Guangxi Key Laboratory of Petrochemical Resource Processing and Process Intensification Technology, School of Chemistry and Chemical Engineering, Guangxi University, Nanning 530004, China
b.
State Key Laboratory of Featured Metal Materials and Life-cycle Safety for Composite Structures, Guangxi University, Nanning 530004, China
c.
State Key Laboratory of Green Chemical Engineering and Industrial Catalysis, School of Chemical Engineering, East China University of Science and Technology, Shanghai 200237, China
Received Date:
05 April 2024 Accepted Date:
24 June 2024 Revised Date:
21 June 2024 Available Online:
15 January 2025
Abstract:
Copper (Cu) is widely used in the electrochemical carbon dioxide reduction reaction (ECO2RR) for efficient methane (CH4) product. However, the morphology and valence of Cu-based catalysts are usually unstable under reaction conditions. In this work, we prepared Ce-doped MOF-199 precursor (Ce/HKUST-1) and further obtained nanoparticle electrocatalyst Ce/CuOx-NPs by cyclic voltammetry (CV) pretreatment. The Faradic efficiency of methane () maintains above 62% within a broad potential window of 350 mV and the maximum reaches 67.4% with a partial current density of 293 mA/cm2 at −1.6 V vs. a reversible hydrogen electrode. Catalyst characterization and theoretical calculations revealed that the unique electronic structure and large ionic radius of Cerium (Ce) not only promoted the generation of key intermediate *CO but also lowered energy barrier of the *CO to *CHO step. This study provides a novel and efficient catalyst for methane production in ECO2RR and offers profound insights into constructing high performance Cu-based catalysts.
The excessive emission of carbon dioxide (CO2) has caused global climate change and ocean acidification [1–3]. To solve these issues, the electrochemical CO2 reduction reaction (ECO2RR) is considered as a promising strategy for CO2 utilization [4,5]. A main challenge in electrochemical CO2 reduction is to develop high efferent catalysts with desired products. Among these products, CH4 receives widespread attention as a green chemical fuel and industrial raw material [6]. However, due to multiple electron transfer steps and high-energy barriers involved, this process is slow kinetically [7–10]. Therefore, efficient electrocatalysts are required to lower the reaction overpotential, promote kinetics, and achieve commercially significant rates.
Various metal catalysts (Bi [11], Cu [12], Au [13], Ag [14], Pd [15], amongst other) have been evaluated as electrodes for ECO2RR. Among them, Cu is a unique metal catalyst can convert CO2 to hydrocarbons, methanol, and multicarbon oxygenates [16,17]. However, the dynamic structural transformations, the presence of mixed Cu+/Cu0 states, the microenvironment, and metal-support interactions lead to a very complex structure of Cu-based catalysts and low selectivity for single products [18–21]. For the ECO2RR to CH4, the hydrogenation of *CO to *CHxO (x = 1, 2 or 3) is believed to be the key reaction step that determines the selectivity, which is followed by the production of the gaseous CH4 through the breakage of the C−O bond in *CH3O [22,23]. Nonetheless, the methane formation pathway on Cu-based catalysts shares many intermediates (i.e., *CO, *CH3O) with other ECO2RR byproducts, such as ethylene (C2H4) and/or methanol (CH3OH) at industrial current densities [24,25].
Several strategies have been reported to improve the CH4 selectivity of Cu-based catalysts. For instance, Qiao and colleagues enhanced the adsorption stability of the *CO intermediate by stabilizing Cu2+ in the catalyst, which facilitated further hydrogenation of *CO and improved CH4 selectivity [24]. Yang and coworkers directed a robust reconstruction of copper metal during electrocatalysis by adding ethylenediamine tetramethylenephosphonic acid to the electrolysis, which greatly facilitated the methane selectivity of commercial polycrystalline copper, with CH4 Faraday efficiencies of 64% in an alkaline flow cell [26]. Oh et al. constructed a high concentration of oxygen vacancies which attached to Cu+ sites on the metal oxide surface, and the highly hydroxylated surface formed at high pH accelerated *CO protonation toward CH4 production with a Faraday efficiency of 58% [27]. Among them, introducing another metal dopant was found an attractive approach to modulate the adsorption energy of intermediates [28–30]. Introducing metal with high oxophilicity into Cu may enhance the bonding of O in *CHxO onto the oxophilic metal and enhance the metal-oxygen (M–O) bonding on the pro-oxidized metal, thereby facilitating the breakage of the C–O bonding in *CH3O and increasing CH4 production [30–32]. Reducing the concentration of the dopant and dispersing it as individual atoms in the host lattice, it can be prepared with greater ECO2RR activity and stability through geometrical and electronic effects [33,34]. The ionic radius difference between the substrate and the metal dopant can also cause lattice distortion [25]. It affects the d-band center of the catalyst, which can optimize the binding energy of the reaction intermediates (e.g., *CHO), leading to changes in product selectivity [35,36]. Cerium (Ce) possesses a unique 4f electronic structure and unfilled 5d orbitals, which confer upon its strong spin-orbit coupling and lanthanide contraction effects [37,38]. These characteristics enable Ce to effectively modulate the local electron densities of surrounding atoms and sustain them in a high-valence chemical state. Meanwhile, the introduction of large-size Ce atoms leads to a distortion of the main atomic lattice of Cu catalyst [25,32]. However, the effective doping of Ce at the atomic scale usually need high temperature. Therefore, developing a simple and effective process for atomic Ce doping into the lattice of Cu is highly desired.
Herein, we prepared the catalyst precursor Ce/HKUST-1 by doping trace amount of Ce into HKUST-1 via a simple wet-chemical method. Next, the electrocatalysts for ECO2RR were prepared by further treating HKUST-1 and Ce/HKUST-1 using electrochemical processes (cyclic voltammetry (CV) and timed-current (I-T)). CuOx-NPs and Ce/CuOx-NPs were obtained through CV treatment, while CuOx-MDs and Ce/CuOx-MDs were obtained through I-T treatment. Electrochemical performance tests, physical characterization, and density-functional-theory (DFT) calculations revealed that atomic Ce doping induced the lattice stretching strain and effectively modulated the local electron densities of surrounding Cu atoms, which enhanced the generation of *CO in the catalyst surface, lowered the energy barriers from *CO to *CHO, and facilitated the breakage of the C–O bond in *CH3O, which effectively enhanced the selectivity of methane preparation by ECO2RR.
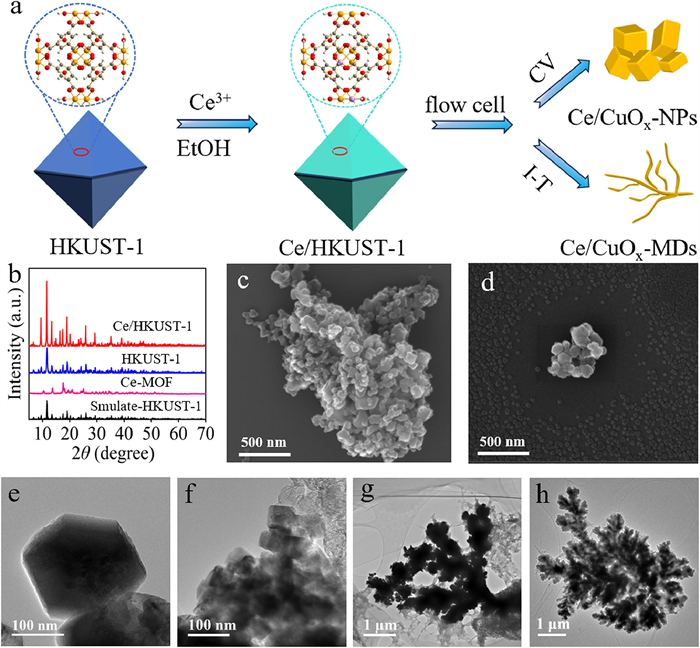
Two types of MOFs, HKUST-1and Ce/HKUST-1 were synthesized using the referenced methods [39]. First, HKUST-1 and Ce/HKUST-1 were uniformly coated onto carbon paper as the cathode electrodes of the flow cell. Subsequently, the prepared cathode electrodes underwent CV electrochemical pretreatment in a flow cell to produce nanoparticle catalysts for ECO2RR (Fig. 1a, and more details can be found in Supporting information). The powder X-ray diffraction (PXRD) patterns of the prepared HKUST-1 closely matched the simulated PXRD patterns of HKUST-1, confirming the successful synthesis of MOF-199 at room temperature (Fig. 1b). Furthermore, no significant phase difference was observed in the PXRD patterns between Ce/HKUST-1 and HKUST-1. However, PXRD patterns of the reference sample Ce-MOF displayed an amorphous phase (Fig. 1b). The difference may be due to the stronger Lewis acid properties of Ce3+ ions compared to Cu2+ ions, which accelerated the dissolution of Ce-MOF in aqueous solution [39,40]. Scanning electron microscopy (SEM) (Figs. 1c and d, Fig. S1 in Supporting information) was used to investigate the morphology of the prepared MOFs, and revealed that both HKUST-1 and Ce/HKUST-1 exhibit similar nanoparticle morphologies, while Ce-MOF exhibits micrometer-sized straw-bundle three-dimensional structure. This could be attributed to the introduction of CTAB surfactant which cause defects in HKUST-1, imparting an anionic framework characteristic that facilitated cation exchange, leading to the successful incorporation of Ce3+ by partially replacing Cu2+ in HKUST-1 [39]. Generally, Cu-based catalysts often undergo uncontrolled structural rearrangements during electrochemical testing processes [36]. To investigate the effect of electrochemical pretreatment on the morphology of the catalysts, transmission electron microscopy (TEM) was employed to determine the catalyst morphology after electrochemical pretreatment. Figs. 1e and f depict the morphology of Ce/CuOx-NPs and CuOx-NPs obtained after CV pretreatment. Moreover, the average diameters (Figs. S2 and S3 in Supporting information) of Ce/CuOx-NPs (75.02 nm) and CuOx-NPs (72.48 nm) were calculated. The size of Ce/CuOx-NPs is relatively larger, probably due to the larger Ce atoms substituting for Cu atoms in the lattice. However, Ce/CuOx-MDs (Fig. 1g) and CuOx-MDs (Fig. 1h) obtained through I-T pretreatment exhibit a micrometer-scale dendritic morphology. This indicates that different electrochemical pretreatment processes can result in distinct morphologies.
Figure 1
Figure 1.
(a) Schematic synthesis procedure for MOFs-derived Ce-doped Cu catalysts. (b) PXRD patterns of Ce/HKUST-1, HKUST-1 and Ce-MOF. (c, d) SEM images of Ce/HKUST-1 and HKUST-1. (e-h) TEM images of Ce/CuOx-NPs, CuOx-NPs, Ce/CuOx-MDs and CuOx-MDs.
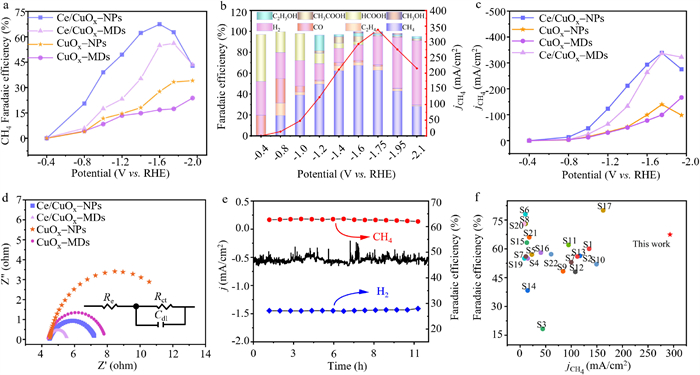
The catalytic performance of as-prepared catalysts for ECO2RR was evaluated in a flow cell (Fig. S4 in Supporting information) using a CHI760E workstation. All catalysts electrochemical performance tests were performed using the timed current method (Fig. S5 in Supporting information). Gas-phase products were quantitatively analyzed by gas chromatography (GC), while liquid-phase products were quantitatively analyzed by nuclear magnetic resonance (NMR). Hydrogen evolution reaction (HER) is a competitive reaction to ECO2RR, and effectively inhibiting H2 generation can enhance the selectivity of ECO2RR products [41]. Additionally, inhibiting the generation of by-products such as CO and C2H4 is key to improving methane selectivity [42]. As shown in Fig. S6 (Supporting information), all four catalysts show significant inhibition of HER. Furthermore, Figs. S7 and S8 (Supporting information) indicate that Ce doping and nanoparticle morphology can effectively inhibit the generation of CO and C2H4. The methane selectivity of several catalysts was further evaluated. Ce/CuOx-NPs exhibits the highest methane catalytic activity (Fig. 2a), with a highest FECH4 of 67.4% at –1.6 V, surpassing Ce/CuOx-MDs (54.8%), CuOx-NPs (27.6%), and CuOx-MDs (16.8%) by 1.2, 2.5, and 4 times respectively. As shown in Fig. 2b, the FECH4 of Ce/CuOx-NPs exhibits a volcano-like trend with negative shifting test potential, reaching its maximum at –1.6 V. With a further negative shift of the cathodic potential, the FEH2 of Ce/CuOx-NPs increases, leading to abundant H2 generation and a decrease in FECH4. Moreover, the FECH4 of Ce/CuOx-NPs at the potential range from –1.4 V to –1.75 V exceeds 62%, a remarkable catalytic performance rarely reported in the literature. At lower cathodic potentials, the predominant liquid-phase product of Ce/CuOx-NPs is formic acid, and the selectivity of formic acid decreases as the cathodic potential decreases. As depicted in Fig. S9 (Supporting information), the liquid-phase products of Ce/CuOx-NPs include formic acid, acetic acid, and ethanol at –1.6 V. The detailed results of electrochemical performance test results for Ce/CuOx-NPS can be referred to Table S1 (Supporting information). Methane partial current density (jCH4 > 100 mA/cm2) is an important evaluation criterion for the industrial application of electrocatalysts [43]. As shown in Fig. 2c, Ce/CuOx-NPs exhibits a maximum jCH4 (338 mA/cm2) at –1.75 V, surpassing Ce/CuOx-MDs (336 mA/cm2) and CuOx-NPs (139 mA/cm2). Although the jCH4 of CuOx-MDs shows an upward trend, its methane selectivity is too low, limiting its further applications. These results indicate that Ce/CuOx-NPs is the optimal catalyst for electrochemical CO2 methanation and possess industrial application potential.
Figure 2
Figure 2.
ECO2RR performance evaluation of the catalysts. (a) FECH4 of Ce/CuOx-NPs, Ce/CuOx-MDs, CuOx-NPs and CuOx-MDs. (b) Faradaic efficiency and methane partial current density of Ce/CuOx-NPs. (c) jCH4 tested at different potentials of Ce/CuOx-NPs, Ce/CuOx-MDs, CuOx-NPs and CuOx-MDs. (d) Nyquist plots in the flow cell at a potential of −0.4 V in 1.0 mol/L KOH. (e) Time-dependent total current density, FECH4 and FEH2 for Ce/CuOx-NPs recorded at −1.6 V in 1.0 mol/L KOH for 10 h. (f) Comparison of FECH4 for Ce/CuOx-NPs with the reported Cu-based catalysts (Table S2 in Supporting information).
To further investigate the kinetic behavior in ECO2RR, electrochemical impedance spectroscopy (EIS) was performed to determine the charge transfer capability. The Nyquist plots show a depressed semicircle corresponding to the charge transfer reactance. The magnitude of the radius curvature of the semicircle is usually used to estimate the charge transfer rate [44,45]. As shown in Fig. 2d, the charge transfer resistance of Ce/CuOx-NPs and Ce/CuOx-MDs were significantly lower than those of CuOx-NPs and CuOx-MDs. This confirms that the doping of Ce effectively improves the charge transfer rate of electrons on the catalyst surface. We adopted the double-layer capacitance (Cdl) method to measure the electrochemical active surface area (ECSA) of the catalysts. The catalyst Cdl value was determined by testing the CV data at different scan rates (Figs. S10 and S11 in Supporting information). As shown in Table S3 (Supporting information), the ECSA of Ce/CuOx-NPs is 96 cm2, the highest one among the tested catalysts, indicating that Ce/CuOx-NPs possess a larger electrochemical reaction area and expose more active sites [11,32]. The stability test of Ce/CuOx-NPs was conducted at –1.75 V, sampling every 30 min and quantitatively analyzing CH4 and H2 using GC. As shown in Fig. 2e, the FECH4 of Ce/CuOx-NPs remained above 60% over 10 h, the morphology of Ce/CuOx-NPs and the valence state of Cu also did not show any significant changes (Figs. S12 and S13 in Supporting information), indicating a good durability in the long-term electrocatalysis. The performance of Ce/CuOx-NPs was compared with reported Cu-based catalysts for ECO2RR, where Ce/CuOx-NPs exhibited excellent electrochemical catalytic performance, with FECH4 (67.4%) and jCH4 (293 mA/cm2) at a testing potential of –1.6 V (Fig. 2f).
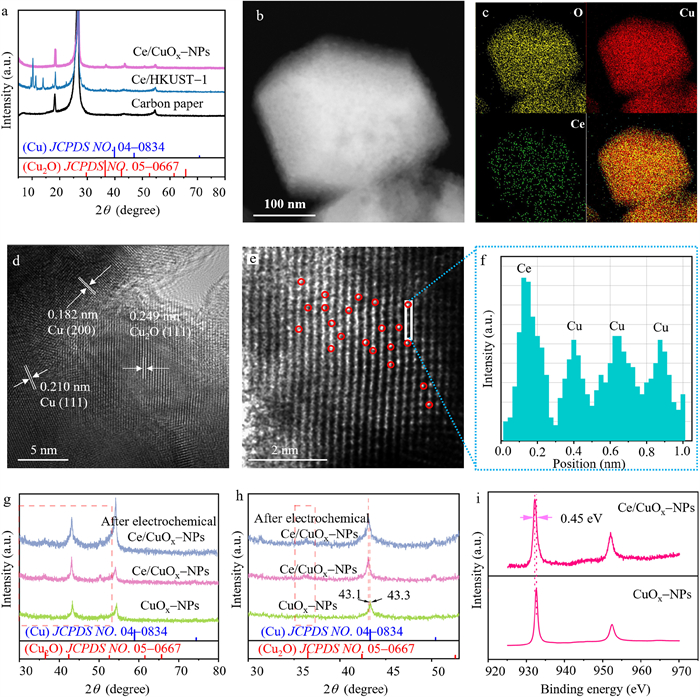
To further investigate the effect of Ce doping on the selectivity of electrochemical CO2 methanation, we first tested the selectivity of Ce-MOF in ECO2RR, as shown in Fig. S14 (Supporting information), Ce-MOF exhibits selectivity only for H2, indicating that monometallic Ce catalysts do not have the catalytical activity in ECO2RR. Then we have investigated Ce/CuOx-NPs and CuOx-NPs using electron microscopy, XRD and XPS. As shown in Fig. 3a, the PXRD patterns of Ce/CuOx-NPs does not exhibit diffraction peaks consistent with Ce/HKUST-1, while its diffraction peaks matched those of Cu (JCPDS No. 04–0836) and Cu2O (JCPDS No. 05–0667). This indicates that the organic framework structure of Ce/HKUST-1 was disrupted after CV treatment, with Cu2+ electrochemically reduced to Cu and Cu2O. Furthermore, the atomic percentages of Cu and Ce were determined by Inductively Coupled Plasma Optical Emission Spectroscopy (ICP-OES) for Ce/HKUST-1, Ce/CuOx-NPs and Ce/CuOx-MDs. Table S4 (Supporting information) confirms the trace amount doping of Ce and further reduction of Ce content after electrochemical pretreatment. The decrease could be attributed to the dissolution of Ce in the electrolyte and its subsequent discharge from the flow cell along with the electrolyte during the electrochemical pretreatment. Additionally, scanning transmission electron microscopy (STEM) images (Fig. 3b) and energy-dispersive X-ray spectroscopy (EDX) spectra (Fig. 3c) demonstrated the uniform distribution of Ce and Cu elements in Ce/CuOx-NPs. Similar phenomena were observed in Ce/CuOx-MDs (Fig. S15 in Supporting information). High-resolution transmission electron microscopy (HR-TEM) images (Fig. 3d) revealed that the lattice spacings of 0.249, 0.182, and 0.210 correspond to the Cu2O (111), Cu (200), and Cu (111) crystal planes. Compared with pure Cu2O (0.246 nm) and Cu (0.208 nm), the larger lattice spacings of Cu2O (111) and Cu (111) indicated a lattice expansion due to the incorporation of larger Ce atoms at the atomic level into the Cu lattice [42]. Atomic dispersion of Ce was identified using aberration-corrected high-angle annular dark-field scanning transmission electron microscopy (AC—HAADF-STEM), and the light spots marked with red circles in Fig. 3e could be identified as Ce species. Intensity distribution analysis (Fig. 3f) further confirmed the dispersion of Ce atoms, verifying their effective doping into the Cu lattice. To validate the effect of Ce doping on Cu lattice, Cu samples without a Ce precursor were synthesized using the same method. HR-TEM measurement of Cu lattice spacings (Fig. S16 in Supporting information) revealed that the Cu lattice spacing is 0.208 nm, corresponding to reported lattice spacing of Cu (111).
Figure 3
Figure 3.
(a) PXRD patterns before and after CV pre-reduced Ce/HKUST-1. (b) STEM image, (c) element mappings, (d) HRTEM image, (e) AC-HAADF-STEM image of Ce/CuOx-NPs, and (f) corresponding intensity profile along the line. (g) PXRD patterns of Ce/CuOx-NPs, CuOx-NPs, and Ce/CuOx-NPs after electrochemical tested. (h) The enlarged image of (g). (i) XPS spectra of Cu 2p.
Copper is inevitably reduced to Cu0 during electrochemical testing, leading to a decline in catalytic performance. To understand the origin of high electrocatalytic performance of Ce/CuOx-NPs, PXRD and XPS were performed to investigate the crystal structure and chemical valence of Ce/CuOx-NPs and Cu-NPs. All the catalysts (Fig. 3g) exhibited diffraction peaks matching well with the planes of Cu (111) and Cu (200) (JCPDS No. 04–0836). However, a distinct diffraction peak appeared at 36.1° in the PXRD pattern of Ce/CuOx-NPs, assignable to the Cu2O (111) plane (JCPDS No. 05–0667). This diffraction peak remained observable after electrochemical testing, indicating that Cu2O phase remained after electrochemical testing of Ce/CuOx-NPs. Furthermore, as shown in Fig. 3h, the diffraction peak corresponding to the Cu (111) plane of Ce/CuOx-NPs (43.1°) is significantly shifted to a lower angle compared to that of CuOx-NPs (43.3°), attributed to lattice distortion caused by atomic-level Ce doping. In the XPS spectra of Cu 2p in Ce/CuOx-NPs (Fig. 3i), the peak at 932.3 eV could be identified as Cu0 or Cu+, Cu LMM XPS spectra (Fig. S17 in Supporting information) proves the existence of Cu+, without any observable satellite peaks, indicating the absence of Cu2+. Meanwhile, the XPS spectra of Cu 2p of Ce/CuOx-NPs is negatively shifted by 0.45 eV in binding energy compared with CuOx-NPs. This shift may be attributed to the electron enrichment around Cu atoms due to atomic-level Ce doping [25]. Bader charge calculations based on density functional theory (DFT) were used to evaluate the changes in the electronic environment around Cu atoms before and after Ce doping. As shown in Figs. S18 and S19 (Supporting information), there is a noticeable difference in electron counts around Cu atoms on the CuOx and Ce/CuOx surfaces. The Bader charges on the surface of Ce/CuOx was +0.05 e higher than that on the surface of CuOx, indicating that atomic-level Ce doping increased the electron density around the Cu atoms and modulated the valence state of Cu between Cu0 and Cu+. This modulation could influence the adsorption of intermediates on the catalysts' surface, thereby enhancing the selectivity of electrochemical CO2 methanation.
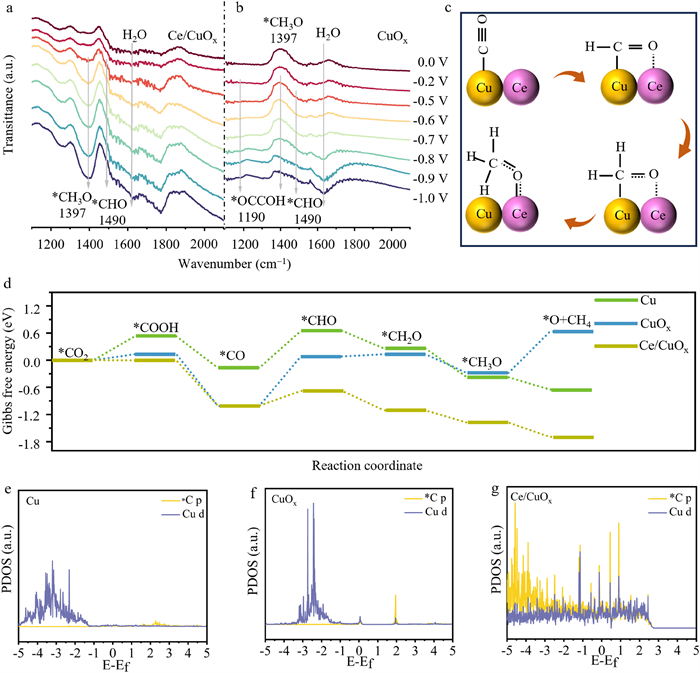
To provide insight into the underlining mechanism of the promotive effect of atomic-level Ce doping, an in-situ attenuated total reflection surface-enhanced infrared absorption spectroscopy (in-situ ATR-SEIRAS) was conducted in a homemade cell to identify the reactive species during ECO2RR. The peak at ~1631 cm–1 indicates the H2O in the electrolyte (Figs. 4a and b) [24,46]. The peaks at ~1490 cm–1 and 1392 cm–1 correspond to the *CHO intermediate and *CH3O intermediate [32,47]. Comparing with Cu-NPs, much stronger peaks can be obtained on the Ce/CuOx-NPs during the negative scan, which may result from the strong adsorption of *CHO and *CH3O by doping oxophilic metal as discussed previously. No obvious signals of top-adsorbed CO (*COtop) appeared in both Ce/CuOx-NPs and CuOx-NPs throughout the potential test interval. Meanwhile, a peak at 1190 cm–1 of CuOx-NPs from –0.5 V to –1.0 V is consistent with *OCCOH [48,49]. *OCOOH is a key intermediate toward producing C2H4, in agreement with the results of electrochemical tests (Fig. S20 in Supporting information). Combined with the results of in-situ ATR-SEIRAS, we propose that on the Ce/CuOx-NPs surface, the methane reaction pathway of CO2 → *CO → *CHO → *CH2O → *CH3O → CH4 (Fig. 4c) [50].
Figure 4
Figure 4.In-situ ATR-SEIRAS spectra on (a) Ce/CuOx-NPs and (b) CuOx-NPs at different potentials in CO2-saturated 1 mol/L KOH electrolyte. (c) Proposed reaction pathways of ECO2RR to CH4 on the Ce/CuOx-NPs surface. (d) The calculated Gibbs free energy for Cu, CuOx, and Ce/CuOx. Projected density of states (PDOS) for d orbitals of the Cu atom and p orbitals of the C atom on (e) Cu, (f) CuOx, and (g) Ce/CuOx with adsorbed *CHO.
To further understand the enhanced activity of ECO2RR to CH4 by Ce/CuOx-NPs, DFT calculations were performed. The relevant theoretical models, including Cu, CuOx, and Ce/CuOx were first constructed (Fig. S21 in Supporting information). Gibbs free energies were calculated for Cu, CuOx, and Ce/CuOx. The optimized ensemble shapes of *CO2, *COOH, *CO, *CHO, *CH2O, and *CH3O intermediates adsorbed on the surfaces of different catalysts are shown in Figs. S22–S24 (Supporting information). From the Gibbs free energy diagram shown in Fig. 4d, the rate-limiting step for Cu, CuOx, and Ce/CuOx is the further hydrogenation of *CO to *CHO. The ΔG (*CO → *CHO) of 0.33 eV for Ce/CuOx is lower than that of Cu (0.82 eV) and CuOx (1.10 eV). Besides, from the energy profile, *CH3O is more inclined to break the C–O bond and form gaseous methane on the Ce/CuOx surface. In contrast, the CuOx surface shows the opposite trend. The projected density of states (PDOS) value was calculated to investigate the interaction between the catalyst surface and the key intermediate *CHO (Figs. 4e–g). Ce/CuOx exhibit a higher overlap of Cu d and C p orbitals in the key intermediate *CHO compared to Cu and CuOx, suggesting the presence of strong electronic interactions and binding between *CHO and surface [51,52]. Moreover, we calculated the PDOS values after the adsorption of intermediate *CO using the same method (Fig. S25 in Supporting information). The appropriate adsorption of *CO on Ce/CuOx was also confirmed in DFT calculations. The above studies suggest that the superior ECO2RR performance of Ce/CuOx-NPs is due to atomic Ce doping that modulates the adsorption of *CO and *CHxO, lowers the energy barrier for the step of *CO to *CHO, and promotes the breaking of C−O bonds in *CH3O.
In summary, Ce/CuOx-NPs with nanoparticle morphology were successfully prepared and validated as an efficient catalyst for converting CO2 to CH4. The FECH4 of 67.4% could be achieved with a partial current density of 293 mA/cm2. XPS measurement demonstrated that atomic-level Ce doping increased the electron density around the Cu atoms. In situ spectroscopy analysis and theoretical calculations show that the Ce atoms doping enhances the stability of the key intermediate of *CHO and reduces the energy barrier for the conversion of *CO to *CHO, thus significantly increasing the activity of the CO2-to-CH4. This work demonstrates that electrochemical pretreatment can well control the catalyst morphology and atomic Ce doping can effectively improve the ECO2RR performance of Cu-based catalysts, which would inspire the design of electrochemical catalysts with high performance.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
CRediT authorship contribution statement
Xiangyu Chen: Writing – review & editing, Writing – original draft, Methodology, Data curation. Aihao Xu: Data curation. Dong Wei: Data curation. Fang Huang: Investigation. Junjie Ma: Data curation. Huibing He: Project administration. Jing Xu: Writing – review & editing, Supervision, Project administration, Funding acquisition.
Acknowledgments
We acknowledge the funding support from the National Natural Science Foundation of China (No. 22308066), the Science and Technology Major Program of Guangxi (No. Guike AA23062018), the Guangxi Science and Technology Base and Talent Special Project (Nos. 2021AC19353, 2022AC20018, AD23026311), the Natural Science Foundation of Guangxi Province (No. 2024GXNSFAA010271), the Innovation Project of Guangxi Graduate Education (No. YCBZ2022012).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.110175.
Y. Katayama, F. Nattino, L. Giordano, et al., J. Phys. Chem. C 123 (2019) 5951–5963. doi: 10.1021/acs.jpcc.8b09598
[48]
T.C. Chou, C.C. Chang, H.L. Yu, et al., J. Am. Chem. Soc. 142 (2020) 2857–2867. doi: 10.1021/jacs.9b11126
[49]
C.M. Gunathunge, V.J. Ovalle, Y. Li, et al., ACS Catal. 8 (2018) 7507–7516. doi: 10.1021/acscatal.8b01552
[50]
G. Wang, J. Chen, Y. Ding, et al., Chem. Soc. Rev. 50 (2021) 4993–5061. doi: 10.1039/d0cs00071j
[51]
W. Wang, Z. Wang, R. Yang, et al., Angew. Chem. Int. Ed. 60 (2021) 22940–22947. doi: 10.1002/anie.202110000
[52]
R. Yang, J. Duan, P. Dong, et al., Angew. Chem. Int. Ed. 61 (2022) e202116706. doi: 10.1002/anie.202116706
Figure 1
(a) Schematic synthesis procedure for MOFs-derived Ce-doped Cu catalysts. (b) PXRD patterns of Ce/HKUST-1, HKUST-1 and Ce-MOF. (c, d) SEM images of Ce/HKUST-1 and HKUST-1. (e-h) TEM images of Ce/CuOx-NPs, CuOx-NPs, Ce/CuOx-MDs and CuOx-MDs.
Figure 2
ECO2RR performance evaluation of the catalysts. (a) FECH4 of Ce/CuOx-NPs, Ce/CuOx-MDs, CuOx-NPs and CuOx-MDs. (b) Faradaic efficiency and methane partial current density of Ce/CuOx-NPs. (c) jCH4 tested at different potentials of Ce/CuOx-NPs, Ce/CuOx-MDs, CuOx-NPs and CuOx-MDs. (d) Nyquist plots in the flow cell at a potential of −0.4 V in 1.0 mol/L KOH. (e) Time-dependent total current density, FECH4 and FEH2 for Ce/CuOx-NPs recorded at −1.6 V in 1.0 mol/L KOH for 10 h. (f) Comparison of FECH4 for Ce/CuOx-NPs with the reported Cu-based catalysts (Table S2 in Supporting information).
Figure 3
(a) PXRD patterns before and after CV pre-reduced Ce/HKUST-1. (b) STEM image, (c) element mappings, (d) HRTEM image, (e) AC-HAADF-STEM image of Ce/CuOx-NPs, and (f) corresponding intensity profile along the line. (g) PXRD patterns of Ce/CuOx-NPs, CuOx-NPs, and Ce/CuOx-NPs after electrochemical tested. (h) The enlarged image of (g). (i) XPS spectra of Cu 2p.
Figure 4In-situ ATR-SEIRAS spectra on (a) Ce/CuOx-NPs and (b) CuOx-NPs at different potentials in CO2-saturated 1 mol/L KOH electrolyte. (c) Proposed reaction pathways of ECO2RR to CH4 on the Ce/CuOx-NPs surface. (d) The calculated Gibbs free energy for Cu, CuOx, and Ce/CuOx. Projected density of states (PDOS) for d orbitals of the Cu atom and p orbitals of the C atom on (e) Cu, (f) CuOx, and (g) Ce/CuOx with adsorbed *CHO.

 DownLoad:
DownLoad:

 下载:
下载:





 下载:
下载:
