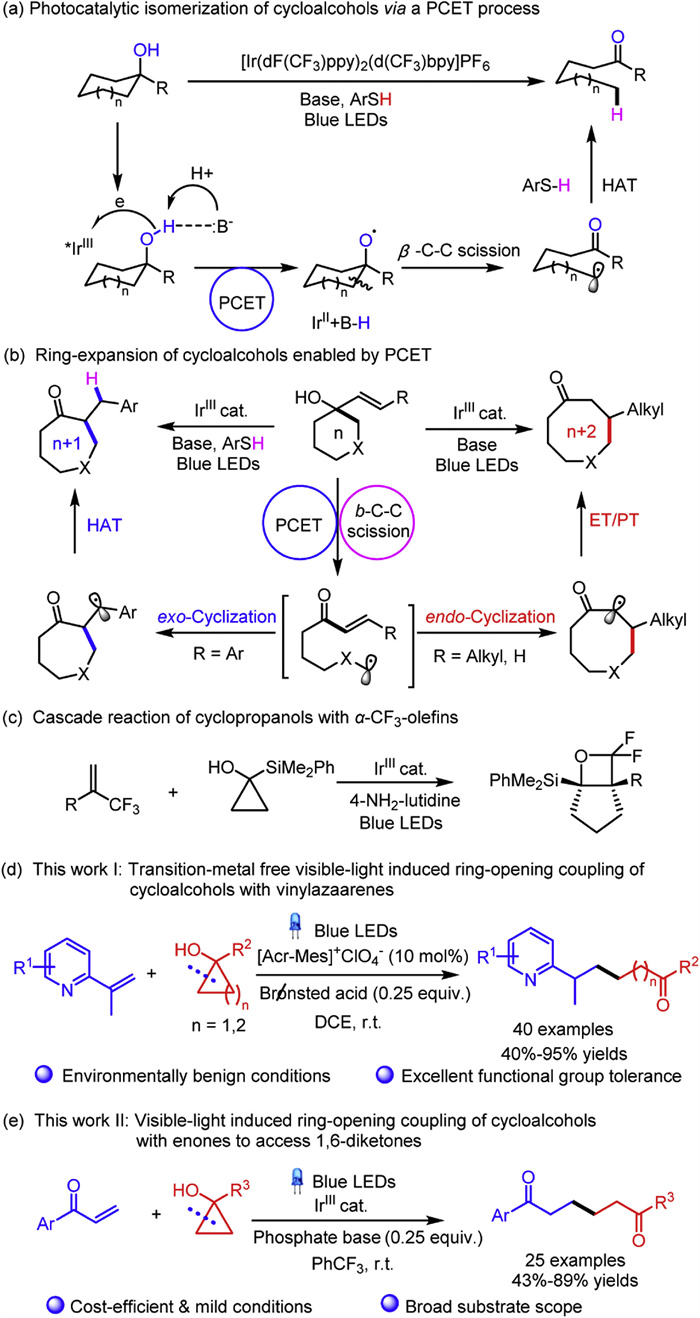
Scheme 1.
Strategies for the C—C bond cleavage of cycloalcohols enabled by PCET.
Visible-light-induced ring-opening cross-coupling of cycloalcohols with vinylazaarenes and enones via β-C-C scission enabled by proton-coupled electron transfer
Qinghong Zhang , Qiao Zhao , Xiaodi Wu , Li Wang , Kairui Shen , Yuchen Hua , Cheng Gao , Yu Zhang , Mei Peng , Kai Zhao

The selective C—C bond cleavage has been considered as a unique and robust synthetic tool to streamline the construction of complex molecules via the rapid and intriguing skeleton reconstruction and functionalization [1-9]. Nevertheless, the inherent abundance and inertness of C—C bonds in organic molecules render the above task a formidable challenge. In this context, the selective β-C-C scission and functionalization of cycloalcohols via the formation of homoenolates [10-19] or keto radical [20-28] has emerged as a burgeoning and prosperous research area over the past few decades. In particular, the later cases involving the β-C-C fragmentation of O-centered alkoxy radical to give the distal alkyl radicals have gathered increasing attention among the chemical community [29-33]. Nevertheless, the formation of the alkoxy radical through the homolysis of the alcoholic O—H bond was challengeable owing to the relative high bond dissociation energy. It was not until recently that a number of significant breakthroughs were achieved by the groups of Lectka [34], Chen [23,35,36], Zhu [37-40], Knowles [41-43], Melchiorre [44], Rueping [45,46], Zuo [47-51], Lei [52] and many others [25,53-55] through photocatalytic manner. Noteworthily, the visible-light promoted proton-coupled electron transfer (PCET) by virtue of transferring an electron and proton in a synergistic step, has provided a competent protocol for the homolytic activation of O—H bonds of cycloalcohols under mild and redox-neutral conditions [56-59]. For instance, Knowles and coworkers pioneeringly demonstrated the photocatalytic isomerization reactions (Scheme 1a) [41,42] and an intramolecular ring-expansion [43] of cycloalcohols (Scheme 1b), respectively. As compared to the intramolecular ring-opening reactions of cycloalcohols, the intermolecular scenario has been considered as a more intriguing yet challengeable task. Very recently, Shen and coworkers elegantly reported the visible-light-induced cascade reaction of cyclopropanols with α-trifluoromethylated olefins to access fused gem-difluorooxetanes (Scheme 1c) [60]. Despite those aforementioned innovative and prominent advancements, the development of intermolecular ring-opening cross-coupling of cycloalcohols with distinctive olefins such as vinylazaarenes and enones bearing a multitude of crucial functionalities, trigged by PCET mechanism under environmentally benign and cost-effective conditions, remained an unmet and fascinating challenge.
Pyridine-derived scaffolds are widespread subunits in a multitude of natural products, pharmaceuticals, agrochemicals, ligands and functional materials [61-65]. Therefore, the structural modification of pyridine derivatives has been at the forefront of intense research among synthetic community [66-70]. Of note, pyridyl-based carbonyl motifs are of particular synthetic interest owing to their prevalence in myriad bioactive lead molecules. Consequently, the development of straightforward and generally applicable platforms to access such frameworks represents a practical and conceptually crucial pursuit with paramount synthetic value. Recently, Hong and coworkers have developed a regioselective cross-coupling of N-amidopyridinium salts with cyclopropanols to access β-carbonyl alkyl-substituted pyridines [66]. The group of Jiang has demonstrated a reductive coupling-enantioselective protonation of α-branched vinylketones with vinylpyridines, delivering pyridyl substituted ketones bearing remote stereogenic centers [67]. Enlightened by those seminal advancements, we herein disclosed a visible-light induced ring-opening cross-coupling of cycloalcohols with vinylpyridines to access a diverse range of δ- or ε-pyridyl substituted ketones with excellent functional group compatibility under environmentally benign metal-free conditions (Scheme 1d).
1, 6-Diketones served as pivotal building blocks in the construction of various bioactive carbocyclic and heterocyclic compounds [71-75]. Moreover, the versatile utilities of functionalized 1, 6-diketones in a multitude of organic transformations have leveraged their practical synthesis as a highly desired topic of interest. Particularly, unsymmetrical 1, 6-diketones are recognized as more appealing yet challenging synthetic targets in contrast to their symmetrical analogues [76-79]. The representative state-of-the-art synthetic methods in this arena involved the C—C bond forming reactions of electrophilic enones with metal-homoenolates [80,81] or β-keto radicals [82-86] derived from siloxycyclopropanes or cyclopropanols. Nevertheless, those methodologies posed a handful of challenges arising from the employment of toxic heavy metals [83,84] or radical initiators [85] as well as the inevitable homocoupling of enones. As such, the quest for catalytic and highly selective protocols for the synthesis of structural diverse unsymmetrical 1, 6-diketones under environmentally benign reaction conditions is of inarguably importance. Recently, we have developed a manganese-catalyzed radical mediated methodology to attain various unsymmetrical 1, 6-diketones, albeit relative harsh conditions were required [82]. Encouraged by this achievement, we herein demonstrated a photocatalytic ring-opening cross-coupling of cycloalcohols with enones, providing a significant complement to the synthesis functionalized 1, 6-diketones with broad substrate scope under mild conditions (Scheme 1e).
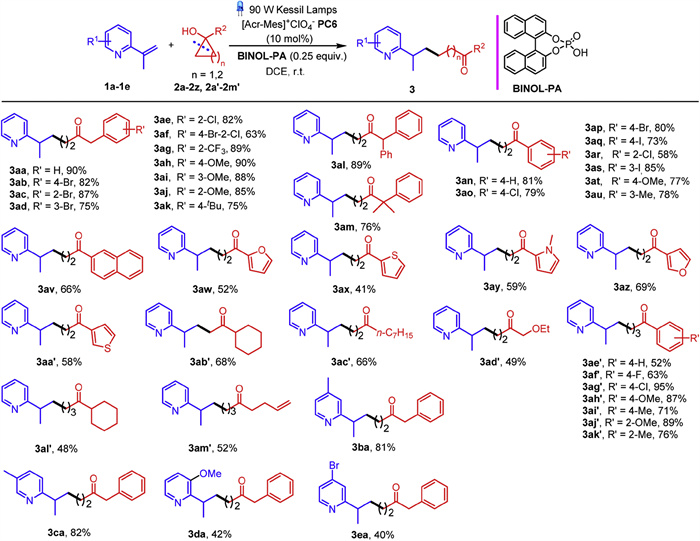
We commenced our investigation by screening the optimal reaction conditions using α-methylated 2-vinylpyridine 1a and 1-benzylcyclopropan-1-ol 2a as the model substrate (Table S1 in Supporting information for details). To our delight, the transformation proceeded smoothly in the presence of [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) (PC1) as a photocatalyst and BINOL phosphoric acid (BINOL-PA) as an additive using (trifluoromethyl)benzene as the solvent under the irradiation of 90 W Kessil Lamps, furnishing desired product 3aa in 58% yield (Table S1, entry 1). Other photocatalysts including [Ir(dtbbpy)(ppy)2](PF6) (PC2), fac-Ir(ppy)3 (PC3) and Eosin Y (PC4) gave inferior results attributed to the lower oxidative potentials in comparison with PC1 (Table S1, entries 2–4). Gratifyingly, transition-metal free photosensitizers such as 4CzIPN (PC5) and acridinium salt [Acr-Mes]+ClO4− (PC6) with higher oxidizing abilities proved to be efficacious photocatalysts, furnishing 3aa in 63% and 68% yields, respectively (Table S1, entries 5 and 6). A screening of solvents showed that 1, 2-dichloroethane (DCE) was the optimal choice, giving 3aa in 75% yield (Table S1, entry 14 vs. entries 7–13). Further optimization by increasing the loading of PC6 to 10 mol% resulted a significant improvement to the reaction efficiency, forging 3aa in 87% yield (Table S1, entry 15). Adjusting the reaction concentration to 0.033 M gave the optimal yield of 90% (Table S1, entry 16). Notably, a gradient decrease of the product yields was observed upon lowering the power of the light source (Table S1, entries 17 and 18). Control experiments showed that a dramatic attenuation of the reaction efficiency was observed without the aid of phosphoric acid (Table S1, entry 19), and the presence of the photocatalyst and irradiation proved to be indispensable for the transformation (Table S1, entries 20 and 21).
Taking advantage of the optimal reaction conditions, we investigated the substrate scope of the cross-coupling reaction of vinylazaarenes with cycloalcohols, as summarized in Scheme 2. A range of benzyl-substituted cyclopropanols were examined, it was observed that a plethora of electron-withdrawing halides on the benzene ring were well tolerated, affording 3ab-3af in 63%−87% yields. Biologically crucial -CF3 moiety was amenable, giving 3ag in 89% yield. Substrates bearing electron-donating groups such as -OMe and -tBu were viable in the transformation, delivering 3ah-3ak in 75%−90% yields. Steric effect on the phenyl ring of benzyl group did not affect the above transformation, para- (2b, 2h, 2k), orthro- (2c, 2e, 2g, 2j) and meta- (2d, 2i) substituted cyclopropanols as well as disubstituted substrate 2f all reacted with 1a smoothly. Substituents such as phenyl and dimethyl on the benzylic position did not attenuate the reaction efficiency, forging 3al and 3am in 89% and 76% yields, respectively. Next, it was observed that a range of aryl-substituted cycloalcohols coupled with 1a smoothly, regardless of their electronic effects or steric patterns, delivering 3an-3av in 58%−85% yields. Heterocyclic substituents including 2-furyl and 2-thienyl were compatible with the standard conditions, forging 3aw and 3ax in moderate yields. N-Methylpyrrole substituted cyclopropanol also partook in the transformation uneventfully, delivering 3ay in 59% yield. 3-Furyl and 3-thienyl derived substrates also reacted with 1a efficiently, forging desired products 3az and 3aa' in 69% and 58% yields, respectively. Gratifyingly, cyclopropanols bearing aliphatic substituents such as cyclohexyl, fatty n-heptyl and ethoxymethyl were all viable in the ring-opening coupling reaction with 1a, furnishing 3ab', 3ac' and 3ad' in 68%, 66% and 49% yields, respectively. To our delight, aside from cyclopropanols, a diverse array of aryl-substituted cyclobutanols bearing substituents with various electronic peculiarities and steric effects proved to be eligible coupling partners of 1a, delivering 3ae'−3ak' in 52%−95% yields. Cyclobutanols bearing cyclohexyl and unsaturated alkene moieties partook in the reaction smoothly, forging 3al' and 3am' in 48% and 52% yields. Next, we examined the substrate scope of 2-vinylpyridines. It was observed that 4-methyl and 3-methyl pyridyl substituted substrates 2b and 2c coupled with cyclopropanol 2a uneventfully, giving rise to 3ba and 3ca in 81% and 82% yields. Electron-donating 3-OMe and electron-withdrawing 4-Br substituents on the pyridine ring decrease the reaction efficiencies, furnishing desired products 3da and 3ea in moderate yields.
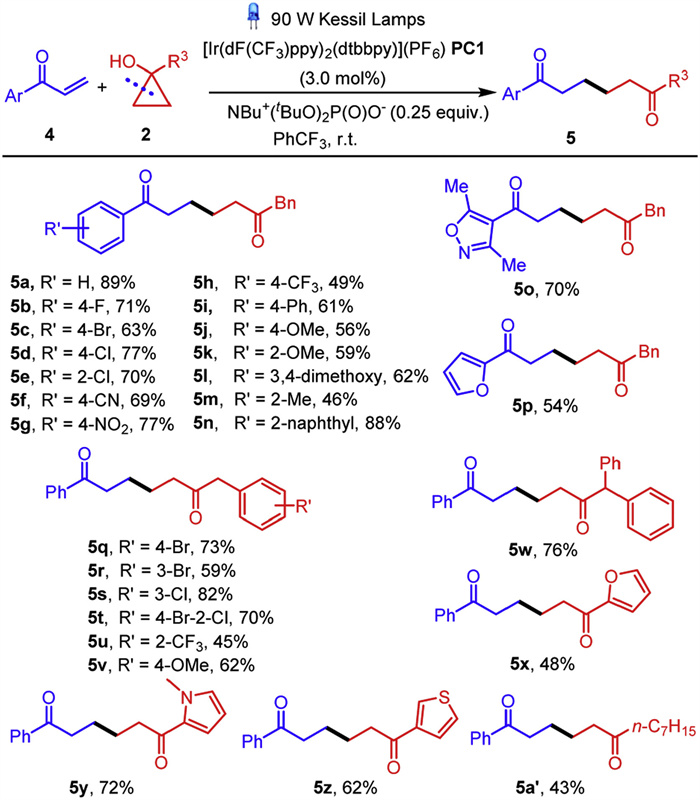
Encouraged by the above conspicuous results, we envisaged that the synthetic strategy could be applied to the ring-opening coupling of cycloalcohols with various enones to access a plethora of 1, 6-diketones. Thus, we commenced to screen the optimal conditions to facilitate the transformation (Table S2 in Supporting information for details). Unfortunately, the reaction of 1-phenylprop-2-en-1-one 4a with cyclopropanol 2a under the standard conditions in Scheme 2 proved to be futile, giving the desired product 5a in only 5% yield (Table S2, entry 1). Gratifyingly, replacing the phosphoric acid BINOL-PA to a Brønsted base tetrabutylammonium di-tert-butyl phosphate NBu4+(tBuO)POO− provided 5a in 43% yield (Table S2, entry 2). While other photocatalysts did not result in significant increase of the reaction efficiency (Table S2, entries 3–6), it was delightful that 5a could be furnished efficiently in 74% yield upon the employment of [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) (PC1) (Table S2, entry 7). The screening of solvents proved that (trifluoromethyl)benzene was the optimal choice, forging 5a in 89% yield (Table S2, entry 12). Decreasing the amount of 2a led to lower product yield (Table S2, entry 12 vs. entries 13 and 14). The reaction efficiency was attenuated when decreasing the power of the light sources (Table S2, entry 12 vs. entries 15 and 16). Control experiments showed that no 5a was formed in the absence of either the photocatalyst, the Brønsted base or the light irradiation (Table S2, entries 17–19).
With the optimal reaction conditions in hand, we explored the substrate scope of enones and cyclopropanols, as demonstrated in Scheme 3. A series of enones bearing electron-withdrawing halides, -CN, -NO2, -CF3 substituents on the phenyl ring were well tolerated, generating 5b-5h in 49%−77% yields. Enones bearing electron-donating groups were also amenable to couple with 2a, generating 5i-5m in satisfying yields. Sterically more hindered 2-naphthyl substituent enone proved to be eligible in the transformation, giving 5n in 88% yield. Delightfully, heterocycle substituted enones bearing 3, 5-dimethylisoxazole and furan skeletons coupled with 2a uneventfully, furnishing 5o and 5p in 70% and 54% yields, respectively. The scope of cyclopropanols were proved to be broad as well. Benzyl-substituted cyclopropanols bearing electron-withdrawing groups coupled with 4a smoothly, forging 5q-5u in moderate to good yields. The presence of electron-donating –OMe group on the phenyl ring or a –pH at the benzylic position were also viable, furnishing 5v and 5w in 62% and 76% yields. Heterocycle-substituted cycloalcohols bearing 2-furyl, N-methyl-2-pyrrolyl and 3-thienyl all showed excellent compatibilities with the standard conditions, furnishing 5x-5z in moderate to good yields. Notably, aliphatic n-heptyl substituted cyclopropanol proved to be eligible coupling partner of 4a, giving rise to 5a' in 43% yield.
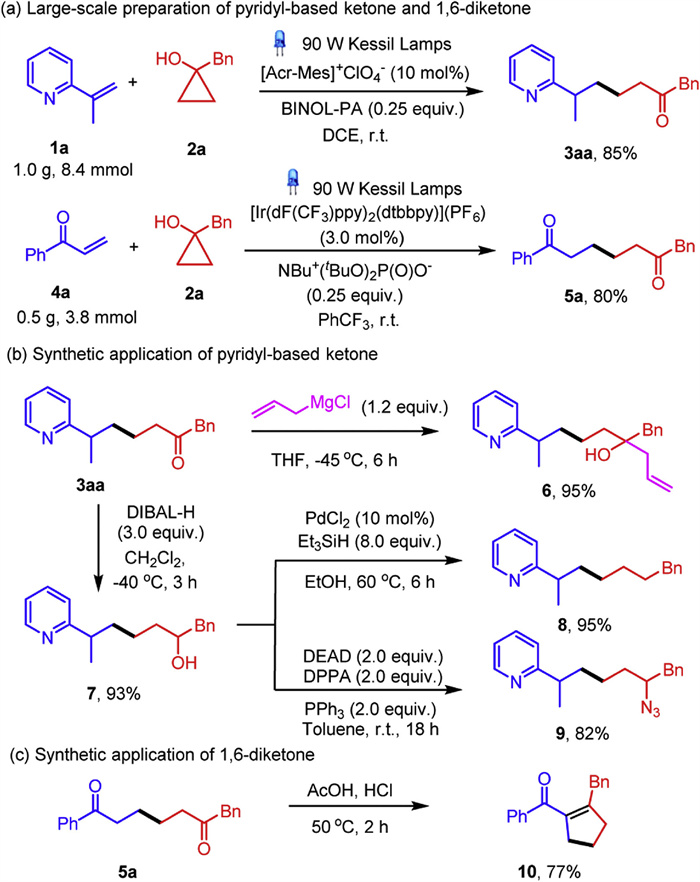
Gratifyingly, the large-scale preparations of pyridyl-based ketone 3aa and 1, 6-diketone 5a proved to be successful without losing the reaction efficiencies (Scheme 4a). Subsequently, a handful of further transformation of 3aa and 5a were carried out to illustrate the synthetic utilities of the aforementioned ring-opening coupling reactions. 3aa could be converted to synthetically useful allylic alcohol 6 in 95% yield. The carbonyl moiety could also be reduced by DIBAL-H to give pyridyl-based tertiary alcohol 7, the hydroxyl group could be reduced in the presence of PdCl2 and Et3SiH to give pyridine derivative 8 in 95% yield. More importantly, the biologically and synthetically prominent pyridyl-containing azide 9 could be prepared efficiently through the Mitsunobu-type SN2 reaction of 7 (Scheme 4b). The synthetic practicality of the 1, 6-diketone 5a could be highlighted through intramolecular aldol condensation, furnishing cyclic enone 10 in 77% yield (Scheme 4c).
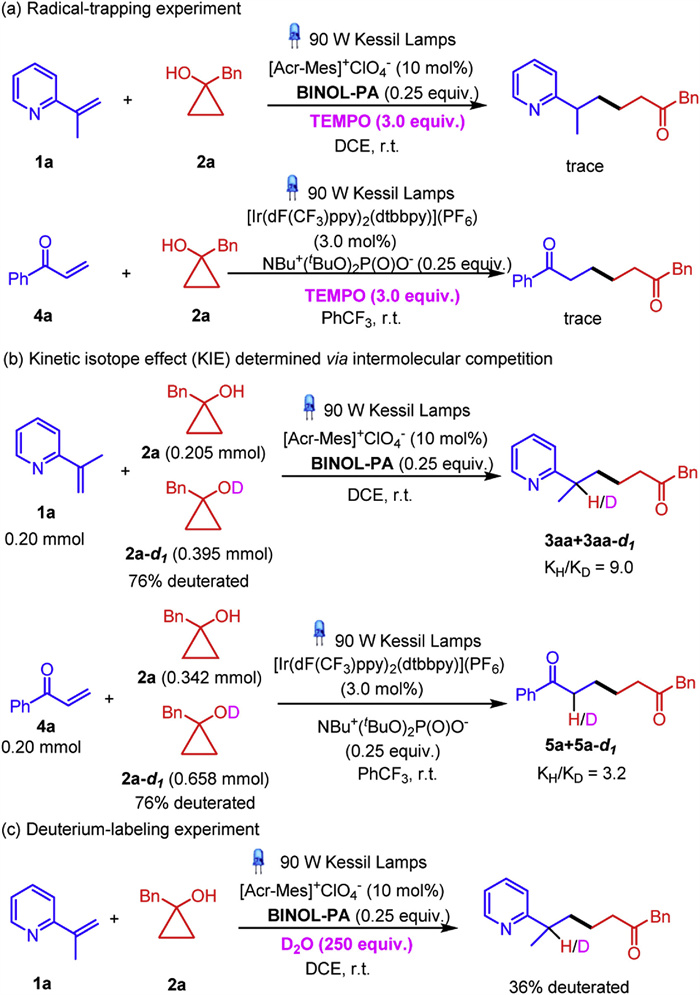
Preliminary mechanistic studies were conducted to elucidate the plausible pathway for the aforementioned ring-opening coupling reactions. It was observed that the reactions of either vinylpyridine 1a or enone 4a with cyclopropanol 2a was hampered in the presence of a radical scavenger 2, 2, 6, 6-tetramethylpiperidine 1-oxyl (TEMPO), along with the interception of the β-keto radical species (see Supporting information for details), implying a plausible radical pathway (Scheme 5a). The kinetic isotope effect (KIE) experiments for the ring-opening coupling reactions of cyclopropanol 2a and deuterium-labeled 2a-d with 1a or 4a were conducted, the KIE values were calculated to be 9.0 and 3.2, indicating that the cleavage of the O—H bond in cycloalcohols was rate-determining in both transformations (Scheme 5b) (see Supporting information for details). It was observed that in the presence of external deuterium source D2O, 36% deuterated 3aa-d was isolated via the reaction of 1a with 2a, suggesting that the C—H bond formation might be attributed to the formation of a radical at the α-position of azaarenes and the ensuing SET reduction and protonation (Scheme 5c). The quantum yields for the ring-opening coupling reaction of cyclopropanol 2a with 1a and 4a were calculated to be 0.36 and 0.25, respectively, implying that both transformations proceeded through a photoredox-catalyzed pathway rather than radical-chain mechanism (see Supporting information for details). The Stern-Volmer experiment for the reaction of 1a with 2a showed that a linear quenching was only observed in the presence of both PC6 and BINOL-PA. Similar result was obtained in the reaction of enone 4a with 2a, wherein the presence of PC1 and NBu4+(tBuO)POO− were both indispensable for the linear quenching. The above observations evidentially supported the PCET process in the ring-opening coupling transformations (see Supporting information for details).
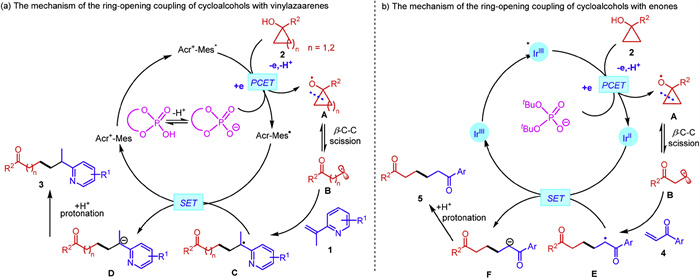
Based on the aforementioned experimental results, we postulated plausible reaction pathways for the ring-opening coupling reactions (Scheme 6). For the ring-opening coupling reaction of vinylpyridines with cycloalcohols (Scheme 6a), the photocatalyst [Acr-Mes]+ClO4− absorbs a photon under the irradiation of blue LEDs to generate a highly oxidative singlet excited state *[Acr-Mes]+ClO4−, which reacts with the phosphate and the cycloalcohols 2 through a concerted PCET process to furnish an alkoxyl radical species A. Then, the β-C-C scission occurs to forge the distal β- or γ-keto alkyl radical B, which would be intercepted by 2-vinylpyridine through a Giese-type radical addition to furnish a tertiary radical C. Subsequently, a single electron reduction of C generates an anion D. Finally, the protonation of D gives rise to the desired product 3. The reaction of cyclopropanols with enones was postulated to proceed via a similar mechanism as depicted in Scheme 6b, involving the formation of alkoxyl radical species A via a PCET process in the presence of excited *Ir(Ⅲ)-complex and a Brønsted base NBu4+(tBuO)POO−, followed by the β-C-C scission to access radical B and its ensuing interception by enones to generate radical E, the formation of anion F via SET, and its protonation to forge 1, 6-diketones.
In summary, we have demonstrated unprecedented visible-light-induced intermolecular ring-opening cross-coupling reactions of cycloalcohols with vinylazaarenes or enones. The photoinduced proton-coupled electron transfer and the following β-C-C cleavage played key roles for the formation of the key distal keto alkyl radicals. The operational ease, excellent functional group tolerance and environmentally and mild reaction conditions of those transformations have paved a new avenue for the facile and efficient synthesis of pharmaceutically and synthetically prominent pyridyl-based ketones or 1, 6-diketones and their related derivatives.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Qinghong Zhang: Methodology, Investigation, Formal analysis. Qiao Zhao: Methodology, Investigation, Formal analysis, Data curation. Xiaodi Wu: Investigation, Formal analysis, Data curation. Li Wang: Investigation, Data curation. Kairui Shen: Formal analysis, Data curation. Yuchen Hua: Formal analysis, Data curation. Cheng Gao: Formal analysis, Data curation. Yu Zhang: Methodology, Formal analysis. Mei Peng: Formal analysis, Data curation. Kai Zhao: Writing – review & editing, Writing – original draft, Supervision, Project administration, Investigation, Conceptualization.
We gratefully acknowledge the financial support from National Natural Science Foundation of China (Nos. 21801129, 22078153 and 22378201), National Key Research and Development Program of China (No. 2022YFB3805603). Natural science research projects in Jiangsu Higher Education Institutions (No. 18KJB150018), Open Research Fund of School of Chemistry and Chemical Engineering, Henan Normal University (No. 2024Y16) and Nanjing Tech University (Start-up Grant Nos. 39837137, 39837101 and 3827401739) for financial support.
Supplementary material associated with this article can be found, in the online version, at doi:
X.Y. Yu, J.R. Chen, W.J. Xiao, Chem. Rev. 121 (2021) 506–561. doi: 10.1021/acs.chemrev.0c00030
I. Marek, A. Masarwa, P. Delaye, M. Leibeling, Angew. Chem. Int. Ed. 54 (2015) 414–429. doi: 10.1002/anie.201405067
S.P. Morcillo, Angew. Chem. Int. Ed. 58 (2019) 14044–14054. doi: 10.1002/anie.201905218
Y.F. Liang, M. Bilal, L.Y. Tang, et al., Chem. Rev. 123 (2023) 12313–12370. doi: 10.1021/acs.chemrev.3c00219
C.Y. Huang, J. Li, C.J. Li, Chem. Sci. 13 (2022) 5465–5504. doi: 10.1039/d2sc00202g
O.O. Sokolova, J.F. Bower, Chem. Rev. 121 (2021) 80–109. doi: 10.1021/acs.chemrev.0c00166
R. Vicente, Chem. Rev. 121 (2021) 162–226. doi: 10.1021/acs.chemrev.0c00151
S. Zhu, P. Zhu, M. Guo, et al., Chin. Chem. Lett. 35 (2024) 108835–108838. doi: 10.1016/j.cclet.2023.108835
H. Yan, G.S. Smith, F.E. Chen, Green. Synth. Catal. 3 (2022) 219–226. doi: 10.1016/j.gresc.2022.05.007
Q. Zhang, S. Zhou, C. Shi, L. Yin, Angew. Chem. Int. Ed. 60 (2021) 26351–26356. doi: 10.1002/anie.202110709
S.B. Park, J.K. Cha, Org. Lett. 2 (2000) 147–149. doi: 10.1021/ol991250r
Z. Ye, X. Cai, J. Li, M. Dai, ACS Catal. 8 (2018) 5907–5914. doi: 10.1021/acscatal.8b00711
D.C. Davis, K.L. Walker, C. Hu, R.N. Zare, R.M. Waymouth, M. Dai, J. Am. Chem. Soc. 138 (2016) 10693–10699. doi: 10.1021/jacs.6b06573
X. Zhou, S. Yu, L. Kong, X. Li, ACS Catal. 6 (2016) 647–651. doi: 10.1021/acscatal.5b02414
M. Lee, J. Heo, D. Kim, S. Chang, J. Am. Chem. Soc. 144 (2022) 3667–3675. doi: 10.1021/jacs.1c12934
K. Cheng, P.J. Walsh, Org. Lett. 15 (2013) 2298–2301. doi: 10.1021/ol4008876
H. Zhang, G. Wu, H. Yi, et al., Angew. Chem. Int. Ed. 56 (2017) 3945–3950. doi: 10.1002/anie.201612138
P. Wu, M. Jia, W. Lin, S. Ma, Org. Lett. 20 (2018) 554–557. doi: 10.1021/acs.orglett.7b03637
S. Zhai, S. Qiu, S. Yang, et al., Chin. Chem. Lett. 34 (2003) 107657–107660.
Y.F. Wang, S. Chiba, J. Am. Chem. Soc. 131 (2009) 12570–12572. doi: 10.1021/ja905110c
H.J. Zhao, X.F. Fan, J.J. Yu, C. Zhu, J. Am. Chem. Soc. 137 (2015) 3490–3493. doi: 10.1021/jacs.5b00939
R. Ren, H. Zhao, L. Huan, C. Zhu, Angew. Chem. Int. Ed. 54 (2015) 12692–12696. doi: 10.1002/anie.201506578
K. Jia, F. Zhang, H. Huang, Y. Chen, J. Am. Chem. Soc. 138 (2016) 1514–1517. doi: 10.1021/jacs.5b13066
S. Wang, L. Guo, H. Wang, X.H. Duan, Org. Lett. 17 (2015) 4798–4801. doi: 10.1021/acs.orglett.5b02353
Z.G. Ren, W.L. Yu, H.X. Zheng, P.F. Xu, Org. Lett. 25 (2023) 93–98. doi: 10.1021/acs.orglett.2c03894
L. Zhao, Q. Zhong, J. Tian, et al., Org. Lett. 24 (2022) 4421–4426. doi: 10.1021/acs.orglett.2c01649
C. Lou, X. Wang, L. Lv, Z. Li, Org. Lett. 23 (2021) 7608–7612. doi: 10.1021/acs.orglett.1c02824
P. Gao, H. Wu, J.C. Yang, L. Guo, Org. Lett. 21 (2019) 7104–7108. doi: 10.1021/acs.orglett.9b02675
L. Chang, Q. An, L. Duan, K. Feng, Z. Zuo, Chem. Rev. 122 (2022) 2429–2486. doi: 10.1021/acs.chemrev.1c00256
J. Xuan, X.K. He, W.J. Xiao, Chem. Soc. Rev. 49 (2020) 2546–2556. doi: 10.1039/c9cs00523d
X. Wu, C. Zhu, Chem. Commun. 55 (2019) 9747–9756. doi: 10.1039/c9cc04785a
A.A.M.A. El Gehani, H.A. Maashi, J. Harnedy, L.C. Morrill, Chem. Commun. 59 (2023) 3655–3664. doi: 10.1039/d3cc00302g
N. Jha, P. Mishra, M. Kapur, Org. Chem. Front. 10 (2023) 4941–4971. doi: 10.1039/d3qo01090b
S. Bloom, D.D. Bume, C.R. Pitts, T. Lectka, Chem. Eur. J. 21 (2015) 8060–8063. doi: 10.1002/chem.201501081
Y. Ge, Y. Shao, S. Wu, et al., ACS Catal. 13 (2023) 3749–3756. doi: 10.1021/acscatal.3c00230
S. Wu, J. Li, R. He, K. Jia, Y. Chen, Org. Lett. 23 (2021) 9204–9209. doi: 10.1021/acs.orglett.1c03526
M. Ji, Z. Wu, C. Zhu, Chem. Commun. 55 (2019) 2368–2371. doi: 10.1039/c9cc00378a
X. Fan, H. Zhao, J. Yu, X. Bao, C. Zhu, Org. Chem. Front. 3 (2016) 227–232. doi: 10.1039/C5QO00368G
D. Wang, J. Mao, C. Zhu, Chem. Sci. 9 (2018) 5805–5809. doi: 10.1039/c8sc01763h
R. Ren, Z. Wu, Y. Xu, C. Zhu, Angew. Chem. Int. Ed. 55 (2016) 2866–2869. doi: 10.1002/anie.201510973
E. Ota, H. Wang, N.L. Frye, R.R. Knowles, J. Am. Chem. Soc. 141 (2019) 1457–1462. doi: 10.1021/jacs.8b12552
H.G. Yayla, H.J. Wang, K.T. Tarantino, H.S. Orbe, R.R. Knowles, J. Am. Chem. Soc. 138 (2016) 10794–10797. doi: 10.1021/jacs.6b06517
K. Zhao, K. Yamashita, J.E. Carpenter, et al., J. Am. Chem. Soc. 141 (2019) 8752–8757. doi: 10.1021/jacs.9b03973
Ł. Woźniak, G. Magagnano, P. Melchiorre, Angew. Chem. Int. Ed. 57 (2018) 1068–1072. doi: 10.1002/anie.201711397
L. Huang, T. Ji, M. Rueping, J. Am. Chem. Soc. 142 (2020) 3532–3539. doi: 10.1021/jacs.9b12490
T. Ji, X.Y. Chen, L. Huang, M. Rueping, Org. Lett. 22 (2020) 2579–2583. doi: 10.1021/acs.orglett.0c00493
A. Hu, Y. Chen, J.J. Guo, et al., J. Am. Chem. Soc. 140 (2018) 13580–13585. doi: 10.1021/jacs.8b08781
K. Zhang, L. Chang, Q. An, X. Wang, Z. Zuo, J. Am. Chem. Soc. 141 (2019) 10556–10564. doi: 10.1021/jacs.9b05932
L. Wen, J. Ding, L. Duan, et al., Science 382 (2023) 458–464. doi: 10.1126/science.adj0040
Y. Chen, J. Du, Z. Zuo, Chem 6 (2020) 266–279. doi: 10.1016/j.chempr.2019.11.009
J. Guo, A. Hu, Y. Chen, et al., Angew. Chem. Int. Ed. 55 (2016) 15319–15322. doi: 10.1002/anie.201609035
Z. Yang, D. Yang, J. Zhang, et al., J. Am. Chem. Soc. 144 (2022) 13895–13902. doi: 10.1021/jacs.2c05520
B. Wang, H. Li, L. Wang, Y.G. Liu, J. Wu, Chem. Catal. 2 (2022) 2096–2105. doi: 10.1016/j.checat.2022.07.002
T. Xue, Z. Zhang, R. Zeng, Org. Lett. 24 (2022) 977–982. doi: 10.1021/acs.orglett.1c04365
J. Wang, B. Huang, C. Shi, C. Yang, W. Xia, J. Org. Chem. 83 (2018) 9696–9706. doi: 10.1021/acs.joc.8b01225
D.R. Weinberg, C.J. Gagliardi, J.F. Hull, et al., Chem. Rev. 112 (2012) 4016–4093. doi: 10.1021/cr200177j
P.R.D. Murray, J.H. Cox, N.D. Chiappini, et al., Chem. Rev. 122 (2022) 2017–2291. doi: 10.1021/acs.chemrev.1c00374
E. Tsui, H. Wang, R.R. Knowles, Chem. Sci. 11 (2020) 11124–11141. doi: 10.1039/d0sc04542j
Z. Zhou, X. Kong, T. Liu, Chin. J. Org. Chem. 41 (2021) 3844–3879. doi: 10.6023/cjoc202106001
Y. Zhang, Y. Niu, Y. Guo, et al., Angew. Chem. Int. Ed. 61 (2022) e202212201. doi: 10.1002/anie.202212201
J.J. Li, Heterocyclic Chemistry in Drug Discovery, John Wiley & Sons, Hoboken, NJ, 2013.
E. Vitaku, D.T. Smith, J.T. Njardarson, J. Med. Chem. 57 (2014) 10257–10274. doi: 10.1021/jm501100b
Z. Ma, D.C.H. Lin, R. Sharma, et al., Bioorg. Med. Chem. Lett. 26 (2016) 15–20. doi: 10.1016/j.bmcl.2015.11.050
A.Y. Guan, C.L. Liu, X.F. Sun, Y. Xie, M.A. Wang, Bioorg. Med. Chem. 24 (2016) 342–353. doi: 10.1016/j.bmc.2015.09.031
A.E. Goetz, N.K. Garg, Nat. Chem. 5 (2013) 54–60. doi: 10.1038/nchem.1504
M. Vellakkaran, T. Kim, S. Hong, Angew. Chem. Int. Ed. 61 (2022) e202113658. doi: 10.1002/anie.202113658
M. Kong, Y. Tan, X. Zhao, et al., J. Am. Chem. Soc. 143 (2021) 4024–4031. doi: 10.1021/jacs.1c01073
Y. Yin, Y. Dai, H. Jia, et al., J. Am. Chem. Soc. 140 (2018) 6083–6087. doi: 10.1021/jacs.8b01575
K. Cao, S.M. Tan, R. Lee, et al., J. Am. Chem. Soc. 141 (2019) 5437–5443. doi: 10.1021/jacs.9b00286
K.N. Lee, Z. Lei, M.Y. Ngai, J. Am. Chem. Soc. 139 (2017) 5003–5006. doi: 10.1021/jacs.7b01373
D.S. Hays, G.C. Fu, J. Org. Chem. 63 (1998) 6375–6381. doi: 10.1021/jo9809130
Y. Miyahara, Y.N. Ito, J. Org. Chem. 79 (2014) 6801–6807. doi: 10.1021/jo5006137
S. Sarkar, A. Banerjee, W. Yao, E.V. Patterson, M.Y. Ngai, ACS Catal. 9 (2019) 10358–10364. doi: 10.1021/acscatal.9b03570
J. Tormo, D.S. Hays, G.C. Fu, J. Org. Chem. 63 (1998) 201–202. doi: 10.1021/jo971574y
M. Larsen, H. Kromann, A. Kharazmi, S.F. Nielsen, Bioorg. Med. Chem. Lett. 15 (2005) 4858–4861. doi: 10.1016/j.bmcl.2005.07.012
B.V. Pati, A. Ghosh, P.C. Ravikumar, Org. Lett. 22 (2020) 2854–2860. doi: 10.1021/acs.orglett.0c00967
T. Fujiwara, Y. Tsuruta, K. Arizono, T. Takeda, Synlett (1997) 962–964. doi: 10.1055/s-1997-970
I. Ryu, M. Ikebe, N. Sonoda, et al., Tetrahedron Lett. 41 (2000) 5689–5692. doi: 10.1016/S0040-4039(00)00925-4
H.Q. Xiao, X.Z. Shu, K.G. Ji, C.Z. Qi, Y.M. Liang, Catal. Commun. 10 (2009) 1824–1827. doi: 10.1016/j.catcom.2009.06.008
I. Ryu, K. Matsumoto, Y. Kameyana, et al., J. Am. Chem. Soc. 115 (1993) 12330–12339. doi: 10.1021/ja00079a013
I. Ryu, M. Ando, A. Ogawa, S. Murai, N. Sonoda, J. Am. Chem. Soc. 105 (1983) 7192–7194. doi: 10.1021/ja00362a041
Y.H. Zhang, W.W. Zhang, Z.Y. Zhang, K. Zhao, T.P. Loh, Org. Lett. 21 (2019) 5101–5105. doi: 10.1021/acs.orglett.9b01703
B. Giese, H. Horler, W. Zwick, Tetrahedron Lett. 23 (1982) 931–934. doi: 10.1016/S0040-4039(00)86986-5
B. Giese, H. Horler, Tetrahedron 41 (1985) 4025–4037. doi: 10.1016/S0040-4020(01)97181-9
N. Iwasawa, S. Hayakawa, M. Funahashi, K. Isobe, K. Narasaka, Bull. Chem. Soc. Jpn. 66 (1993) 819–827. doi: 10.1246/bcsj.66.819
C. Liu, J. Wang, X. Liu, J. Feng, D. Du, Chem. Commun. 59 (2023) 13175–13178 doi: 10.1039/d3cc04765b

Scheme 2 Substrate scope of the ring-opening coupling reaction of cycloalcohols with vinylazaarenes. Reaction conditions: 1a-1e (0.2 mmol), 2a-2z, 2a'−2m' (0.6 mmol), [Acr-Mes]+ClO4− (PC6) (0.02 mmol), BINOL-PA (0.05 mmol), DCE (6.0 mL), 90 W Kessil Lamps. Isolated yields.

Scheme 3 Substrate scope of the ring-opening coupling reaction of cycloalcohols with enones. Reaction conditions: 4 (0.2 mmol), 2 (1.0 mmol), [Ir(dF(CF3)ppy)2(dtbbpy)](PF6) (PC1) (0.006 mmol), NBu4+(tBuO)2POO− (0.05 mmol), PhCF3 (4.0 mL), 90 W Kessil Lamps. Isolated yields.

Scheme 4 Large-scale ring-opening coupling reactions and their synthetic applications.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:

 下载:
下载:


 下载:
下载: