Received Date:
19 April 2024 Accepted Date:
24 June 2024 Revised Date:
19 June 2024 Available Online:
15 January 2025
Abstract:
Diradicaloid polycyclic hydrocarbons (PHs) own unique open-shell electronic structures and exhibit potential utility in the fields of organic electronics and spintronics. Herein, we disclose precise fusion of B/O-heterocycles onto PHs for control over their electronic structures and diradical properties. We designed and synthesized four B/O-containing diradicaloid isomers that feature the fluoreno[3,2-b]fluorene and fluoreno[2,1-a]fluorene π-skeletons, respectively. The precise B/O-heterocycle fusion modes along with the changed conjugation patterns lead to their modulated electronic structures and properties, such as diradical and aromatic structures, energy levels and band gaps, as well as magnetic, electrochemical and photophysical properties. Notably, the mode A may decrease the open-shell extent, whereas the mode B can enhance the diradical nature, leading to their well-tuned diradical characters in the range of 0.46‒0.70. Moreover, the mode A stabilizes the LUMOs and the mode B obviously increases the HOMO levels, which are remarkably contributed by the B and O atoms, respectively, further giving rise to the decreased band gaps and redshifted absorptions. This study clearly illustrates the electronic effects of B/O-heterocycle fusion on PHs and gains insight into B/O-type organic diradicaloids. These findings will provide an important guideline for the design of more fascinating heteroatom-containing diradicaloids.
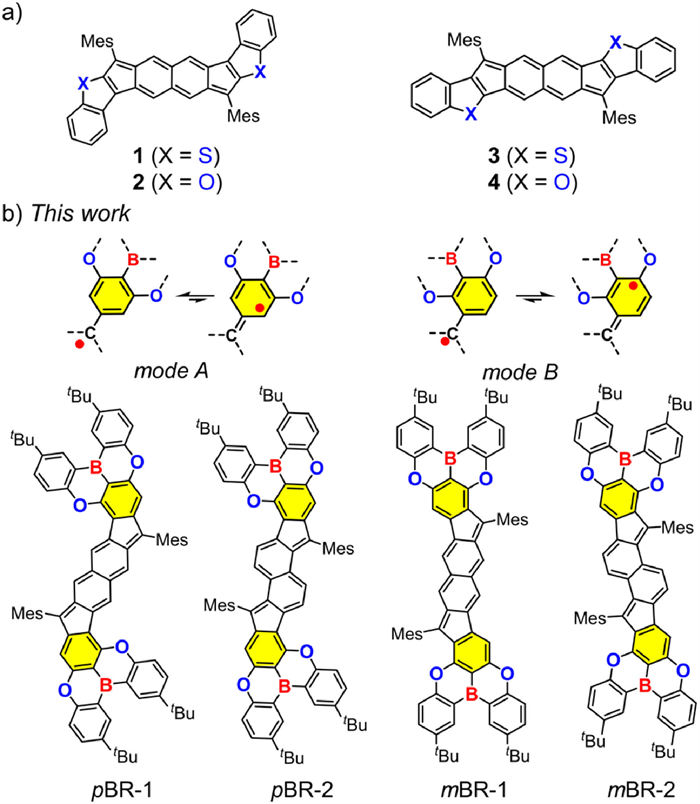
Diradicaloid polycyclic hydrocarbons (PHs), featuring open-shell electronic structures, have received much attention because of their intriguing optical, electronic and magnetic properties, as well as potential utility in the fields of organic electronics and spintronics [1–6]. Modulation of their electronic structures, especially two key parameters including diradical characters (y) and singlet-triplet energy gap (ΔES‒T), is very important for finely tuning physical properties and thereby achieving desirable functions and applications [7–9]. Extending the quinoidal conjugation and altering the conjugation patterns of PHs are two widely used methods, which have promoted the development of various all-carbon PHs with fascinating topological structures and tunable diradical properties, such as bisphenalenyls, anthenes, indenofluorenes and zethrenes, as well as their related derivatives [10–19]. Incorporation of heterocycles into PHs represents another effective strategy, following which a number of diradicaloid PHs containing the sulfur, oxygen, and nitrogen atoms have been developed [20–26]. It is noteworthy that modulating the position relationship between heteroatoms and radical centers can alter the conjugation mode and thereby lead to different electronic effects of heteroatoms. For example, Haley et al. disclosed that the position of the sulfur or oxygen atom relative to the radical center greatly impacts on antiaromatic structures and diradical properties of organic diradicaloids (1‒4) (Fig. 1a) [27,28]. Despite of these advances, studies on the use of heterocycles to rationally modulate the electronic structures of diradicaloid PHs are still rare, which are due to the lack of heterocycles available for these compounds and the challenge of precise incorporation of heteroatoms, thus hampering the expansion of diradicaloid chemistry and materials.
Figure 1
Figure 1.
Schematic illustration of control over the electronic structures of diradicaloid PHs via incorporation of (a) the sulfur or oxygen atom and (b) the boron and oxygen atoms, along with chemical structures of 1‒4, as well as pBR-1, pBR-2, mBR-1 and mBR-2.
The boron atom has one less electron than carbon atom and features a vacant p orbital. Incorporation of the boron atom into the π-skeletons has generated a variety of boron-doped PH systems that display fascinating physical properties and functions, e.g., catalysis, thermochromism, photochromism, charge transport, amplified spontaneous emission, and controllable supramolecular polymerization [29–43]. Recently, we devoted our efforts to the development of boron-containing organic diradicaloids [44–48]. We disclosed the borylation of antiaromatic PHs to be an effective design strategy and thereby developed neutral boron/oxygen- and boron/nitrogen-type organic diradicaloids, which are different from the previously reported organoborane diradicaloids that usually feature ionic nature [49–51]. They have outstanding ambient stability and narrow band gaps, thus exhibiting great potentials as near infrared (NIR) absorption dyes and efficient photothermal conversion materials. Nevertheless, because the boron atom is electron deficient and the cooperative heteroatoms (oxygen and nitrogen) are electron rich, the electronic effects of the heterocycles on PHs are very complicated and remain unclear. Thus, it is needed to gain insight into heterocycle fusion, which would be desirable for rationally tuning the electronic structures and properties of diradicaloid PHs.
Herein, we report four new boron/oxygen-containing diradicaloid isomers that were designed by precise heterocycle-fusion of fluorenofluorenes (Fig. 1b). The fusion modes A and B lead to the molecules pBRs (pBR-1 and pBR-2) and mBRs (mBR-1 and mBR-2), respectively, in which the boron and oxygen atoms are located at the different positions relative to the radical centers. Theoretical and experimental studies reveal that the mode A may decrease the open-shell extent, whereas the mode B can enhance the diradical character, thus achieving large-scale control over electronic structures of and successively modulating physical properties of PHs. In this article, we disclose their synthesis, magnetic, electrochemical and photophysical properties, together with in-depth elucidation of their electronic structures.
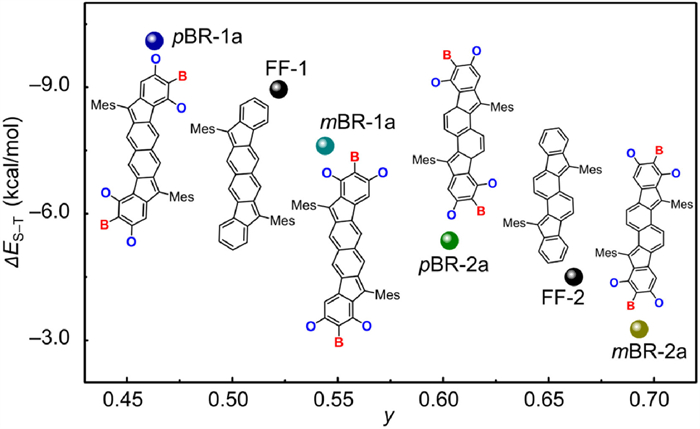
As shown in Fig. 1, pBR-1 and mBR-1 have a fluoreno[3,2-b]fluorene (FF-1) π-skeleton, whereas pBR-2 and mBR-2 contain a fluoreno[2,1-a]fluorene (FF-2) π-framework [52,53]. The fusion mode A leads to pBRs, whose B and O atoms are located at the para- and meso-positions relative to the radical centers, respectively. For mBRs with the mode B, the B atoms are located at the meso-positions and the O atoms are located at the para- and ortho-positions relative to the radical centers. We first performed the density functional theory (DFT) calculations (UB3LYP/6-311G(d) level) on pBR-1a, mBR-1a, pBR-2a and mBR-2a that omit the tert-butyl groups, as well as their all-carbon π-skeletons FF-1 and FF-2 to obtain their diradical characters and singlet-triplet energy gaps. As shown in Fig. 2, in contrast to FF-1 (y = 0.523; ∆ES–T = ‒8.95 kcal/mol), pBR-1a has a lower y of 0.462 and a larger ∆ES–T of ‒10.10 kcal/mol, whereas mBR-1a has a higher y of 0.544 and a smaller ∆ES–T of ‒7.61 kcal/mol. Similar change tendency is observed from FF-2 (y = 0.662; ∆ES–T = ‒4.50 kcal/mol) to pBR-2a (y = 0.603; ∆ES–T = ‒5.36 kcal/mol) and mBR-2a (y = 0.693; ∆ES–T = ‒3.26 kcal/mol). These theoretical results may lead to three conclusions: (1) All of these six compounds are organic diradicaloids with open-shell singlet diradical ground states. (2) In comparison to the all-carbon analogues, mBRs have enhanced open-shell extent but pBRs exhibit reduced open-shell nature. Namely, the fusion modes of B/O-heterocycles onto fluorenofluorenes impact on the diradical electronic states, and the fusion mode B is more desirable for diradical structures. (3) Such large-scale control over diradical structures may further lead to tuned physical properties. Inspired by these analyses, we conducted the synthesis and characterizations of these B/O-type diradicaloid PHs.
Figure 2
Figure 2.
Diradical characters and singlet-triplet energy gaps of all-carbon and B/O-type diradicaloid PHs, obtained from DFT calculations at the UB3LYP/6-311G(d) level.
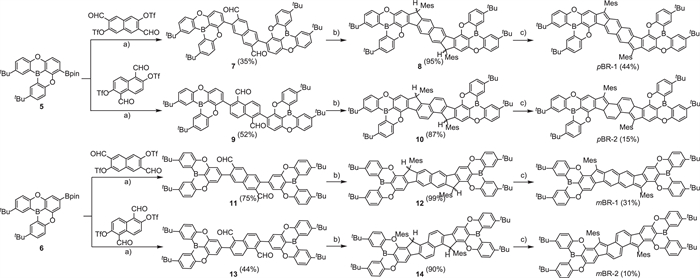
As shown in Scheme 1, these B/O-type diradicaloid PHs were synthesized from the para- and meso-boronate-ester-functionalized dioxa-bridged triphenylboran precursors 5 and 6, as well as 3,7-diformylnaphthalene-2,6-diyl bis(trifluoro-methanesulfonate) and 1,5-diformylnaphthalene-2,6-diyl bis(tri-fluoromethanesulfonate) [54], following the well-established synthesis method. The Suzuki–Miyaura cross-coupling, nucleophilic addition, intramolecular Friedel–Crafts alkylation and oxidative dehydrogenation reactions afforded the four target isomers, which were purified by silica gel column chromatography (Figs. S15‒S34 in Supporting information). The formation of pBR-1, pBR-2, mBR-1 and mBR-2 were verified using high-resolution mass spectrometry (HRMS). Their experimental molecular mass and isotopic distributions are in good agreement with the simulated data, confirming their molecular formula of C82H76B2O4 (Figs. S1 and S2 in Supporting information).
Scheme 1
Scheme 1.
Synthesis of pBR-1, pBR-2, mBR-1 and mBR-2. Reagents and conditions: (a) Pd(PPh3)4, K2CO3, toluene:ethanol:H2O (25:3:3), 80 ℃. (b) (i) Mesitylmagnesium bromide, THF, 25 ℃; (ii) BF3·Et2O, CH2Cl2, 25 ℃. (c) 2,3-Dichloro-5,6-dicyano-1,4-benzoquinone, toluene, 80 ℃.
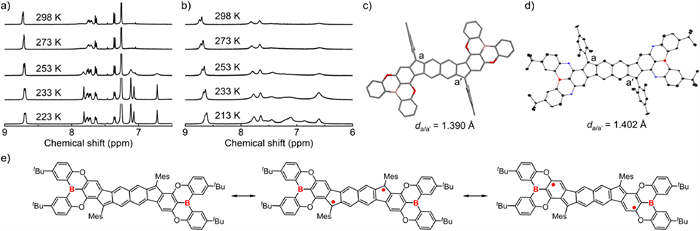
To experimentally investigate their singlet diradical nature, variable-temperature (VT) 1H NMR measurements were performed on pBR-1, pBR-2 and mBR-1, and room-temperature 1H NMR measurement was conducted on mBR-2 (Figs. 3a and b, Fig. S3 in Supporting information). The CDCl3 solutions of pBR-1 and pBR-2, as well as the CD2Cl2 solution of mBR-1 at 298 K exhibit partial and broadened resonance signals for the aryl protons, which gradually became sharp and intensive when decreasing the temperature. For mBR-2, the C6D6 solution at 298 K displays almost no resonance signals of the aryl protons. These behaviors are usually observed for open-shell singlet diradicaloid PHs [6,10–19,55–57], and can be ascribed to the presence of thermally excited triplet species due to the relatively small singlet-triplet energy gap.
Figure 3
Figure 3.
Aromatic region of the variable-temperature 1H NMR spectra of (a) pBR-1 in CDCl3 and (b) mBR-1 in CD2Cl2. (c) Optimized structure of pBR-1a obtained from DFT calculation at the UB3LYP/6-311G(d) level. (d) X-ray crystal structure of mBR-1 (50% probability for thermal ellipsoids). Hydrogen atoms are omitted for clarify. (e) The resonance structures of mBR-1 in close-shell and open-shell forms.
We then conducted structural analyses because the bond lengths can reflect the contributions of the closed/open-shell structures. In the optimized geometries, the lengths of bond a/a′ from the apical methine carbon to the central hexagon are 1.390 Å for pBR-1a, 1.407 Å for mBR-1a, 1.405 Å for pBR-2a and 1.427 Å for mBR-2a (Fig. 3c and Fig. S4 in Supporting information). The single crystals suitable for X-ray diffraction (XRD) analysis for mBR-1 were obtained, and the a/a′ bond lengths (1.402 Å) observed in the single-crystal structure of mBR-1 well reproduce the theoretical data for mBR-1a (Fig. 3d and Fig. S7 in Supporting information). These a/a′ bond lengths are much longer than that (∼1.33 Å) of a typical double C–C bond, indicating the contribution of singlet diradical forms to their ground electronic states (Fig. 3e and Fig. S6 in Supporting information). In addition, the a/a′ bond lengths for mBR-1a are longer than that of pBR-1a, which are also observed for mBR-2a and pBR-2a. The opposite transition is observed for the length of bonds adjacent to a/a' bonds on the pentagonal ring. The analyses thus suggest that the fusion mode B may enhance the diradical nature of fluorenofluorenes.
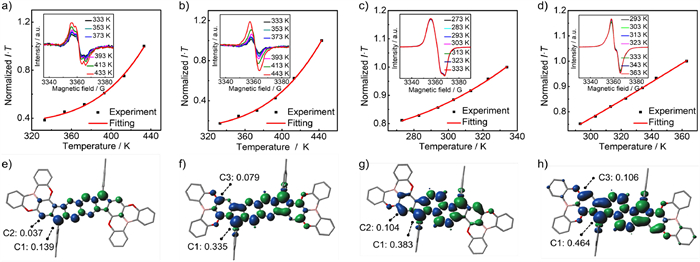
Variable-temperature electron paramagnetic resonance (VT EPR) measurements were performed on their powders to further reveal their diradical nature and magnetic properties. All of them display unresolved EPR signals with g-factor of 2.0029 for pBR-1, 2.0032 for mBR-1, 2.0029 for pBR-2 and 2.0036 for mBR-2. Upon increasing the temperature, the intensity of the EPR signals for pBR-1 and mBR-1 obviously enhances, whereas the EPR intensity for pBR-2 and mBR-2 slightly increases, probably owing to the larger open-shell extent of pBR-2 and mBR-2 [19]. These EPR changes indicate that thermally accessible triplet states contribute more significantly at higher temperature, which unambiguously prove the singlet diradical ground states of these four isomers (Figs. 4a‒d). By fitting the curves of I·T versus T based on the Bleaney-Bowers equation, the experimental ∆ES–T is estimated to be ‒7.60 kcal/mol for pBR-1, ‒6.78 kcal/mol for mBR-1, ‒3.67 kcal/mol for pBR-2 and ‒1.97 kcal/mol for mBR-2, well reproducing the variation trend of the theoretical values and further proving that the fusion mode B in mBRs may decrease the ∆ES–T and enhance the diradical nature of fluorenofluorenes.
Figure 4
Figure 4.
Experimental I·T–T plots (black dots) with Bleaney-Bowers fitting curves (red line) of (a) pBR-1, (b) mBR-1, (c) pBR-2, and (d) mBR-2. Insets are the variable-temperature EPR spectra for their solids. Spin-density distributions (isovalue: 0.001) of (e) pBR-1a, (f) mBR-1a, (g) pBR-2a, and (h) mBR-2a, along with the spin-density values for the specific carbon atoms.
The spin-density distributions provide more details of their open-shell structures. The spin densities of pBR-1a and pBR-2a are mainly localized on the dicyclopentanaphthalene cores of the fluorenofluorene skeletons, along with a small population of the peripheral hexagon rings (Figs. 4e‒h). For mBR-1a and mBR-2a, the spin densities are delocalized over the entire fluorenofluorene skeletons and the O atoms, thus exhibiting more delocalization to a certain extent compared with that of the all-carbon analogues FF-1 and FF-2 (Fig. S5 in Supporting information). Moreover, the spin density population of the C1 atom that represents the radical center is in the sequence of pBR-1a ˂ FF-1 ˂ mBR-1a and pBR-2a ˂ FF-2 ˂ mBR-2a. These results clearly reveal that the mode B for mBRs is positive for enhancing spin delocalization and open-shell nature, which can be attributed to the O atoms (Fig. 1b). For pBRs, the fusion mode A inhibits the spin density delocalization and further decrease the open-shell nature, which is probably due to the electron-deficient B atoms, but the explicit reason is unclear at this stage. The resonance structures in closed-shell and open-shell forms for these four isomers are provided in Fig. S6.
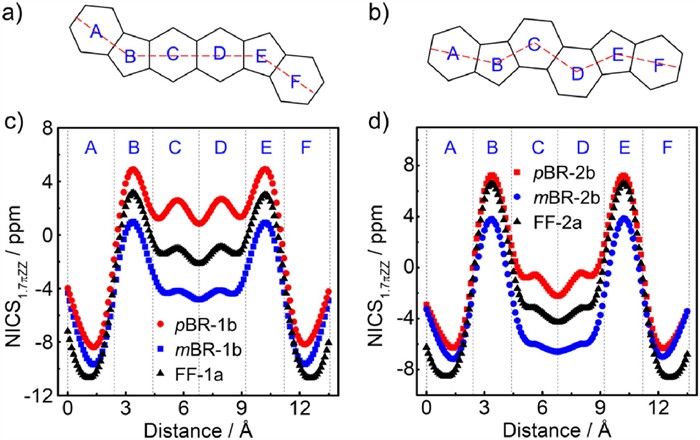
To illustrate the diradical structures in terms of the aromaticity, nucleus-independent chemical shift (NICS)-XY scan calculations with σ-only method at the GIAO-UB3LYP/6-311+G(d) level were performed on these four isomers and their all-carbon analogues that omit the tert-butyl and mesityl groups for simplified calculations (Fig. 5). In comparison to FF-1a, pBR-1b exhibits the reduced aromatic characters of rings C and D, as supported by the observed positive NICS values, indicating that the quinoidal closed-shell resonance form contributes more significantly on the ground state of pBR-1b. In contrast, from FF-1a to mBR-1b, the more negative NICS values and enhanced aromaticity of rings C and D are observed, which provide additional driving force for recovery of the Clar sextet, leading to the enhanced open-shell structure of mBR-1b. Similar variations in aromaticity and diradical character from FF-1a to pBR-1b and mBR-2b are also observed. The analyses on aromatic structures are fully consistent with the discussions on their open-shell structures.
Figure 5
Figure 5.
(a, b) Dashed lines are the path of the ghost atoms used in NICS calculations. (c, d) NICS-XY scans of pBR-1b, mBR-1b, pBR-2b and mBR-2b, as well as FF-1a and FF-2a.
The cyclic voltammetry measurements were performed on their CH2Cl2 solutions with nBu4-NPF6 as supporting electrolyte. As shown in Fig. 6a, pBR-1, mBR-1 and mBR-2 exhibit three reversible redox processes, whereas pBR-2 has four reversible redox waves. The half-wave oxidation and reduction potentials (E1/2ox and E1/2red) are +0.44/‒1.19/‒1.44 V for pBR-1, +0.04/+0.28/‒1.34 V for mBR-1, +0.32/+0.80/‒1.02/‒1.20 V for pBR-2, and +0.04/+0.28/‒1.11 V for mBR-2. According to the first reduction and oxidation potentials, the HOMO and LUMO levels (EHOMO/ELUMO) and HOMO‒LUMO gap (Eg) are estimated to be ‒5.24/‒3.61 eV and 1.63 eV for pBR-1, ‒4.84/‒3.46 eV and 1.38 eV for mBR-1, ‒5.12/‒3.78 eV and 1.34 eV for pBR-2, as well as ‒4.84/‒3.69 eV and 1.15 eV for mBR-2 (Table 1). From pBR-1 to mBR-1, the HOMO and LUMO levels are enhanced by 0.40 and 0.15 eV, respectively, leading to the reduced HOMO‒LUMO gap. Similar changes are observed from pBR-2 to mBR-2. Therefore, the fusion mode B is more positive for increasing energy levels and reducing energy gaps of PHs.
Figure 6
Figure 6.
(a) Cyclic voltammograms of pBR-1, mBR-1, pBR-2 and mBR-2 in CH2Cl2 (Fc = ferrocene). (b) Absorption spectra of pBR-1, mBR-1, pBR-2 and mBR-2 in toluene. The insets show the photographs for their solutions.
Fig. 6b and Fig. S8 show the UV–vis-NIR absorption spectra of these four isomers in toluene. The main absorption peaks (λabs) are observed at 652/607 nm for pBR-1, 736/673 nm for mBR-1, 781/729 nm for pBR-2, and 827/919 nm for mBR-2. The absorption coefficients are gradually lowered from pBR-1/mBR-1 (87,745 and 95,545 M‒1 cm‒1) to pBR-2 (47,134 M‒1 cm‒1) and mBR-2 (29,028 M‒1 cm‒1) (Table 1). Thus, the well-tuned absorption properties are achieved in these organic diradicaloids that feature isomeric structures. For mBR-2, the obvious shoulder peak at 919 nm can be attributed to the diradical contributions to the ground electronic states, as reported for other diradical PHs with significant open-shell nature. It has been reported that the main absorption peaks of FF-1 and FF-2 bearing the anthracene groups appeared at 600 nm and 712 nm, respectively [52,53]. Thus, the red-shifted absorbance is observed for these B/O-type organic diradicaloids, namely, both fusion modes A and B can lead to the red-shifted absorption properties, and moreover, the mode B is more desirable for this electronic effect.
Time-dependent absorption measurements were further performed to evaluate their stabilities. The toluene solutions of pBR-1 and mBR-1 left at ambient conditions for ten days exhibit almost no spectral changes, indicating their excellent stabilities (Fig. S9 in Supporting information). For pBR-2 and mBR-2, the gradual decompositions are observed within ten days and four hours, respectively. The half-life estimated by fitting the time-dependent absorbance is 4.98 days for pBR-2 and 101 min for mBR-2. The decreased stabilities are in consistence with their enlarged open-shell nature. It is notable that FF-2 has never been successfully synthesized, and the bulky anthracene group or extended π-conjugation is needed to enhance its stability [53,58]. It is suggested that the B/O-heterocycle fusion can enhance the stability of diradical PHs, even along with simultaneously increasing open-shell nature for the mode B.
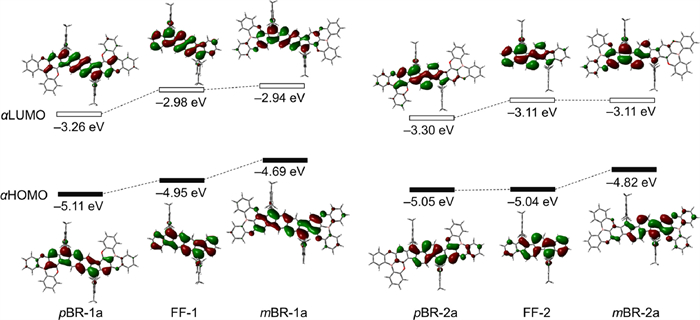
Finally, we would like to gain insight into the electronic effects of the B/O-heterocycle fusion on PHs based on DFT calculations. Fig. 7 displays the αHOMOs and αLUMOs and their energy levels of pBR-1a, mBR-1a, pBR-2a and mBR-2a, as well as FF-1 and FF-2. pBR-1a has the lower αHOMO and αLUMO levels than FF-1 by 0.16 and 0.28 eV, respectively, leading to its slightly decreased band gap of 1.85 eV. This is owing to the p orbitals of the B atoms that contribute to the αHOMO and αLUMO of pBR-1a to a significant extent. From FF-1 to mBR-1a, while the αLUMO level is slightly elevated, the αHOMO level is obviously increased, and thus mBR-1a has a much smaller band gap of 1.75 eV, agreeing with the variation tendency of the electrochemical gaps. These differences may be attributed to the electron-donating effects of the O atoms. Therefore, the fusion modes A and B have different effects on energies, to which the B and O atoms contribute significantly, respectively. This conclusion in the electronic effects of these heteroatoms and fusion modes are further proved by comparing FF-2, pBR-2a and mBR-2a. On the other hand, according to time-dependent DFT (TD-DFT) calculations (UB3LYP/6-311G(d) level), the theoretical absorption patterns, including wavelengths, energies and oscillator strengths, well reproduce the experimental spectra (Figs. S10‒S13 in Supporting information). It is notable that their low-energy absorption bands all involve the αLUMOs and αHOMOs, demonstrating the contributions of the B and O atoms to their NIR absorption properties.
Figure 7
Figure 7.
Pictorial representations of the Kohm-Sham molecular orbitals and their energy levels for pBR-1a, mBR-1a, pBR-2b and mBR-2b, as well as FF-1 and FF-2.
In conclusion, we designed and synthesized four B/O-containing diradicaloid isomers that feature the fluoreno[3,2-b]fluorene and fluoreno[2,1-a]fluorene π-skeletons, respectively. The precise B/O-heterocycle fusion modes along with the changed conjugation patterns effectively control their electronic structures and thereby successively modulate physical properties, such as diradical and aromatic structures, energy levels and band gaps, as well as magnetic, electrochemical and photophysical properties. Notably, the mode A may decrease the open-shell extent, whereas the mode B can enhance the diradical nature, leading to their well-tuned diradical characters in the range of 0.46‒0.70. Moreover, the mode A stabilizes the LUMOs and the mode B obviously increases the HOMO levels, which are remarkably contributed by the B and O atoms, respectively, further giving rise to the decreased energy gaps and gradually redshifted absorptions. This study clearly illustrates the electronic effects of B/O-heterocycle fusion on PHs and gains insight into B/O-type organic diradicaloids. These findings will provide an important guideline for the design of more fascinating heteroatoms-containing diradicaloids.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
This work was supported by National Natural Science Foundation of China (Nos. 52373182 and 22175074), Jilin Scientific and Technological Development Program (No. 20220101054JC), and Department of Education of Jilin Province (No. JJKH20221046KJ).
Supplementary materials
Supplementary material associated with this article can be found, in the online version, at doi:10.1016/j.cclet.2024.110174.
[1]
Z. Feng, S. Tang, Y. Su, et al., Chem. Soc. Rev. 51 (2022) 5930–5973. doi: 10.1039/d2cs00288d
J. Melidonie, E. Dmitrieva, K. Zhang, et al., J. Org. Chem. 85 (2019) 215–223.
Figure 1
Schematic illustration of control over the electronic structures of diradicaloid PHs via incorporation of (a) the sulfur or oxygen atom and (b) the boron and oxygen atoms, along with chemical structures of 1‒4, as well as pBR-1, pBR-2, mBR-1 and mBR-2.
Figure 2
Diradical characters and singlet-triplet energy gaps of all-carbon and B/O-type diradicaloid PHs, obtained from DFT calculations at the UB3LYP/6-311G(d) level.
Figure 3
Aromatic region of the variable-temperature 1H NMR spectra of (a) pBR-1 in CDCl3 and (b) mBR-1 in CD2Cl2. (c) Optimized structure of pBR-1a obtained from DFT calculation at the UB3LYP/6-311G(d) level. (d) X-ray crystal structure of mBR-1 (50% probability for thermal ellipsoids). Hydrogen atoms are omitted for clarify. (e) The resonance structures of mBR-1 in close-shell and open-shell forms.
Figure 4
Experimental I·T–T plots (black dots) with Bleaney-Bowers fitting curves (red line) of (a) pBR-1, (b) mBR-1, (c) pBR-2, and (d) mBR-2. Insets are the variable-temperature EPR spectra for their solids. Spin-density distributions (isovalue: 0.001) of (e) pBR-1a, (f) mBR-1a, (g) pBR-2a, and (h) mBR-2a, along with the spin-density values for the specific carbon atoms.
Figure 5
(a, b) Dashed lines are the path of the ghost atoms used in NICS calculations. (c, d) NICS-XY scans of pBR-1b, mBR-1b, pBR-2b and mBR-2b, as well as FF-1a and FF-2a.
Figure 6
(a) Cyclic voltammograms of pBR-1, mBR-1, pBR-2 and mBR-2 in CH2Cl2 (Fc = ferrocene). (b) Absorption spectra of pBR-1, mBR-1, pBR-2 and mBR-2 in toluene. The insets show the photographs for their solutions.
Figure 7
Pictorial representations of the Kohm-Sham molecular orbitals and their energy levels for pBR-1a, mBR-1a, pBR-2b and mBR-2b, as well as FF-1 and FF-2.

 DownLoad:
DownLoad:



 下载:
下载:









 下载:
下载:
