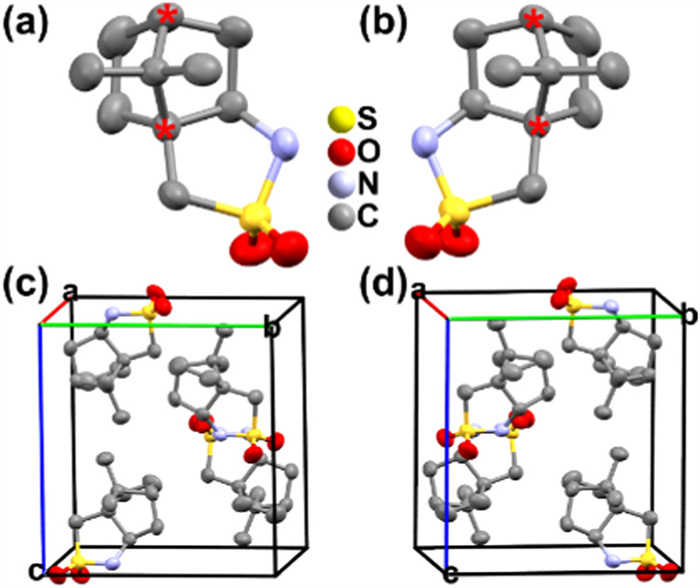
Figure 1.
Asymmetric unit of the molecular structure of (a) S-CPS and (b) R-CPS. Crystal structural packing view of (c) S-CPS and (d) R-CPS in one unit cell. Hydrogen atoms are omitted for clarity.
Homochiral organic ferroelastics with plastic phase transition
Yan-Ran Weng , Wen-Fu Tian , Wen-Jing Ding , Bi-He Ren , De-Hou Liu , Jia-Ying Tang , Feng Zhou , Xiao-Gang Chen , Xian-Jiang Song , Hui-Peng Lv , Yong Ai

Ferroelastic generally refers to materials that exhibit spontaneous strain that can be reversed or reoriented under an external mechanical stress [1,2]. Since the ferroelastic hysteresis was firstly reported in an inorganic crystal of lead(Ⅱ) phosphate (Pb3(PO4)2) [3,4], the investigation of ferroelastic has been dominated mainly by inorganic perovskites and hybrid organic-inorganic perovskites for a long time, such as BaTiO3, BiFeO3, SrBi2Ta2O9, VO2, and CH3NH3PbI3 [5-9]. However, the inorganic ferroelastic materials have disadvantages such as high energy consumption and toxic heavy metals. In contrast, organic ferroelastic crystals have advantages, including easy preparation, softness, low molecular mass, and low material cost, and may show great potential for the supplementary of the inorganic ones [10].
In 1979, the first work of organic single ferroelastic crystal squaric acid (C4H2O4) was reported [11]. Then, a series of organoferroelastics and organic-inorganic hybrid ferroelastics with excellent properties were reported [12-16]. For instance, Takamizawa and his co-workers found ferroelasticity and spontaneous strain in organic molecules 5‑chloro-2-nitroaniline [17], adipic acid [10], 4, 4′-dicarboxydiphenyl ether [18], trans-1, 4-cyclohexane-dicarboxylic acid [19], and 2-methyl-5-nitrobenzoic acid [20]. Radical TEMPO crystal [21] and bis(triisopropylsilylethynyl)-pentacene organic semiconductor [22] also show ferroelasticity. Whereas, the development of targeted organic compounds with ferroelastic phase transitions is still challenging. It is worth mentioning that plastic crystals hold great potential as candidates for phase transition materials, because they possess spherical molecular structures that are prone to trigger solid-solid phase transitions accompanying dramatic symmetry breaking [23]. In the high temperature phase, the highly disordered spherical rotation gives rise to favorable crystal plasticity and highly symmetric structure, which can be considered as the prototype paraelastic phase [24-27]. The solid-solid phase transition in plastic crystals leads to a dramatic break in symmetry, which in turn promotes the formation of ferroelastic phases. In addition, it is concurrent with the occurrence of significantly large changes in entropy as well [28]. Plastic crystals hold great opportunities for achieving ferroelasticity, and also possess tremendous potential in solid-state heat storage or refrigeration applications [29].
Although great progress has been achieved in the respective fields of plastic crystals and organoferroelastic materials, single-component organoferroelastics with plastic features have rarely been reported. In this work, we discovered a pair of enantiomeric single-component organoferroelastics, namely, (1S and/or 1R)−2,10-camphorsultam (S-CPS and R-CPS), which crystallizes in the P212121 space group, chiral 222 point group, at room temperature. The enantiomers undergo plastic phase transition at high Tc of 385 K, showing large entropy gain (∆St = 45 J mol-1 K-1), and evidently ductile deformation in plastic phase. Although the crystal structure, phase transition properties of S- and R-CPS were reported elsewhere [30,31], the high-temperature phase structure was not elucidated and ferroelastic behaviors were not discovered, which have been revealed in detail here. The plastic phase transition with large symmetry breaking is beneficial to the paraelastic-ferroelastic transition, and these single-component organic ferroelastic materials will provide intriguing application potential for future wearable smart materials and biocompatible materials.
The colorless rod-like crystals were obtained by slowly evaporating the n‑butyl alcohol solution of the S-CPS and R-CPS enantiomers at room temperature for one week (Fig. S1 in Supporting information). At 298 K, the crystal structure of S-CPS was refined in the P212121 space group, and the unit cell parameter of S-CPS were a = 9.2630(2) Å, b = 10.4109(3) Å, c = 11.1921(3) Å, α = β = γ = 90°, and V = 1079.32(5) Å3 (Table S1 in Supporting information). R-CPS shows similar crystal structure to that of S-CPS (Table S1), which is in good agreement with literature [30-32]. The asymmetric unit contains one single molecule of R-CPS or S-CPS (Figs. 1a and b), with the chiral carbon atom marked with red asterisk '*'. Figs. 1c and d show the packing view of S-CPS and R-CPS in a unit cell, showing a mirror-image relationship. The adjacent molecules in the stacking rely on the weak hydrogen bonds N—H···O with identical donor-acceptor distance of 3.192 Å and van der Waals forces (Fig. S2 in Supporting information).
To effectively identify chiral enantiomer relationships between R-CPS and S-CPS, the vibrational circular dichroism (VCD) measurements were conducted. In Fig. S3 (Supporting information), the VCD spectra of R-CPS and S-CPS enantiomers exhibit a few pairs of VCD signals (Δε) at around 1022, 1067, 1136, 1162, 1184, 1214, 1244, 1260, 1295, 1338, and 1412 cm-1, which match well with the corresponding specific IR vibration peaks. The fact that VCD spectra of the R-CPS and S-CPS enantiomers have a near mirror relationship with each other, further confirms their enantiomorphic nature.
Differential scanning calorimetry (DSC) is an efficient way to measure the structural phase transition behavior. Thermal anomalies were observed during the heating-cooling traces for S- and R-CPS (Fig. 2a), and the phase transition points (Tc) in the heating runs are 385 K, which is higher than that of [QUI]ReO4 (Tc = 367 K) [33], [3-F-Q]ReO4 (Tc = 370 K) [34], [AH][ReO4] (Tc = 322 K) and [AH][IO4] (Tc = 258 K) (AH = 1-azabicyclo[2.2.1]heptanium) [35]. The Tc value is a bit lower than that reported before [31], due to the lower heating rate of 5 K/min here. For convenience, the phase above 385 K was labeled as the high-temperature phase (HTP) and the phase below as the low-temperature phase (LTP). Note that there are two peaks at 362 and 352 K in the DSC cooling trace but only one peak in the heating trace. Apparently, there exist additional a metastable state during the cooling process. Such complicated phase transition behaviors are also observed in other plastic crystals, such as [tetra-n-butylammonium]2[Ni(maleonitriledithiolate)2] [36], (‒)-camphoric acid [23]. Marek Szafrański explained the unexpected shoulder peak of the endothermic peak is due to the pretransitional effect [37,38].
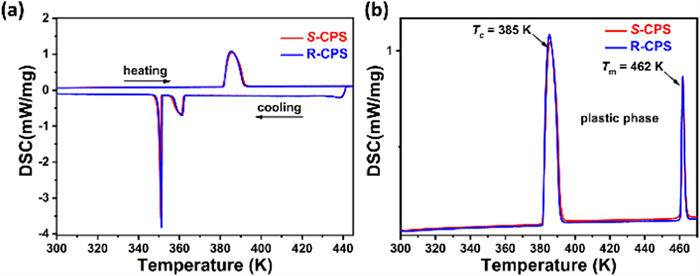
For S-CPS in the heating run (Fig. 2b), the entropy change of the phase transition (∆St = 44.6 J mol-1 K-1) is much higher than that of its melting process (∆Sm = 7.6 J mol-1 K-1), close to the reported values of 42.33 and 7.354 J mol-1 K-1 [31]. R-CPS exhibits similar ∆St (43.4 J mol-1 K-1) and ∆Sm (7.5 J mol-1 K-1). Such large ∆St value and ∆St/∆Sm ratio are typical characteristics of the plastic phase transition [26,39]. According to the Boltzmann rule, ΔS can be described as Rln(nH/nL), where R is the gas constant, n is the number of geometrically distinguishable molecular orientations in HTP and LTP, respectively. The ratio of nH/nL was calculated as high as 215 for S-CPS, which is much higher than that of other plastic crystals such as (R)−3-quinuclidinol (nH/nL = 65) [40], and comparable to the C60 (nH/nL = 240) [41], (-)-camphanic acid (nH/nL = 326) [23].
Furthermore, the plastic phase transition behavior was evidenced by compression tests as shown in Fig. 3. Below Tc, the R-CPS powder crystals underwent fracture and collapsed into tiny fragments, when subjected to compression force under glass slide imposed by fingers (Figs. 3a and b). Whereas, after structural phase transition, the powder crystals merged into an intact tablet with flat and smooth surface (Figs. 3c and d), indicating favorable ductility, which is typical characteristic of plastic deformation. The compression tests of the S-CPS have also been performed (Fig. S4 in Supporting information), exhibiting similar feature to that of the R-CPS. The mechanical performance tests together with DSC results clearly demonstrate the plastic phase transition behavior of the enantiomers.

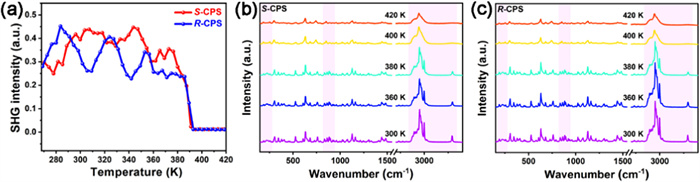
Solid materials with second harmonic generation (SHG) activity should crystallize in the noncentrosymmetric point groups (except for 432, 422, and 622), according to Kleinman's symmetry rules [42]. For this reason, SHG measurements are widely used for detecting symmetry variations and measuring the polar properties of single crystals. The S-CPS and R-CPS powder crystal samples possess nearly 20% SHG intensity of that of the potassium dihydrogen phosphate (KDP) at room temperature (Fig. S5 in Supporting information). Fig. 4a shows the temperature-dependent SHG signals of the camphorsultam enantiomers and the SHG intensity of S-CPS and R-CPS suddenly decreases to the background value at around Tc. Such an abrupt drop in SHG intensity matches well with the phase transition behavior observed on DSC curves. Raman spectrum serves as another powerful tool to investigate structural phase transition via detecting variations in chemical bonds and groups [43], hence variable temperature Raman spectra of S- and R-CPS have been conducted (Figs. 4b and c). Below Tc, the peaks remain relatively stable as temperature increases from 300 K to 380 K, while there are obvious decreases of intensities of all the peaks after undergoing structural phase transition, with further weakening trend as temperature raises from 400 K to 420 K. The descending of peak intensities especially the peaks around 220 cm-1 corresponding to lattice vibrations indicates elongation of chemical bonds and lattice softening in more loosely packed HTP. The C—H stretching peaks around 3000 cm-1 degenerate in HTP, declaring that the chemical environments surrounding H atoms of C—H bonds tend to be more identical. Also, the N—H stretching peak around 3300 cm-1 shrinks into a tiny bump in HTP, corresponding to weakening of N—H···O hydrogen bonds and raised disorder degree of N—H bonds [43]. The alicyclic vibration peaks in the range between 800 and 900 cm-1 partially merge, which manifests that alicyclic ring becomes more disordered. Both S-CPS and R-CPS exhibit similar variation trends in the variable temperature Raman spectra, proving the enantiomeric feature.
We have tried single-crystal X-ray diffraction measurements for HTP, but it is difficult to determine the structure due to the highly disordered nature. Further, PXRD measurements at various temperatures were performed to probe the structure of S-CPS and R-CPS in HTP. In Figs. S6a and b (Supporting information), at room temperature, the experimental PXRD matches well with the simulated PXRD obtained from the single crystal structure. At high temperatures, the number of peaks was decreasing compared to that in LTP, as shown in Figs. S6c and d (Supporting information). The disappearance of most of the PXRD peaks in HTP indicates a highly symmetric phase, being similar to that reported before [31], nevertheless, which is not enough to gain a deep understanding of HTP structure. Therefore, to further reveal the HTP structure, Pawley refinements of the PXRD of S-CPS and R-CPS have been performed, suggesting a plausible cubic lattice system for HTP (Figs. S7 and S8 in Supporting information). As there are only two chiral point groups of cubic lattice, namely 23 and 432, considering the non-active SHG responses of S-CPS and R-CPS in HTP, S-CPS and R-CPS should be refined in the 432 point group.
It is noticed that structural phase transition may trigger ferroelastic phase transition [44], therefore, we have investigated the ferroelastic behaviours of the compound. A single crystal of R-CPS with smooth surface (Fig. 5a) was chosen to perform the ferroelastic domain observations. Initially, the ferroelastic domain patterns could not be observed (Fig. S9 in Supporting information), probably due to its as-grown single-domain structure or smaller domain width than optical resolution. By raising temperature above Tc, the image under cross-polarized light became totally dark (Fig. S10 in Supporting information). This phenomenon demonstrates that the birefringence feature of the crystal disappears in HTP, and the HTP phase should belong to the cubic system which holds isotropic refraction index [45,46]. When cooling down below Tc, multiple strip-type ferroelastic domains appeared (Fig. 5b), with intersection angles of 180° and 90°, respectively. Ferroelastic behaviors have been further investigated via nanoindentation technology. The stress field imposed by the pyramid-shaped Berkovich indenter could drive ferroelastic domain movements and resultant step-like bursts appear on the load-displacement curves [47-49]. In some cases, step-like bursts also appear due to plastic deformation [50], whereas, those "pop-in" steps only arise in the loading runs while the unloading process do not present "pop-out" steps. For our case (Fig. 5c), both the loading and unloading segments present the "pop-in" and "pop-out" serrations, respectively, indicating the bidirectional ferroelastic domain switching process. This bidirectional ferroelastic domain switching process observed at other testing sites for both enantiomers (Fig. S11 in Supporting information) exclude the possibility of occasional error of the instruments, further demonstrating the ferroelastic properties of the enantiomers. The total displacements of R- and S-CPS are not always the same due to the fact that local ferroelastic domain distributions are different to each other, as well as domain movements. The organoferroelastic CPS enantiomers may exhibit application potential in flexible acousto-optic, shape memory, strain modulation and data storage devices.

In summary, we have discovered a single-component organoferroelastic, (1S and/or 1R)−2,10-Camphorsultam, by taking advantage of its plastic phase transition behavior. The enantiomers exhibit a high Tc of 385 K and a large entropy gain ∆St of 45 J K-1 mol-1, and obviously plastic deformation in HTP. SHG and Raman spectra reveals the highly-disordered structure in HTP, and Pawley refinements of the PXRD further infer that the S- and R-CPS should be refined in 432 point group in HTP. Strip-like ferroelastic domain structure and domain movements have been observed by cross-polarized light microscopy and nanoindentation technique. In this work, homochirality and plastic phase transition have been combined to produce a single-component organoferroelastic, which may serve as promising candidate for multipurpose biocompatible materials.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Yan-Ran Weng: Data curation. Wen-Fu Tian: Methodology, Data curation. Wen-Jing Ding: Data curation. Bi-He Ren: Data curation. De-Hou Liu: Data curation. Jia-Ying Tang: Data curation. Feng Zhou: Data curation. Xiao-Gang Chen: Supervision, Data curation. Xian-Jiang Song: Supervision, Data curation. Hui-Peng Lv: Supervision, Data curation. Yong Ai: Writing – review & editing, Writing – original draft, Supervision, Funding acquisition.
This work was supported by the National Natural Science Foundation of China (No. 22271131) and the Department of Science and Technology in Jiangxi Province (No. 20225BCJ23029).
The X-ray crystallographic structures have been deposited at the Cambridge Crystallographic Data centre (deposition numbers CCDC 2257, 435-2257, 436) and can be obtained free of charge from the CCDC via www.ccdc.cam.ac.uk/getstructures.
Supplementary material associated with this article can be found, in the online version, at doi:
E.K.H. Salje, Annu. Rev. Mater. Res. 42 (2012) 265–283. doi: 10.1146/annurev-matsci-070511-155022
M. Wojciechowska, A. Gagor, A. Piecha-Bisiorek, et al., Chem. Mater. 30 (2018) 4597–4608. doi: 10.1021/acs.chemmater.8b00962
E. Salje, G. Hoppmann, Mater. Res. Bull. 11 (1976) 1545–1550.
M. Chabin, F. Gilletta, J. Ildefonse, J. Appl. Crystallogr. 10 (1977) 247–251.
F. Meschke, A. Kolleck, G. Schneider, J. Eur. Ceram. Soc. 17 (1997) 1143–1149.
A. Bhatnagar, A.R. Chaudhuri, Y.H. Kim, et al., Nat. Commun. 4 (2013) 1–8.
S. Kamba, J. Pokorný, V. Porokhonskyy, et al., Appl. Phys. Lett. 81 (2002) 1056–1058.
A. Tselev, V. Meunier, E. Strelcov, et al., ACS Nano 4 (2010) 4412–4419. doi: 10.1021/nn1004364
Y. Liu, L. Collins, R. Proksch, et al., Nat. Mater. 17 (2018) 1013–1019. doi: 10.1038/s41563-018-0152-z
S.H. Mir, Y. Takasaki, S. Takamizawa, Phys. Chem. Chem. Phys. 20 (2018) 4631–4635. doi: 10.1039/c7cp07206f
I. Suzuki, K. Okada, Solid State Commun. 29 (1979) 759–762.
C.Y. Su, Y.F. Yao, Z.X. Zhang, et al., Chem. Sci. 13 (2022) 4794–4800. doi: 10.1039/d1sc07045b
X. Zheng, L. Zhou, P.P. Shi, et al., Chem. Commun. 53 (2017) 7756–7759.
Y.J. Cao, L. Zhou, P.P. Shi, et al., Chem. Commun. 55 (2019) 8418–8421. doi: 10.1039/c9cc03857d
Z.B. Liu, L. He, P.P. Shi, J. Phys. Chem. Lett. 11 (2020) 7960–7965. doi: 10.1021/acs.jpclett.0c02235
J.X. Gao, X.N. Hua, P.F. Li, J. Phys. Chem. C 122 (2018) 23111–23116. doi: 10.1021/acs.jpcc.8b07853
S.H. Mir, Y. Takasaki, E.R. Engel, et al., Angew. Chem. Int. Ed. 56 (2017) 15882–15885. doi: 10.1002/anie.201707749
E.R. Engel, Y. Takasaki, S.H. Mir, et al., Roy. Soc. Open Sci. 5 (2018) 171146. doi: 10.1098/rsos.171146
S.H. Mir, Y. Takasaki, E.R. Engel, et al., CrystEngComm 20 (2018) 3807–3811. doi: 10.1039/c8ce00295a
S.H. Mir, Y. Takasaki, E.R. Engel, et al., RSC Adv. 8 (2018) 21933–21936. doi: 10.1039/c8ra02499e
S. Ranjan, S. Takamizawa, Cryst. Growth Des. 22 (2021) 585–589.
H. Sun, S.K. Park, Y. Diao, et al., Chem. Mater. 33 (2021) 1883–1892. doi: 10.1021/acs.chemmater.1c00080
Y. Ai, P.F. Li, M.J. Yang, et al., Chem. Sci. 13 (2022) 748–753. doi: 10.1039/d1sc06781h
H.Y. Ye, J.Z. Ge, Y.Y. Tang, et al., J. Am. Chem. Soc. 138 (2016) 13175–13178. doi: 10.1021/jacs.6b08817
J. Harada, APL Mater. 9 (2021) 020901.
D. Chandra, W. Ding, R.A. Lynch, et al., J. Less-Common Met. 168 (1991) 159–167.
A. Mondal, B. Bhattacharya, S. Das, et al., Angew. Chem. Int. Ed. 59 (2020) 10971–10980. doi: 10.1002/anie.202001060
Y. Kaneko, M. Sorai, Phase Trans. 80 (2007) 517–528. doi: 10.1080/01411590701339401
B. Li, Y. Kawakita, S. Ohira-Kawamura, et al., Nature 567 (2019) 506–510. doi: 10.1038/s41586-019-1042-5
A.M. Piatek, A. Chojnacka, C. Chapuis, et al., Helv. Chim. Acta 88 (2005) 2441–2453. doi: 10.1002/hlca.200590181
T.T. Ying, Y.Z. Tang, Y.H. Tan, et al., Cryst. Growth Des. 22 (2022) 7501–7507. doi: 10.1021/acs.cgd.2c01047
A. Piatek, C. Chapuis, J. Jurczak, Helv. Chim. Acta 85 (2002) 1973–1988.
J. Harada, T. Shimojo, H. Oyamaguchi, et al., Nat. Chem. 8 (2016) 946–952. doi: 10.1038/nchem.2567
Y. Xie, Y. Ai, Y.L. Zeng, et al., J. Am. Chem. Soc. 142 (2020) 12846 -12492.
J. Harada, Y. Kawamura, Y. Takahashi, et al., J. Am. Chem. Soc. 141 (2019) 9349–9357. doi: 10.1021/jacs.9b03369
Y. Qian, D.S. Shao, W.W. Yao, et al., J. Phys. Chem. C 124 (2020) 20722–20729. doi: 10.1021/acs.jpcc.0c05430
M. Szafranski, Cryst. Growth. Des. 16 (2016) 3771–3776. ´ doi: 10.1021/acs.cgd.6b00279
P. Szklarz, A. Ingram, Z. Czapla, et al., Phase Trans. 90 (2017) 610–617. doi: 10.1080/01411594.2016.1252980
J. Timmermans, J. Phys. Chem. Solids. 18 (1961) 1–8.
P.F. Li, W.Q. Liao, Y.Y. Tang, et al., Proc. Natl. Acad. Sci. U. S. A. 116 (2019) 5878–5885. doi: 10.1073/pnas.1817866116
Y. Miyazaki, M. Sorai, R. Lin, A. Dworkin, et al., Chem. Phys. Lett. 305 (1999) 293–297. doi: 10.1539/sangyoeisei.KJ00001991052
D. Kleinman, Phys. Rev. 126 (1962) 1977.
M. Ptak, A. Sieradzki, M. Šimenas, et al., Coordin. Chem. Rev. 448 (2021) · 214180.
K. Aizu, J. Phys. Soc. Jpn. 27 (1969) 387–396.
M. Bari, A.A. Bokov, Z.G. Ye, J. Mater. Chem. C 9 (2021) 3096–3107. doi: 10.1039/d0tc05618a
C. Xu, Q. Li, Q. Yan, et al., J. Am. Ceram. Soc 99 (2016) 2706–2712. doi: 10.1111/jace.14267
J. Li, Q. Zhou, L. Yang, et al., J. Alloys Compd. 889 (2021) 161557.
N. Orlovskaya, Y. Gogotsi, M. Reece, et al., Acta Mater. 50 (2002) 715–723.
E. Withey, M. Jin, A. Minor, et al., Mater. Sci. Eng. A 493 (2008) 26–32.
I.A. Olson, A.G. Shtukenberg, B. Kahr, et al., Rep. Prog. Phys. 81 (2018) 096501. doi: 10.1088/1361-6633/aac303

Figure 1 Asymmetric unit of the molecular structure of (a) S-CPS and (b) R-CPS. Crystal structural packing view of (c) S-CPS and (d) R-CPS in one unit cell. Hydrogen atoms are omitted for clarity.

Figure 2 Phase transition behaviours of both camphorsultam enantiomers. (a) DSC curves in the heating-cooling runs and (b) DSC curves in the heating runs until melting. Scan rate: 5 K/min.

Figure 3 Morphology of R-CPS powder crystals (a) before and (b) after compression by glass slide at 303 K, and those (c) before and (d) after compression at 413 K.

Figure 4 (a) Temperature dependence of SHG intensities of S- and R-CPS, and variable temperature Raman spectra of (b) S-CPS and (c) R-CPS.

Figure 5 (a) Morphology of the single crystal of R-CPS, (b) strip-type ferroelastic domains after heating and cooling down to low-temperature phase under cross-polarized light, and (c) load-displacement curve of R-CPS film, with serrations on both loading and unloading segments indicating ferroelastic domain movements.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:
 下载:
下载:
 下载:
下载: