature pyrolysis for the precursor mixture of urea and Vulcan XC-72R carbon black. During the pyrolysis process, urea can facilitate the successful in-situ N-doping into carbon black. Characterization of the Brunauer−Emmett−Teller (BET) surface area (BSA) and pore structure of N-C and Vulcan XC-72R is performed through the N2 adsorption-desorption (Fig. 1A) and pore size distribution (Fig. 1B) measurements. The values for surface areas and total pore volume value (TPV) are listed in Table S1 (Supporting information). The BSA and TPV values of N-C are determined to be 267.6 m2/g and 0.65 cm3/g, respectively, which are larger than those of Vulcan XC-72R (238.4 m2/g and 0.50 cm3/g). Additionally, the pore size distribution results obtained by the Barrett-Joiner-Halenda (BJH) method reveals a significant increase in micro/meso-pores. It appears reasonable to conclude that urea can contribute to an activation effect, improving the BSA and increasing the TPV and pore density of N-C. Based on such advantages of this N-C material, it is utilized as the functional carbon support for the construction of Pt/N-C catalyst. The synthetic process for Pt NPs is remained consistent with our previous work [33]. Typically, Pt NPs are produced through the microwave reduction of H2PtCl6·6H2O in alkaline ethylene glycol (EG). By mixing Pt NPs with N-C support and dialyzing the suspension with deionized water, the expected Pt/N-C catalyst is obtained. Scanning electron microscopy (SEM) image shows the amorphous carbon-based nanostructure (Fig. S1A in Supporting information) and the corresponding energy-dispersive spectroscopy (EDS) result provides a Pt content in Pt/N-C catalyst of almost 20 wt% (Fig. S1B in Supporting information).
Transmission electron microscopy (TEM) images display that the resulting Pt/N-C (Fig. 2A) has a similar morphology with commercial Pt/C (Fig. S2 in Supporting information), with Pt NPs uniformly dispersed on the surface of the carbon support. The average diameter of Pt NPs in Pt/N-C is ~2.8 nm, slightly smaller than the ~3.0 nm of Pt/C. High-resolution TEM (HRTEM) image (Fig. 2B) shows the well-defined lattice fringe spaces of 0.227, 0.197 and 0.146 nm, which are corresponding to the (111), (200) and (220) planes of Pt. High-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) and elemental mapping images (Fig. 2C) reveal the uniform distribution of C, N and Pt elements.
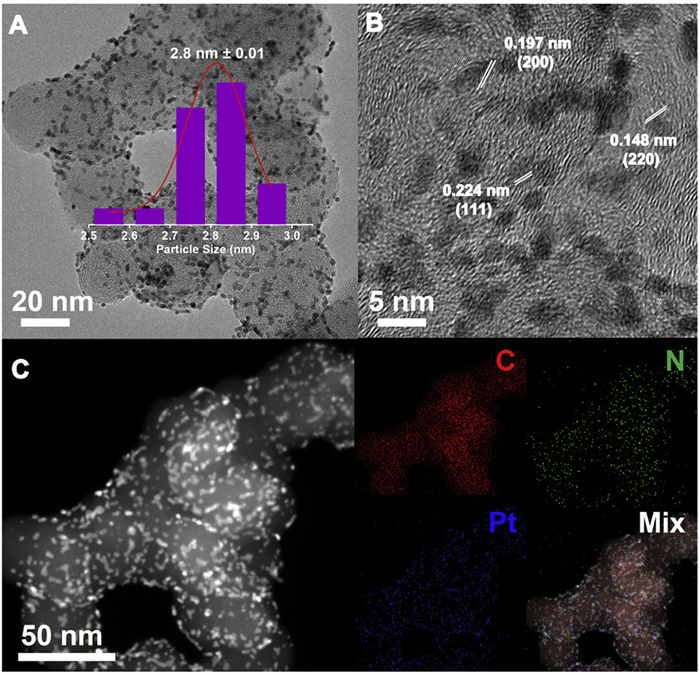
Fig. 3A shows the X-ray diffraction (XRD) patterns of Pt/N-C and Pt/C catalysts, indicating the typical diffraction signals of carbon support and Pt crystal phase. The peak at 24.58° is assigned to C(002) planes and four diffraction peaks located at 39.66°, 46.48°, 67.94°, 81.4° are indexed to the (111), (200), (220) and (311) planes of Pt crystal (JCPDS No. 87-0636), respectively. X-ray photoelectron spectroscopy (XPS) investigation was used to characterize the chemical compositions and valence states of surface elements in Pt/N-C and Pt/C. Both survey spectra (Fig. 3B) exhibit the characteristic peaks of C, O and Pt. In contrast, the N 1s characteristic peak can be found in Pt/N-C, with an atomic concentration of N being 1.3 at%. Furthermore, the high-resolution N 1s spectrum of Pt/N-C (Fig. 3C) can be deconvoluted into five different bands at 398.4, 399.2, 400.0, 401.0, and 403.5 eV, which correspond to pyridinic-N, Pt-N, pyrrolic-N, graphitic-N, and oxidized-N, respectively. The high-resolution Pt 4f XPS spectra (Fig. 3D) can be divided into three peaks at 71.83, 72.75 and 74.85 eV, corresponding to three paired Pt0, Pt2+ and Pt4+, respectively. Compared with Pt/C, there is a negative shift of ~0.35 eV for Pt/N-C, indicating the less formation of Pt oxides. This shift can be ascribed to the electron transfer from N-C support to Pt NPs through Pt-N as a bridge. The Pt0 content in Pt/N-C is calculated to be 58.1%, higher than that of 48.0% in Pt/C.
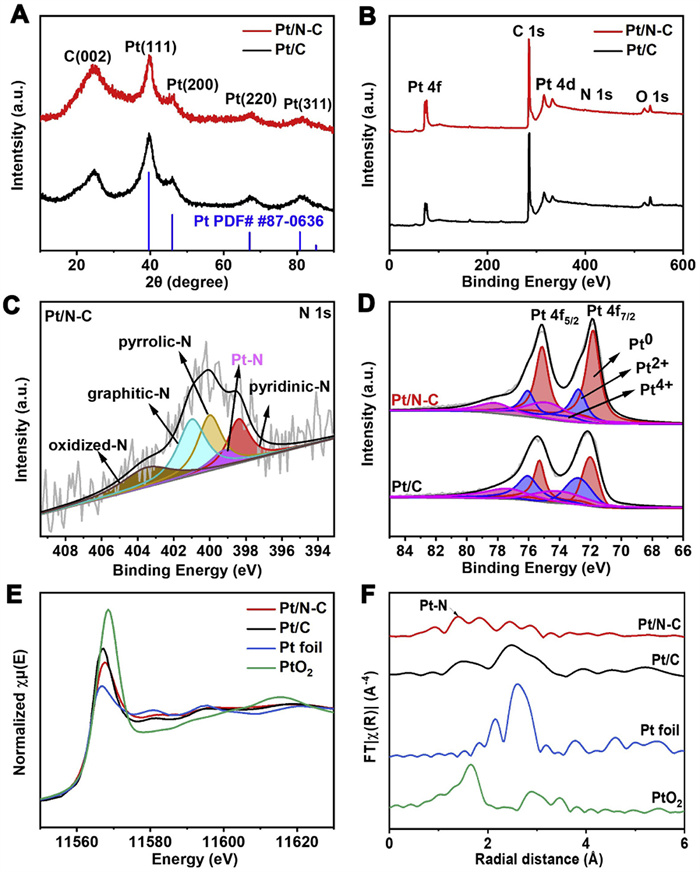
Furthermore, X-ray absorption fine structure (XAFS) spectroscopy at the Pt L3-edge was employed to discern the electronic structures and the coordination environment of Pt/N-C. As indicated in Fig. 3E, the white line (WL) intensity of Pt/N-C from Pt L3-edge X-ray absorption near edge structure (XANES) is located between PtO2 and Pt foil, suggesting that the Pt valence state in Pt/N-C catalyst should have more positive charges than Pt foil but fewer than PtO2. Moreover, the WL intensity of Pt/N-C is lower than that of Pt/C, indicating a higher content of Pt0 in Pt/N-C compared to Pt/C. This observation aligns well with the XPS result in Fig. 3D, providing clear evidence that the N-doping in the carbon support can significantly inhibit the oxidation of Pt [33]. Typically, the Fourier transformed extended X-ray absorption fine structure (EXAFS) spectra of Pt L3-edge (Fig. 3F) reveal a characteristic Pt-N bond contribution. The detailed fitting results and parameters are shown in Fig. S3 (Supporting information) and Table S2 (Supporting information), respectively. These findings provide direct evidence of the interaction between N-C support and Pt NPs, which remarkably contributes to the improvement in the ORR activity and stability of Pt/N-C.
To optimize the synthesis parameters of N-C support, electrochemical tests were conducted to investigate the effects of different urea addition ratio (Fig. S4 in Supporting information), pyrolysis temperature (Fig. S5 in Supporting information) and pyrolysis time (Fig. S6 in Supporting information). The best electrocatalytic ORR activity can be observed when the XC-72R/urea ratio is 1:10, pyrolysis temperature is 950 ℃, and pyrolysis time is 1 h, at which the optimal N-C support can be formed. This optimal N-C material was used as the functional carbon support for the microwave-reduced Pt NPs to construct Pt/N-C catalyst. Being consistent with our previous research results [33,34], the optimal synthesis conditions for microwave reduction were based on microwave power of 160 W, microwave temperature of 160 ℃, microwave time of 2 min, NaOH concentration of 0.4 mol/L, and H2PtCl6 concentration of 38.6 mmol/L.
The electrocatalytic ORR performance of Pt/N-C catalyst was assessed using the commercial Pt/C catalyst as the reference. Cyclic voltammetry (CV) measurements of Pt/N-C and Pt/C catalysts were conducted in an Ar-saturated 0.1 mol/L HClO4 solution (Fig. 4A). The electrochemical active surface area (ECSA) values of catalysts were calculated by measuring the charges collected in the hydrogen adsorption/desorption peak areas (0.02-0.4 V) after double-layer correction. The Pt/N-C catalyst exhibits a greater ECSA of 87.2 m2/gPt than that of the Pt/C catalyst (78.6 m2/gPt). Linear sweep voltammetry (LSV) analysis for Pt/N-C and Pt/C catalysts was performed at 1600 rpm in an O2-saturated 0.1 mol/L HClO4 solution. As shown in Fig. 4B, the half-wave potential (E1/2) of Pt/N-C catalyst is 0.888 V, representing a positive shift of 15 mV relative to that of the Pt/C catalyst (0.873 V). To demonstrate the superiority of the N-doping in carbon black support, the CV and LSV curves of Pt/N-C and Pt/XC-72R were compared (Fig. S7 in Supporting information). Although the two catalysts have similar ECSA values (Fig. S7A), the Pt/N-C catalyst shows the positive E1/2 than that (~0.88 V) of Pt/XC-72R catalyst (Fig. S7B). Additionally, the corresponding Tafel plots (inset of Fig. 4B) indicate that Pt/N-C affords a smaller Tafel slope (65 mV/dec) compared to Pt/C (69 mV/dec), implying the improved kinetic performance for catalytic ORR. According to the Koutecký-Levich (K-L) equation, the kinetic current density (jk) at 0.9 V (vs. RHE) was found to be 3.98 and 2.84 mA/cm2 for Pt/N-C and Pt/C, respectively. Furthermore, as shown in Figs. 4C and D, the initial mass activity and specific activity of Pt/N-C catalyst are 0.13 A/mgPt and 0.149 mA/cm2, respectively, which are about 1.4 and 1.3 times compared to those of Pt/C catalyst (0.093 A/mgPt and 0.118 mA/cm2).
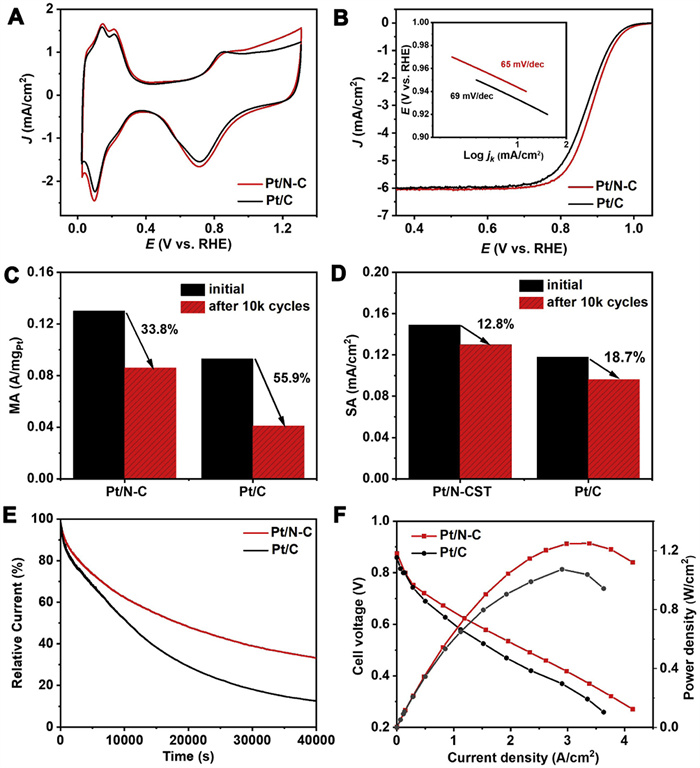
The electrochemical stability of two catalysts was investigated by accelerated durability tests (ADTs) through a continuous potential scanning between 0.6 V and 1.1 V in O2-saturated 0.1 mol/L HClO4 solution. Fig. S8 (Supporting information) shows the curve evolutions of CV (Figs. S8A and C) and LSV (Figs. S8B and D). It can be seen that after 10000 continuous potential cycles, the ECSA of Pt/N-C is dropped by 25.9% to reach 64.6 m2/gPt, while Pt/C experiences a 45.5% drop to 42.8 m2/gPt. In the ORR polarization curves, Pt/N-C shows only a 16 mV negative shift in the E1/2 (Fig. S8B, whereas Pt/C exhibits a 28 mV of E1/2 loss (Fig. S8D). As shown in Figs. 4C and D, the mass activity and specific activity of Pt/N-C are declined by 33.8% and 12.8% at 0.90 V, respectively. In contrast, Pt/C experiences a more significant decline with mass and specific activities dropping by 55.9% and 18.7%, respectively. Additionally, the current-time (i-t) test was carried out in an O2-saturated 0.1 mol/L HClO4 at a constant voltage of 0.7 V (vs. RHE). As depicted in Fig. 4E, Pt/N-C catalyst can maintain 33.3% of the initial current density after 40000 s of continuous operation, surpassing the 12.5% retention observed for Pt/C catalyst. All the comparative electrochemical data for the two catalysts are listed in Table S3 (Supporting information). These results suggest that the as-prepared Pt/N-C catalyst exhibits a superior electrochemical stability compared to the commercial Pt/C catalyst.
Furthermore, H2-O2 PEMFC performances of Pt/N-C and Pt/C as the cathode catalysts with a loading of 0.1 mgPt/cm2 were measured at the temperature of 80 ℃ and full humidity of H2 and O2 with a flow rate of 600 mL/min. As can be seen from their current-voltage (I-V) polarization and power density plots in Fig. 4F, Pt/N-C fuel cell exhibits the higher current and power densities than Pt/C one. The peak power density of the as-prepared Pt/N-C is 1.25 W/cm2 which is higher than that of the commercial Pt/C (1.16 W/cm2), meaning its potential for commercial substitution.
To explore the origins of high catalytic ORR activity and stability of Pt/N-C catalyst, the density functional theory (DFT) calculations and ab initio molecular dynamics (AIMD) simulations were carried out. As shown in Fig. 5A, the same initial distance of 6.72 Å between two Pt13 NPs is set on two different supports of pure carbon and N-C, respectively. As time goes on, two particles on the pure carbon support gradually approach and fuse at about 5400 fs, ultimately aggregating into one large particle at about 7200 fs. While the two particles anchored on N-C support are hard prone to agglomeration even at 7200 fs. These results indicate that the N-C support has a strong affinity with the Pt NPs owing to the interfacial Pt-N bond observed in experiments, which can prevent the particles from aggregation and maintain their sizes for the maximum Pt utilization, so as to obtain higher ORR activity and stability. Based on previous study shown that the stability of Pt NPs increases linearly with size [35], it is believed that the property of smaller Pt NPs can be extrapolated to that of larger one, and the Pt38 NPs are used to discuss the following properties.
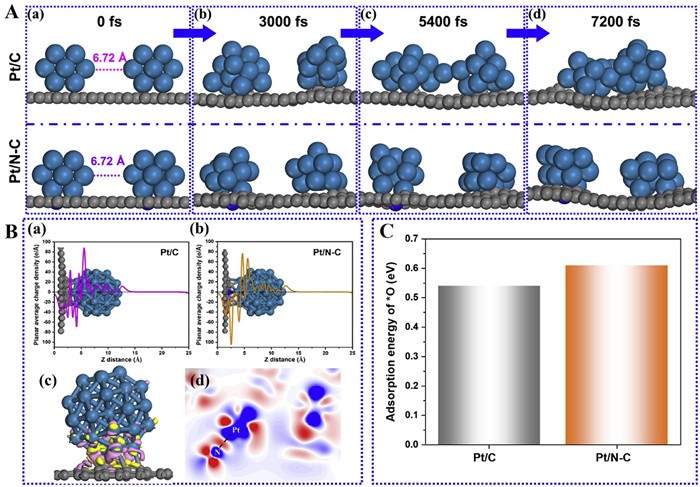
To further clarify the distinctive role of Pt-N bond in Pt/N-C, the planar average charge densities of Pt/C and Pt/N-C are shown in Fig. 5B (a and b). It is evident that both the pure carbon and N-C supports can donate electrons to the Pt NPs with the Pt NPs gaining significantly more electrons after the Pt-N bond formation. This can make a lower Pt binding energy for Pt/N-C compared to Pt/C, which is consistent with the XPS result in Fig. 3. As the differential charge density shown in Fig. 5B (c and d), there are electron accumulations between Pt and N, indicating the strong interaction of the N-C support and Pt NPs through the bridged Pt-N bond. Therefore, the calculation results prove that N-doping of carbon support can greatly suppress the migration of Pt NPs for the enhanced stability and reduced activity loss.
The stability of pure carbon and N-C supports are also discussed in this part. The results obtained in this work have shown that the carbon supports with some defects are easy to be oxidized under the ORR condition, but the oxidation energy increases more than 1.12 eV after pyridinic-N doping into the carbon layer (oxidation energy of defect carbon and N-C are -2.28 and -1.16 eV, respectively), indicating that N-doping can improve the stability of carbon support. Furthermore, after the support is loaded with Pt NPs, both the pure carbon and N-C supports become stable and hard to be oxidized (support oxidation energy of Pt/C and Pt/N-C are 2.17 and 2.41 eV, respectively). These results indicate that the Pt NPs can promote the stability enhancement of supports especially the N-C.
The ability of Pt-oxides formation on Pt/C and Pt/N-C catalysts was estimated by evaluating the adsorption strength of atomic oxygen (*O) on the Pt surface. Fig. S9 (Supporting information) provides the possible adsorption sites and corresponding adsorption energies, it indicates that the different adsorption sites could obtain different adsorption energies of *O. Fig. 5C shows the adsorption energy of *O on Pt38 NPs supported by pure carbon and N-C supports for the most stable adsorption configuration corresponding to site 1 shown in Fig. S9. It can be found that the *O adsorption is weaker on Pt/N-C (0.61 eV) than that on Pt/C (0.54 eV). Hence, the N-doping of carbon support can weaken *O adsorption strength on the Pt surface, resulting in the alleviated Pt-oxides formation. It is believed that this should be another reason for the high catalytic ORR activity and stability of Pt/N-C.
In summary, an in-depth study on the promotion effect of interfacial Pt-N coordination on oxygen reduction electrocatalysis in a Pt/N-C catalyst system is carried out in this work. Successful N-doping of commercial carbon black for a functional N-C support has been achieved by one-step pyrolysis with urea as the nitrogen source. Microwave-reduction of chloroplatinic acid in alkaline ethylene glycol can produce Pt NPs with an average size of ~2.8 nm. The integrating Pt/N-C catalyst exhibits uniform dispersion of Pt NPs on the N-C support. The experimental results from XPS and XAFS indicate the formation of Pt–N bonds at the interface of Pt NPs and N-functional support. This bridged Pt–N coordination facilitates the electron transfer from N-C support to Pt NPs, leading to a high proportion of metallic Pt and a robust metal-support integration in the Pt/N-C catalyst. Consequently, the N-doping of carbon support can not only weaken the adsorption strength of oxygen intermediates on the Pt surface to improve the ORR activity, but also induce the strong Pt-support interaction to enhance the electrochemical stability. The achieved Pt/N-C catalyst shows a superior ORR activity with ~1.4 times higher in mass activity and outstanding electrochemical stability to the commercial Pt/C catalyst. As the cathode catalyst in a single H2-O2 fuel cell, such a Pt/N-C achieves a peak power density of 1.25 W/cm2, higher than the 1.16 W/cm2 of Pt/C. This study can provide meaningful insight into the interfacial Pt-N coordination for ORR electrocatalysis, and will offer valuable guidance for the rational design of Pt-based catalysts supported on the modified/novel supports and the deep study of their interfacial properties.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Jialin Cai: Writing – original draft, Methodology, Investigation, Formal analysis, Data curation. Yizhe Chen: Writing – original draft, Investigation, Data curation. Ruiwen Zhang: Investigation, Formal analysis, Data curation. Cheng Yuan: Investigation, Formal analysis. Zeyu Jin: Formal analysis. Yongting Chen: Writing – original draft, Supervision, Methodology, Funding acquisition. Shiming Zhang: Writing – review & editing, Supervision, Project administration, Methodology, Funding acquisition, Conceptualization. Jiujun Zhang: Writing – review & editing.
This work was supported by the National Natural Science Foundation of China (Nos. 22272105 and 22002110) and Natural Science Foundation of Shanghai (No. 23ZR1423900). The numerical calculations are supported by High-Performance Computing Center of Wuhan University of Science and Technology.
Supplementary material associated with this article can be found, in the online version, at doi:
S. Mo, L. Du, Z. Huang, et al., Electrochem. Energy Rev. 6 (2023) 28.
G. Fisseha, Y. Hu, Y. Yu, et al., Chin. Chem. Lett. 35 (2024) 108445.
M. Song, Q. Zhang, T. Shen, G. Luo, D. Wang, Chin. Chem. Lett. 35 (2024) 109083.
C. Yuan, S. Zhang, J. Zhang, Front. Energy 18 (2024) 206–222. doi: 10.1007/s11708-023-0907-3
M. Tang, S. Zhang, S. Chen, Chem. Soc. Rev. 51 (2022) 1529–1546. doi: 10.1039/d1cs00981h
H. Liu, J. Zhao, X. Li, Electrochem. Energy Rev. 5 (2022) 13.
S. Li, T. Wang, Q. Li, Sci. China Chem. 66 (2023) 3398–3414. doi: 10.1007/s11426-023-1760-x
Y. Chen, S. Zhang, J. Jung, J. Zhang, Prog. Energy Combust. Sci. 98 (2023) 101101.
C. Durante, Curr. Opin. Electrochem. 36 (2022) 101119.
X. Yu, S. Ye, J. Power Sources 172 (2007) 133–144.
X. Yu, S. Ye, J. Power Sources 172 (2007) 145–154.
J. Long, X. Zheng, B. Wang, et al., Chin. Chem. Lett. 35 (2024) 109354.
L. Liu, X. Rao, S. Zhang, J. Zhang, Chem 10 (2024) 1994–2030.
S. Zhang, M. Chen, X. Zhao, et al., Electrochem. Energy Rev. 4 (2021) 336–381.
Q. Li, L. Li, X. Yu, et al., Chem. Eng. J. 399 (2020) 125827.
Y. Xia, X. Zhao, C. Xia, et al., Nat. Commun. 12 (2021) 4225.
D. Liu, S. Gao, J. Xu, et al., Appl. Surf. Sci. 604 (2022) 154466.
J.L. Fiorio, M.A.S. Garcia, M.L. Gothe, et al., Coord. Chem. Rev. 481 (2023) 215053.
W. Ni, J.L. Meibom, N.U. Hassan, et al., Nat. Catal. 6 (2023) 773–783. doi: 10.1038/s41929-023-01007-1
Q. Huang, L. Hu, X. Chen, et al., Adv. Funct. Mater. 33 (2023) 2302582.
X. Zhu, X. Tan, K.H. Wu, et al., Angew. Chem. Int. Ed. 60 (2021) 21911–21917. doi: 10.1002/anie.202107790
W. Tian, Y. Wang, W. Fu, et al., J. Mater. Chem. A 8 (2020) 20463–20473. doi: 10.1039/d0ta06566h
Y. Zhang, J. Zhao, J. Lian, et al., ChemistrySelect 8 (2023) e202301359.
N. Zhou, R. Wang, K. Liu, J. Colloid Interface Sci. 654 (2024) 1186–1198.
V. Perazzolo, R. Brandiele, C. Durante, et al., ACS Catal. 8 (2018) 1122–1137. doi: 10.1021/acscatal.7b03942
Y. Chen, X. Zheng, J. Cai, et al., ACS Catal. 12 (2022) 7406–7414. doi: 10.1021/acscatal.2c00944
S. Zhang, S. Chen, Catalysts 5 (2015) 1202–1210. doi: 10.3390/catal5031202
M. Chen, J. Chen, C. Jia, et al., Cell Rep. Phys. Sci. 4 (2023) 101204.
X. Rao, S. Zhang, J. Zhang, Curr. Opin. Electrochem. 42 (2023) 101416.
Z. Yang, Y. Chen, S. Zhang, J. Zhang, Adv. Funct. Mater. 33 (2023) 2215185.
F. Han, Z. Liu, J. Jia, et al., Mater. Chem. Phys. 237 (2019) 121881.
S. Zhang, S. Chen, J. Power Sources 240 (2013) 60–65.
J. Cai, J. Chen, Y. Chen, J. Zhang, S. Zhang, iScience 26 (2023) 106730.
J. Cai, Y. Chen, J. Jung, J. Zhang, S. Zhang, Energy Storage Sci. Technol. 11 (2022) 3800–3807.
E.M. Dietze, P.N. Plessow, F. Studt, J. Phys. Chem. C 123 (2019) 25464–25469. doi: 10.1021/acs.jpcc.9b06952

Figure 1 (A) Nitrogen adsorption-desorption isotherms and (B) pore size distribution of N-C and Vulcan XC-72R.

Figure 2 (A) TEM and Pt NPs size distribution in the inset, (B) HRTEM, (C) HAADF-STEM image and corresponding elemental mapping images of C, N, Pt for the Pt/N-C catalyst.

Figure 3 (A) XRD patterns, (B) XPS survey spectra, and high-resolution XPS spectra of (C) N 1s and (D) Pt 4f for the Pt/N-C and Pt/C catalysts. (E) Pt L3-edge XANES spectra, and (F) Fourier-transformed EXAFS spectra for Pt/N-C, Pt/C, PtO2 and Pt foil.

Figure 4 Electrochemical performance of Pt/N-C and Pt/C catalysts: (A) CV curves; (B) ORR polarization curves, and the corresponding Tafel plots and slopes in the inset. Performance evolutions of (C) mass activity and (D) specific activity before and after ADTs. (E) i-t curves at the constant potential of 0.7 V (vs. RHE); (F) I-V and power density curves of H2-O2 single PEMFCs performance (The catalyst loading is 0.1 mgPt/cm2 for cathode and anode, respectively. Test conditions: 80 ℃, 100% RH, 100 kPa absolute pressure).

Figure 5 Theoretical calculations: (A) Ab initio molecular dynamics results showing the agglomeration evolutions of two Pt13 NPs loaded on the pure carbon (top) and N-C (down) supports at 300 K, respectively. (B) The planar average charge densities of (a) Pt/C and (b) Pt/N-C, and the (c) isosurface and (d) slice of differential charge density for Pt/N-C. Pink and red areas represent the electron density increase and the yellow and blue areas represent the electron density decrease in (c) and (d). (C) The adsorption energy of *O on Pt/C and Pt/N-C.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:
 下载:
下载: