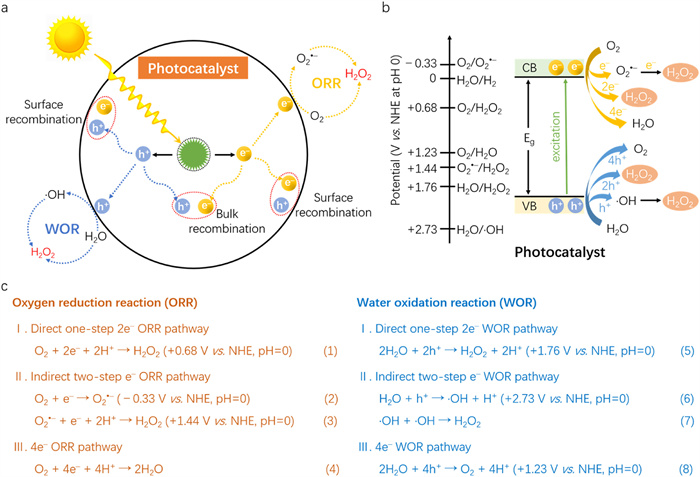
Figure 1.
Schematic illustration of (a) photocatalytic processes for H2O2 production and (b) corresponding energy diagrams. (c) Reaction pathways and corresponding energy diagrams for ORR and WOR pathways.

Since Thenard first discovered H2O2 by reacting BaO2 with HNO3 in 1818, it has been extensively utilized as a highly efficient and environmentally friendly oxidant in various applications such as wastewater treatment, medicine disinfection, and chemical industries [1-3]. The global demand for H2O2 is rapidly increasing. Reports indicate that the global H2O2 market is estimated to grow at a compound annual growth rate of 5.7%, reaching 4.05 billion USD by 2027 from 3.24 billion USD in 2023 [4]. A straightforward route for synthesizing H2O2 involves the reaction of H2 and O2 in the presence of noble metal catalysts, such as Au and Pd (H2 + O2 → H2O2, the standard Gibbs free energy ΔG0 = −120 kJ/mol) [5-7]. Due to safety concerns and the need for optimal selectivity, large quantities of N2 must be added during the actual production, significantly reducing the yield of H2O2 [8]. Currently, H2O2 is primarily produced on an industrial scale through anthraquinone oxidation, which accounts for approximately 95% of the world's total output [1,9]. However, this process requires huge energy input and generates chemical waste. Therefore, the development of an inexpensive and environment-friendly route for H2O2 synthesis is desired.
Photocatalytic H2O2 synthesis offers a potentially more sustainable and cleaner alternative to the anthraquinone oxidation method [10-16]. Due to its utilization of solar energy and green reactants (H2O and O2), the photocatalytic process generates no pollution. Therefore, there has been a significant increase in studies focusing on photocatalytic H2O2 production [17-20]. Among the various investigated photocatalysts, 2D photocatalysts have received tremendous research interest due to their planar structure and ultrathin thickness, which endow several unique physicochemical features [21,22]. For instance, 2D photocatalysts with greatly exposed surfaces offer abundant active sites; the nanoscale thickness dramatically shortens the migration distance of photoinduced charge carriers from the bulk phase to the photocatalysts on the surface; the 2D flexible planar structure facilitates the design and engineering of photocatalysts at molecular-level such as heterojunction construction, defects engineering, surface modification, thereby tuning their electronic structures, active sites number and charge carriers behaviour for photocatalysis [23].
Herein, we review the recent progress in photocatalytic synthesis of H2O2 from H2O and O2 using 2D photocatalysts, focusing on molecular-level regulation strategies. Beginning with the fundamentals of photocatalytic H2O2 synthesis, this review provides a detailed summary of the pathways for producing H2O2 and briefly introduces common methods for H2O2 detection. Subsequently, in alignment with modification strategies, the latest advancements in 2D photocatalysts that significantly facilitate charge separation/transfer to enhance H2O2 generation are exhaustively discussed. Key issues and prospective outlooks for achieving efficient and sustainable generation of H2O2 in scale-up industrial production are also proposed. This review aims to provide comprehensive insight and valuable guidance for the design of high-efficiency, stable and promising photocatalysts for H2O2 production.
The photocatalytic synthesis of H2O2 is based on the fundamental principles of photocatalysis [14]. In general, the light-driven photocatalytic reaction using a photocatalyst primarily involves three steps: photoexcitation, charge carriers separation/migration and surface redox reactions, as illustrated in Fig. 1a. Firstly, photons are absorbed by the photocatalyst. When the energy of photons exceeds the band gap (Eg) of the photocatalyst, electrons (e–) and holes (h+) with certain redox capacity are generated on the conduction band (CB) and valence band (VB), respectively. Subsequently, the two types of charge carriers separate and transfer to the surface of the photocatalyst. During this process, most of these photoinduced electrons and holes will recombine with each other without involving in any chemical reactions, and the remaining charge carriers trigger subsequent redox reactions on the photocatalyst surface. In this context, the light absorption capability and charge carrier separation/transfer of the photocatalyst are critically important for the generation of H2O2.
H2O2 can be generated through the oxygen reduction reaction (ORR) or water oxidation reaction (WOR) (Figs. 1a and b) [18,24]. For example, the ORR (Fig. 1c) pathways can be direct one-step 2e– ORR pathway, indirect two-step e– ORR pathway and 4e– ORR [25,26]. Thermodynamically, the direct 2e– ORR (+0.68 V) is more favourable than 4e– ORR (+1.23 V). In the indirect two-step e– ORR pathway, photoinduced electrons are first captured by electrophilic O2 to form O2•– radicals, which subsequently generate H2O2 through proton-coupled electron transfer (PCET) reactions. Since each step requires only one electron, it is kinetically desirable. However, compared to the direct one-step 2e– ORR pathway, the single-electron reduction of O2 to O2•– radical needs a more negative potential (−0.33 V), necessitating the photocatalyst to have a more negative CB position. Alternatively, H2O2 can be produced via oxidation reactions by photogenerated holes, such as the direct one-step 2e– WOR pathway and indirect two-step e– WOR pathway [27,28]. During the direct 2e– WOR pathway, H2O can be oxidized into H2O2 by two photoexcited holes. In the indirect two-step e– WOR pathway, H2O is oxidized into •OH radical via one photoinduced hole. Subsequently, two •OH radicals combine with each other to form one H2O2 molecule. Due to the indirect two-step e– WOR pathway only requiring one hole, it is kinetically favourable compared to the direct one-step 2e– WOR pathway. However, from a thermodynamic perspective, the direct one-step 2e– WOR pathway (+1.76 V) is more favourable than the indirect two-step e– WOR pathway (+2.73 V). In addition, owing to the more negative reaction potential of the 4e– WOR pathway (+1.23 V) to form O2 (Fig. 1c), it will compete with the direct 2e– WOR pathway, thereby impacting the selectivity of H2O2 production. Hence, finding ways to inhibit the competition of the 4e– water-to-O2 conversion, while also enhancing the efficiency of both the direct one-step 2e– WOR pathway and the indirect two-step e– WOR pathway, is undeniably crucial for addressing these challenges.
Given the presence of multiple ORR and WOR pathways, an indepth understanding of the reaction mechanisms and precise controlling of these pathways are paramount for enhancing the photocatalytic H2O2 production rate and solar-to-chemical conversion (SCC) efficiency. Experimental studies have confirmed the existence of five potential routes for photocatalytic H2O2 synthesis: direct one-step 2e– ORR pathway, indirect two-step e– ORR pathway, direct one-step 2e– WOR pathway, indirect two-step e– WOR pathway, simultaneous ORR and WOR two-channel pathways [25,27–29].
Especially, the simultaneous ORR and WOR two-channel pathways can achieve the photocatalytic generation of H2O2 from both O2 reduction and H2O oxidation. The photoinduced electrons directly reduce O2 to H2O2 via 2e– ORR. Simultaneously, the holes directly oxidize H2O to produce H2O2 via 2e– WOR, providing protons for the 2e– ORR. Since there are no additional sacrificial reagents besides H2O and O2, the theoretical atom utilization efficiency can reach 100% [27]. This process is also defined as a photosynthesis process (2H2O + O2 → 2H2O2, △G0 = 204 kJ/mol), in which the solar energy is converted and stored in H2O2 [25]. However, since the 4e– WOR pathway is thermodynamically more favorable than the direct one-step 2e– WOR, achieving selectivity for H2O2 production becomes a significant issue. Compared with the direct one-step 2e– ORR and direct one-step 2e– WOR dual pathways, the atomic utilization efficiency of this direct one-step 2e– ORR and 4e– WOR pathways is only 68% [30]. From another perspective, if this is realized, oxidation of H2O by the photoinduced h+ provides O2 and H+ for oxygen reduction, which in turn would facilitate the direct one-step 2e– reduction of O2 for the efficient generation of H2O2.
The production rate of H2O2 via photocatalytic H2O2 synthesis is typically low, usually within the micromolar range. Therefore, accurate detection and quantification of H2O2 is crucial for the development of photocatalytic H2O2 synthesis. As early as 1927, Baur et al. used an iodide method (2KI+ H2O2 →2KOH + I2, I2 caused the starch solution to adopt a blue colour) to analyze H2O2 produced over ZnO via a photocatalytic reaction [31]. Up to now, several approaches have been applied to determine the H2O2 concentration in photocatalysis, including colourimetric test strip [32], direct optical absorption [33], colouration [34–37], chemiluminescence [33,38,39], enzymatic assay [11,40], fluorescence probe [41,42], and chemical titration methods [3,43–48]. The advantages and disadvantages of different methods for H2O2 detection are summarized in Table 1.
The colourimetric test strip method is a simple and convenient way to measure the concentration of H2O2 [32,49]. The principle of redox colour change of strip is similar to that of the chemical titration method, but they utilize solid phase assays [32]. When using a colourimetric test strip, it is dipped into the solution containing H2O2 for approximately one second and then exposed to air for a period of time. During this time, the colour of the strip changes from blue to yellow/brown. The concentration of H2O2 can be determined according to the colour of the strip. There are many commercially available H2O2 colourimetric test strips on the market, such as Quantofix has three types of strips with maximum detection limits of 25, 100 and 1000 ppm, respectively. It is worth noting that the concentration of H2O2 is determined by observing the colour of the strip with the naked eye, and the systematic error is large. Additionally, prolonged exposure of the strip to air also affects the accuracy of measurement results. Therefore, the colourimetric test strip method is better as a semi-quantitative method.
For the direct optical absorption method [33], the molar absorption coefficient of H2O2 is 0.01 M–1 cm–1 at the wavelength of 360 nm. Even at a shorter wavelength of 260 nm, the molar absorption coefficient is only 13 M–1 cm–1. Although this method is simple to operate, due to the small absorption coefficient, it is difficult to directly detect the small amount of H2O2 in the reaction system using a UV–vis spectrophotometer.
In photocatalysis, the colouration method is the most widely used for quantifying H2O2. In this method, iodide (I–), Ti4+,N,N–diethyl-p-phenylenediamine (DPD), metavanadate (VO3–) or TiIV-tetrapyridylporphyrin (TiTPyP) are often used as color-development reagents to react with H2O2, forming a compound that strongly absorbs in the ultraviolet or visible range. These compounds are then measured using a UV–vis spectrophotometer to indirectly determine H2O2 concentration. For example, 1 mL of sample is added to the mixture of KI (1 mL, 0.4 mol/L) and potassium hydrogen phthalate (C8H5KO4, 1 mL, 0.1 mol/L) solutions, which is then kept for 30 min. Under acidic conditions, I– can be oxidized by H2O2 to form triiodide anions (I3–), and the resulting amount of I3– (3I– + 2H+ + H2O2 → I3– + 2H2O) can be measured using UV–vis spectrophotometer at 350 nm, which further determines H2O2 concentration [30,50–52]. When using Ti4+ as indicator, H2O2 reacts with Ti4+ to form the yellow peroxo-titanic acid (Ti4+ + 2H2O + H2O2 → H2TiO4 + 4H+). Then the solution containing H2O2 can be monitored indirectly by detecting the yellow H2TiO4 at the wavelength of 410 nm using a UV–vis spectrophotometer [36]. Furthermore, Choi and co-workers detected the concentration of H2O2 using N,N–diethyl-1,4-phenylene-diamine (DPD) and peroxidase (POD), which is based on the POD-catalyzed oxidation of DPD by H2O2 (
In the chemiluminescence method, a luminol chemiluminescence probe is employed to monitor the formation of H2O2. During the experiment, oxidized luminol (5-amino-2,3-diaza-1,4-naphthoquinone) reacts with H2O2 to generate an excited state of ion (3-amino phthalate*), whose chemiluminescence intensity can be measured by a photomultiplier tube. For example, Hirakawa et al. used the chemiluminescence method to determine the amount of H2O2 when investigating the reaction mechanism of selective generation of O2•– and H2O2 by N- and S-doped TiO2 photocatalysts in aqueous solution [39]. It should be noted that the limit of detection for H2O2 by the chemiluminescence method is about 1 nmol/L [33,39].
Enzymatic assay for H2O2 detection is highly sensitive, making them suitable for measuring trace amounts of H2O2 in various applications [56,57]. Horseradish peroxidase is a commonly used enzyme reagent. For example, resorufin, produced from the reaction of H2O2 with Ampliflu Red in the presence of horseradish peroxidase, can be detected using high-performance liquid chromatography with a photo-diode array detector (λabs. = 560 nm) [11,40]. Calibration of the resorufin peak area with a standard H2O2 solution allows for quantitative analysis of H2O2 concentration. At present, this is one of the most accurate ways to determine H2O2 [11]. It should be noted that when horseradish peroxidase catalyzes the reaction of H2O2, there are certain restrictions on the pH and temperature of the reaction solution. During analysis, avoid storing the enzyme for extended periods or exposing it to unfavourable conditions to prevent enzyme deactivation, which affects the accuracy of H2O2 detection.
The fluorescence probe method for the quantification of H2O2 is frequently reported in biological systems [58]. The principle of this method is to convert H2O2 into fluorescence products, and the concentration of fluorescence products is determined by fluorescence spectroscopy. For instance, in the catalysis of horseradish peroxidase (HRP), H2O2 reacts with p-hydroxyphenylacetic acid (POHPAA) to form a fluorescent dimer, which can be measured with a fluorescence spectrophotometer at an emission wavelength of 408.5 nm and an excitation wavelength at 316.5 nm [42].
Chemical titration is the most commonly used method for quantifying H2O2. In this approach, titration standard solutions such as yellow-coloured Ce(SO4)2 and purple-coloured KMnO4 are typically employed [3,43–47]. For yellow-coloured Ce(SO4)2, H2O2 reacts with Ce4+ to form the colourless Ce3+ (2Ce4+ + H2O2 → 2Ce3+ + O2 + 2H+), which can be analyzed by the UV–vis spectrophotometer. Specifically [44,46–48], the yellowcolored Ce(SO4)2 transparent solution (1 mmol/L) is prepared by dissolving Ce(SO4)2 (33.2 mg) in H2SO4 solution (100 mL, 0.5 mol/L). A series of known concentrations of H2O2 are then added to this solution to produce a standard curve, which is measured by UV–vis spectrophotometer at 320 nm. According to the linear relationship between signal intensity and Ce4+ concentration, the H2O2 concentration of the sample can be obtained. When using purple-coloured KMnO4 as the titrant, under acidic conditions, the purple colour of MnO4– will be reduced to colourless Mn2+ by H2O2 (2MnO4– + 6H+ + 5H2O2 → 2Mn2+ + 5O2 + 8H2O) [45]. Before H2O2 is completely consumed, the solution to be tested will quickly turn colourless upon the addition of MnO4–. After a complete reaction of H2O2, further addition of MnO4– leads to the titration endpoint, indicated by a persistent (about 30 s) light pink colour. The concentration of H2O2 in the solution can be determined based on the amount of MnO4– consumed. It should be noted that since the KMnO4 assay relies on the detection of the purple colour produced by a slight excess of KMnO4 to determine the titration endpoint, choosing Ce(SO4)2 as the titration standard solutions will yield more accurate results than KMnO4.
To elucidate the relationship between structure and photocatalytic activity for H2O2 production using 2D photocatalysts, the primarily studied materials have been classified into two types in this review based on their composition and chemical properties: Inorganic materials and organic polymers. To date, inorganic materials such as metal oxides, metal chalcogenides and bismuth-based materials as well as organic polymers including g-C3N4, metal organic frameworks (MOFs) and covalent organic frameworks (COFs) have been explored as photocatalysts to produce H2O2.
Constructing metal oxides, such as TiO2 [59] and ZnO, with 2D structures can effectively increase their specific surface area as well as maximize exposure of active surfaces/sites, thereby improving their photocatalytic activity [21]. The pioneering study dates back to 1927 when Baur et al. first reported the production of H2O2 using ZnO photocatalyst [31]. Approximately 50 years later, Harbour et al. observed that photocatalytic generation of H2O2 via indirect 2e– ORR pathway when using particulate TiO2 as the photocatalysts, yet the presence of H2O2 was transient due to its rapid decomposition [60]. Since then, various strategies have been employed to improve the photocatalytic production performance of H2O2 in terms of efficiency and selectivity. For instance, surface fluorination of TiO2 enhanced the production of H2O2 with steady-state concentration to millimolar levels (1−1.3 mmol/L) by inhibiting the degradation [61]. Additionally, the deposition of Au nanoparticles [62] or silver-gold clusters [63] on TiO2 has been effective in enhancing the rate of H2O2 production. This improvement is attributed to the separation of photogenerated carriers and the transfer of electrons from TiO2 to the loaded metal nanoparticles/clusters. The representative metal oxide photocatalysts [64-86], along with various strategies that enhance H2O2 production at the molecular level are summarized in Table S1 (Supporting information).
The creation of a heterojunction structure is one effective strategy in the pursuit of efficient photocatalytic processes. Such a structure is constructed by combining two kinds of photocatalysts with different band positions and Fermi levels [87]. Typically, heterojunction structures involve conventional type-Ⅰ, type-Ⅱ and type-Ⅲ heterojunctions, p-n heterojunctions, Schottky heterojunctions and Z-scheme heterojunctions [88,89]. The non-uniform distribution of charge at the interface of the heterojunction construction results in the generation of an internal electric field, driving the directional migration of charge carriers to achieve effective charge separation, utilization, and extension of lifetime [90]. For instance, Zhang's group designed a 2D g-C3N4/TiO2 nanocomposites (HT-CN/TiO2) as a vertical Z-scheme heterojunction [64]. Compared with the physically mixed sample, HT-CN/TiO2 generated H2O2 more rapidly. The authors suggested that the enhanced performance was attributed to the Z-scheme transfer mechanism and the integrated effects of this specific face-to-face interfacial morphology. Driven by the internal electric field, photoinduced e– from CB of TiO2 combine with photoinduced h+ of g-C3N4 at the inter-plane contact interface, leading to an accumulation of e– in the CB of g-C3N4 and h+ in the VB of TiO2. As a result, the accumulated holes in the VB of TiO2 oxidized H2O to generate H2O2 via an indirect two-step e– WOR pathway, while electrons in the CB of g-C3N4 reduced O2 to generate H2O2 by indirect two-step e– ORR pathway (Fig. 2a). They also reported a WO3-TiO2 vertical heterojunction via in situ assembling of WO3 nanosheets on TiO2 nanosheets and found the formation of an internal electric field between WO3 and TiO2 [65]. This internal electric field formed at the interface resulted in a Z-scheme photocatalytic mechanism for H2O2 formation without the addition of sacrificial reagents. The Z-scheme charge transfer route enabled the spatial separation of the remaining electrons in the CB of TiO2 and holes in the VB of WO3 with high redox potentials. Therefore, the electrons in CB of TiO2 triggered a powerful production of H2O2 by indirect two-step e– ORR pathway and holes in the VB of WO3 oxidized H2O to yield H2O2 via indirect two-step e– WOR pathway.

In addition to constructing heterojunctions, introducing defects has been proven to be an effective method to modulate the electronic band structure of a photocatalyst, which has a significant impact on its photocatalytic performance [91]. For example, as reported by Zhu et al. [67], C-supported oxygen vacancy (Ov) rich Co3O4 nanoplate (C—OvCo-400) performed selective and efficient H2O2 production (3.78 mmol g–1 h–1) under visible light irradiation (λ ≥ 420 nm) in the absence of sacrificial agent. It has been confirmed that the presence of Ov reduced the band gap and enhanced the donor density. Additionally, Ov can act as the active sites for WOR, while the C support serves as reaction centres for the ORR, thereby markedly facilitating the separation and migration of charge carriers. Consequently, H2O adsorbed on the surface of OvCo was oxidized to form H2O2 via direct one-step 2e– WOR, while O2 was reduced into H2O2 via direct one-step 2e– ORR. As another example, Fu et al. found that the photocatalytic production rate of H2O2 from TiO2 with bulk defects and surface defects was 30 and 45 times higher than that of pristine TiO2 and P25, respectively [68]. A synergistic effect caused by the bulk and surface defects in TiO2 was observed: (1) The bulk defects served as h+ acceptors to drive directional h+ transfer and efficiently boosted the separation and transfer of charge carriers; (2) The surface defects strengthened the adsorption of O2, thereby dramatically improving the generation of H2O2 via indirect two-step e– ORR. Chen et al. also reported that Ov sites in the In2S3@In2O3 can change the H2O2 production pathway from the indirect two-step e– ORR to the direct one-step 2e– ORR, resulting in a significantly improved photocatalytic performance (Fig. 2b) [86].
Surface modification for the photocatalyst is another efficient approach to improve the photocatalytic activity for H2O2 production. Apart from regulating the electronic structure of photocatalysts, the heteroatom on the photocatalyst surface can promote interfacial electron transfer [92]. For instance, Xu's group modified WO3 with Au nanoparticles (NPs) and achieved superior performance for photocatalytic H2O2 production, exhibiting rates 63.6 and 1511.1 times higher than pristine WO3 and Au/SiO2, respectively (Fig. 2c) [69]. When WO3 was excited by visible light (λ ≥ 420 nm), the photogenerated e– transferred from the CB of WO3 to Au NPs, followed by direct one-step 2e– ORR on Au NPs. However, when WO3 was irradiated by light with a wavelength longer than 535 nm, the hot e– transferred from Au NPs to the CB of WO3, thereby generating H2O2 via direct one-step 2e– ORR pathway on WO3. Similarly, Zhou's team reported photocatalytic generation of H2O2 via direct one-step 2e– WOR pathway over 1.0 wt% Pt/WO3 and suggested that Pt loading significantly enhanced the separation and transfer of charge carriers [70]. Additionally, the ratios of Pt0 and PtOx affected the photocatalytic activities. Specifically, Pt0 species acted as active sites to reduce O2 to H2O, while PtOx species served as oxidation centres to oxidize H2O to H2O2.
Doping is also adopted to modulate the electron transfer process. For instance, Wang et al. doped Gd3+ into WO3 for promoted photocatalytic degradation of tetracycline and simultaneous in situ production of H2O2 [76]. They found that doping Gd3+ into WO3 not only expanded the light absorption range of the photocatalyst to the near-infrared range but also greatly facilitated the charge separation/transfer from photocatalyst to O2, thereby stimulating the generation of H2O2 via direct one-step 2e– ORR pathway (Fig. 2d).
As a significant class of 2D materials, layered metal chalcogenides have garnered considerable attention owing to their diverse structures and rich electronic characteristics [93]. Importantly, metal chalcogenides exhibit broad and tunable light absorption bands, which can be modified by changing the morphology and replacing the metal elements. Therefore, chalcogenides exhibit fascinating photocatalytic properties in various photocatalytic reactions such as oxygen or hydrogen evolution from water. In recent years, researchers have discovered the potential of metal chalcogenides in sunlight-driven H2O2 production and have made significant efforts to explore efficient methods for generating H2O2 (Table S2 in Supporting information) [94-108].
Among metal chalcogenides, molybdenum disulfide (MoS2) stands out as one of the most popular photocatalytic materials [109]. It is composed of S-Mo-S modules held together by weak van der Waals interactions, enabling the production of MoS2 ranging from bulk to ultrathin crystals, and even down to atomistic thin layers [110]. It has been reported that the sunlight absorption ability can be effectively improved by decreasing the MoS2 layers [111]. Besides, its electrical conductivity can be enhanced by adding other conducting materials, such as graphene and noble metals [94,112]. For instance, Song et al. modified MoS2 nanosheets with atomic Au0, which not only increased the separation and transfer of photoexcited carriers but also endowed a more negative flat band potential availably to drive the generation of H2O2 from O2 and H2O through a multichannel pathway [94]. Consequently, the photocatalytic H2O2 production rate over MoS2 loaded with 0.50 wt% Au was 2.5 times than that of bare MoS2. Interestingly, doping Mn2+ in MoS2, along with the Au modification, yielded a H2O2 generation twice higher than that of Au@MoS2 (Fig. 3a).
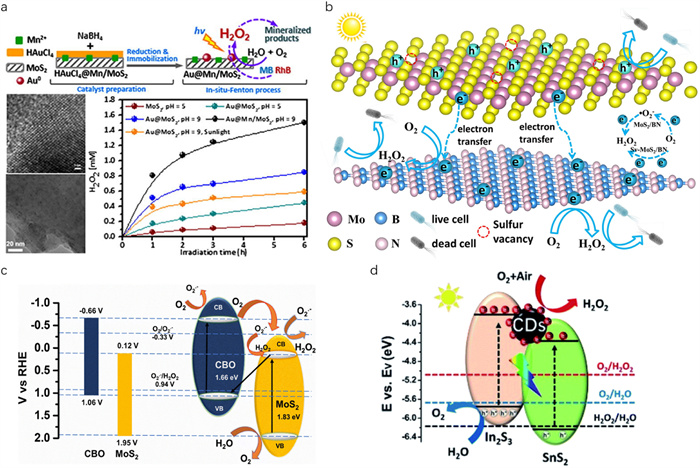
The S-vacancies in metal chalcogenides have been reported to affect the work function and regulate the interfacial barrier of the heterojunction, thereby effectively boosting the separation/transfer of charge carriers [113]. As an example, Jiang et al. prepared a defective MoS2-v/carbon nitride nanotube (TCN) heterojunction and observed that the combination of MoS2-v and TCN enhanced O2 adsorption and facilitated electron exchange between O2 and MoS2-v/TCN, which was beneficial to the ORR pathway [95]. Additionally, the creation of heterojunction between MoS2-v and TCN not only facilitated the rapid separation of charge carriers but also reduced the energy barrier for the production of *OOH and *HO2– toward H2O2, thereby effectively improving the photocatalytic H2O2 generation. Yang et al. adopted defect and interfacial engineering coupled dual strategies to design and synthesize Sv−MoS2/BN heterojunction for sacrificial agent-free photocatalytic H2O2 production, aiming at in situ water disinfection [96]. The experimental and density functional theory (DFT) calculations results demonstrated that the presentence of S-vacancies effectively regulated the interfacial barrier by decreasing the work function of MoS2, which changed the photocatalytic H2O2 production from indirect two-step e– ORR pathway to direct one-step 2e– ORR pathway (Fig. 3b). Therefore, Sv−MoS2/BN exhibited excellent photocatalytic activity for H2O2 production.
In another example, Meduri's team fabricated CBO@MoS2 Z-scheme heterojunction through a facile hydrothermal method [97]. As a result, the photocatalyst exhibited a significantly high H2O2 production rate of 1457 mmol L–1 h–1 under one sun illumination via simultaneous indirect two-step e– ORR and 4e– WOR two-channel pathways (Fig. 3c). The excellent performance was attributed to the efficient separation and transfer of charge carriers, facilitated by the staggered band alignment and the construction of a Z-scheme heterojunction, resulting in a stronger redox capacity.
Apart from MoS2, other metal chalcogenides have also been developed in the field of photocatalytic H2O2 synthesis, such as CdS, In2S3, SnS2, MoSe2, ZrS3 (Table S2). For example, Fang et al. prepared a Bi/BiOBr-CdS heterojunction through the solvothermal method [98]. It has been reported that the synergistic effect of the heterojunction and the introduction of Bi0 decreased the band gap, facilitating the separation and transfer of charge carriers while maintaining a high redox potential for simultaneous ORR and WOR. Consequently, such a Bi/BiOBr-CdS heterojunction showed a high H2O2 production rate of 346.4 µmol L–1 h–1 (λ > 420 nm) via direct one-step 2e– ORR and indirect two-step e– WOR dual channel pathways. Recently, Li et al. reported SnS2/In2S3 type Ⅱ heterostructure modified by carbon dots (CDs) for high-efficiency H2O2 production [99]. CDs not only stored the e– induced by SnS2 and In2S3, thereby regulating the interface photo-charge kinetics of SnS2/In2S3/CDs composite but also acted as the catalytic centres for ORR. As a result, the photocatalytic activity of H2O2 production over SnS2/In2S3/CDs composite increased to 1111.89 µmol h–1 g–1 (λ ≥ 420 nm) without sacrificial agents by simultaneous indirect two-step e– ORR and 4e– WOR two-channel pathways (Fig. 3d).
Bismuth-based materials have received tremendous research interest due to their unique layered structure and tunable electronic properties. These characteristics make them promising candidates for constructing ultrathin 2D structures, which have been demonstrated to have promising photocatalytic abilities [114-117]. Up to now, many bismuth-based materials, including bismuth vanadate (BiVO4), bismuth tungstate (Bi2WO6), bismuth oxyhalides and bismuth oxycarbonate (Bi2O2CO3), have been reported for the photocatalytic generation of H2O2 [11,118-136], and the details are summarized in Table S3 (Supporting information).
As a visible light-responsive photocatalyst, BiVO4 (band gap is about 2.4 eV) has been widely used in photocatalytic water oxidation for O2 evolution [137]. However, the absence of two-electron oxygen reduction active sites in pristine BiVO4 renders it unfavourable for the generation of H2O2 from O2. To address this challenge, various strategies have been adopted, including surface modification, doping, defect engineering and heterojunction construction. For example, Hirakawa et al. deposited metallic Au nanoparticles on BiVO4 (Au/BiVO4) as the cocatalyst, resulting in enhanced H2O2 production from O2 and H2O via direct one-step 2e– ORR and 4e– WOR dual-channel pathways [118]. The formation of a Schottky barrier between BiVO4 and Au facilitated the separation and transfer of photoinduced electrons to Au, thereby hindering their recombination with holes. However, Wang et al. proposed that Au nanoparticle-modified BiVO4 could only enhance the photocatalytic H2O2 production performance to a certain extent because the built-in potential generated by the upward bending of BiVO4 might hamper the transfer of electrons [119]. Additionally, due to the different work functions of Au and BiVO4, when Au interacts with BiVO4, the negative charge density of Au increases, which weakens the adsorption of O2 and HOO* on the Au surface, ultimately leading to a decrease in photocatalytic H2O2 production via direct one-step 2e– ORR. Therefore, they introduced Cu in Au/BiVO4 to form Cu@Au/BiVO4 wherein the metallic Cu core not only promoted the photoinduced e– transfer of BiVO4 but also decreased the negative charge density of the metallic Au shell. As a result, Cu@Au/BiVO4 possessed better photocatalytic activity for H2O2 production compared to Au/BiVO4 (Fig. 4a). In another study, Yu's team used ultrathin g-C3N4 (CN) to modify Au/BiVO4 [121]. The optimized CN-Au/BiVO4 showed a H2O2 yield of 1.35 mmol/L in 2 h (420 nm LED, 50 mW/cm2), which was 2.65 times that of Au/BiVO4. The authors proposed that the modification of BiVO4 with g-C3N4 promoted the hole transfer and restrained H2O2 decomposition, while metallic Au promoted the electron transfer and boosted H2O2 production through a direct one-step 2e– ORR pathway. Wang and co-workers prepared a serial of different Y doping contents BiVO4 (Y: BVO) photocatalysts using a simple hydrothermal method [124]. The doping of Y promoted the adsorption of O2 on the BiVO4 surface and induced the formation of monoclinic/tetragonal BiVO4 heterojunction, which greatly boosted the separation efficiency of charge carriers (Fig. 4b). Consequently, the optimized Y: BVO exhibited much higher activity for H2O2 production (114 µmol g–1 h–1) compared to pristine BiVO4 (26 µmol g–1 h–1) under simulated sunlight irradiation via direct one-step 2e– ORR pathway. Zhang's team prepared defective BiVO4 for the effective generation of H2O2 from O2 [125]. Due to the presence of oxygen vacancies, the adsorption of O2 and interfacial electron transfer rate were 19- and 23-fold higher than that of pristine BiVO4, respectively. Therefore, the coupling of charge carriers to chemisorbed species was effectively boosted, promoting the formation of H2O2 by direct one-step 2e– ORR pathway under visible light (Fig. 4c). Wei et al. constructed NiCo2O4/BiVO4 Z-scheme heterojunction and reported that the H2O2 yield rate over NiCo2O4/BiVO4 under visible light was 16 times than that using pristine BiVO4 [126]. The authors claimed that the desired electronic conductivity and electron transport characteristics of NiⅡ and CoⅡ effectively delayed the recombination of photoinduced e– and h+ and promoted the preferable indirect two-step e– ORR pathway for improved H2O2 generation. Meanwhile, O2•– radical and •OH radical generated by such a heterojunction system improved the efficiency for degradation of methylene blue (Fig. 4d).
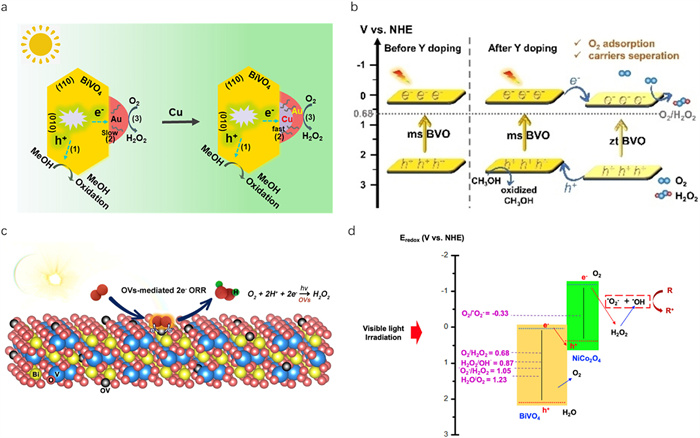
Bi2WO6 (band gap is about 2.8 eV) is another important visible-light-driven bismuth-based photocatalyst [138]. However, pristine Bi2WO6 exhibits unsatisfactory performance in H2O2 production due to poor separation of photoinduced charge carriers and in situ H2O2 consumption [139,140]. In order to enhance the photocatalytic activity of Bi2WO6 for the generation of H2O2, Zeng et al. in situ grew a red phosphorus/black phosphorus (RP/BP) heterostructure on Bi2WO6 nanosheets via a simple ultrasonic agitation method and ultimately constructed RP/BP-Bi2WO6 dual heterostructure photocatalyst [127]. The optimal 20% RP/BP-Bi2WO6 photocatalyst presented the highest H2O2 production rate, being 15-fold greater than that of pristine Bi2WO6 nanosheets under visible light. The enhanced H2O2 production was mainly ascribed to the heterojunctions, which facilitated highly efficient separation and transfer of charge carriers. Moreover, the authors confirmed that the RP/BP-Bi2WO6 photocatalyst produced H2O2 via both indirect two-step e– ORR and indirect two-step e– WOR.
Bismuth oxyhalides are another important photocatalyst with a formula of BiaObXc, where X = Cl, Br and I. Their VB top is mainly occupied by O 2p states as well as a considerable contribution of X δp states (δ = 3, 4 and 5, corresponding to X = Cl, Br, and I, respectively), while their CB bottom is dominated by Bi 6p states [141,142]. Due to the unique electronic structure, the light absorption range of BiaObXc can extend from the ultraviolet to visible light range [143,144]. Especially, the internal electric field induced by BiaObXc can greatly boost the separation and transfer of charge carriers [145]. Therefore, bismuth oxyhalides exhibit excellent photocatalytic performance in pollutant degradation, CO2 reduction and water oxidation. Recently, Xu et al. investigated the facet-dependent photocatalytic H2O2 synthesis on BiOCl and found that the H2O2 production rate of BiOCl with exposed (001) facet is about two times than that of (010) facet, which was attributed to the higher charge separation efficiency of BiOCl (001) facet [128]. Electron paramagnetic resonance (EPR) measurements and trapping experiments revealed that the BiOCl (001) produced H2O2 mainly via indirect two-step e– ORR, while photocatalytic generation of H2O2 on BiOCl (010) involved indirect two-step e– ORR and indirect two-step e– WOR pathways simultaneously. In another study, Gong et al. constructed a BiOCl/BiOBr heterojunction by in situ generating BiOCl nanosheets on the BiOBr surface, followed by NH4Cl etching [129]. This heterojunction boosted the separation of charge carriers, thereby significantly enhancing the productivity of H2O2. Besides, Zhang et al. synthesized BiOBr/COF heterojunction photocatalysts [133], which facilitated a spatial separation and transfer of charge carriers and enhanced redox power. Additionally, by introducing COF, O2 was adsorbed on the TpPaCl2 surface in a lying-down geometry, which led to the reduction and protonation of O2 to form H2O2 via direct one-step 2e– ORR pathway in the presence of ethanol as a sacrificial agent. Consequently, BiOBr/COF displayed the H2O2 production yield of 3749 µmol g–1 h–1 (under 300 W Xe irradiation), which was 27 times higher than BiOBr and 1.85 times higher than COF.
Bi2O2CO3 is also highly promising for photocatalysis owing to its unique layered crystal structure, which induces an internal electric field and enhances the separation and transfer of photogenerated carriers [117,146]. As reported by Dong et al., Bi2O2CO3 possesses a wide band gap of about 3.1−3.5 eV [147]. Additionally, the appropriate band potential of Bi2O2CO3 makes it suitable for the photocatalytic synthesis of H2O2 [136]. Recently, Wang et al. prepared a Bi2O2CO3/Bi-MOF Z-scheme heterojunction, significantly improving the photocatalytic performance of H2O2 formation via the direct one-step 2e– ORR pathway [136]. With the close-contact heterostructure and internal electric field under simulated sunlight irradiation and methanol as a sacrificial agent, the H2O2 yield rate (280.5 µmol g–1 h–1) was 11.23- and 6.06-fold higher than that of pristine Bi2O2CO3 (24.97 µmol g–1 h–1) and Bi-MOF (46.31 µmol g–1 h–1), respectively.
g-C3N4, as one of the important organic polymer materials [148-150], has been thoroughly studied for different photocatalytic applications owing to its awesome optical, electronic, and chemical properties. The photocatalytic generation of H2O2 using g-C3N4 was first reported by Hirai's group as early as 2014 [151]. Efficiently H2O2 was generated via direct one-step 2e– ORR using alcohol as an electron donor. It is also uncovered that the generation of 1,4-endoperoxide species on photoactivated g-C3N4 restrained indirect two-step e– ORR and favored direct one-step 2e– ORR pathway to the generation of H2O2. Nevertheless, pristine g-C3N4 suffered from low photoinduced charge carrier separation efficiency and slow kinetics [152,153]. Therefore, the photocatalytic performance of g-C3N4 was not satisfactory, and the H2O2 produced in a 12-h reaction (λ > 420 nm) was only 30 µmol. Therefore, strategies such as structural modification, doping, defect engineering, and heterojunction construction were applied to improve the photocatalytic ability of g-C3N4 to produce H2O2 (Table S4 in Supporting information) [15,40,151,154-203].
Subsequently, Hirai's group modified g-C3N4 with pyromellitic diimide (PDI) [154], biphenyl diimide (BDI) [155] and mellitic triimide (MTI) [156], achieving efficiently H2O2 production from H2O and O2 without using any sacrificial agents. When modified with PDI, the formation of H2O2 (λ > 420 nm, 48 h) over g-C3N4/PDI51 (50.6 µmol) was more than 250 times that of pristine g-C3N4 (< 0.2 µmol). Such attractive performance originated from the incorporation of PDI, which shifted the redox potentials of g-C3N4, enabling g-C3N4/PDI to oxidize water while maintaining high selectivity for 2e– reduction of O2. Consequently, H2O2 was produced from H2O and O2 over g-C3N4/PDI via direct one-step 2e– ORR pathway coupled with 4e– WOR pathway (Fig. 5a) [154]. When g-C3N4 was modified with BDI instead of PDI, the charge separation was enhanced, and the apparent quantum yield at 420 nm was increased from 2.6% to 4.6% [155]. After incorporating MTI into g-C3N4, the SCC efficiency reached 0.18% and the apparent quantum yield (AQY) was increased to 6.2% at 420 nm. The enhancement was attributed to the creation of a condensed melem layer, which improved intra- and interlayer transfer of the photoinduced charge carriers [156]. Furthermore, the authors prepared g-C3N4/PDI/rGO nanohybrids, which further enhanced the formation of H2O2 with an SCC efficiency of 0.20% [157]. It is revealed that the photoinduced e– from g-C3N4/PDI was transferred to rGO, promoting the separation of photoinduced charge carriers while preserving high selectivity for the 2e– reduction of oxygen. As a result, the h+ in g-C3N4/PDI oxidized H2O through 4e– WOR pathway, while the e– on rGO promoted the reduction of O2 via a direct one-step 2e– ORR pathway. In another study, Zhang's team modified g-C3N4 with polyethylenimine (PEI), which tuned the separation of charge carriers and the pathway of oxygen reduction [159]. The optimal PEI/g-C3N4 photocatalyst showed efficient H2O2 production with a production rate of 208.1 µmol g–1 h–1 (under simulated solar light), being 25 times greater than that of pristine g-C3N4. Specifically, the incorporating of PEI promoted the generation of H2O2 via the indirect two-step e– ORR pathway, which was different from the direct one-step 2e– ORR pathway when using pristine g-C3N4.

Single-atom catalysts, possessing unique electronic characteristics and theoretically enabling 100% metal atom utilization, show great potential as cocatalysts for enhancing the efficiency of photocatalysis when combined with semiconductors [204,205]. Incorporating single atoms into g-C3N4 has been reported to boost the activity and selectivity of the 2e– ORR pathway. In general, there are three types of O2 adsorption on metal surfaces: Pauling-type (end-on), Griffiths-type (side-on) and Yeager-type (side-on) [206]. Among these, side-on adsorption facilitates the cleavage of O−O bond, resulting in high selectivity for 4e– ORR. Conversely, end-on adsorption minimizes O−O bond cleavage, inhibiting 4e– ORR and promoting 2e– ORR [172]. For single atoms, O2 molecules typically undergo end-on adsorption, reducing the possibility of O−O bond cleavage. For instance, Teng et al. deposited various single atoms on g-C3N4 including Fe, Co, Ni, In, Sn and Sb, leading to markedly promoted charge separation and facilitated end-on adsorption of O2 (Fig. 5b) and thus enhanced selectivity of 2e– ORR pathway [172,206]. In another example, Zhang et al. prepared highly loaded Ni single atoms on g-C3N4 and observed the structural evolution of active sites (Ni-N3 → O1Ni-N2 → HOO—Ni-N2) upon O2 adsorption. As the energy barrier of key *OOH intermediate was remarkably reduced, the formation of H2O2 was promoted [15]. Very recently, Ren et al. prepared an Mn atomically dispersed on aryl amino-substituted g-C3N4 photocatalyst (Mn/AB-C3N4), exhibiting excellent photocatalytic activity and stability for the generation of H2O2 from seawater without the addition of sacrificial agents [163]. By introducing aryl amino units, the band gap was shortened, and the light absorption was boosted. Additionally, the presence of coordinatively unsaturated Mn-Nx sites not only stimulated the adsorption and activation of O2 but also promoted the separation of photoinduced charge carriers and suppressed their recombination. Hence, the Mn/AB-C3N4 exhibited outstanding photocatalytic performance of H2O2 production. Most importantly, different from the usual direct or indirect ORR pathway using g-C3N4-based photocatalysts, H2O2 produced over Mn/AB-C3N4 via a less common WOR route in which direct one-step 2e– WOR and indirect two-step e– WOR pathways occurred simultaneously (Fig. 5c).
Apart from surface modification, the production of H2O2 over g-C3N4 can also be enhanced by ion doping. Choi's team demonstrated that in situ doping of K, P and O into g-C3N4 (KPD-CN-x, x represents the amount of substance added dibasic potassium phosphate) remarkably improved the photocatalytic performance of H2O2 formation via direct one-step 2e– ORR without loading noble-metal cocatalysts [169]. The AQY at 420 nm for the formation of H2O2 obtained by using the optimized KPD-CN-7.5 photocatalyst was 25-fold higher than that of pristine g-C3N4. The authors also prepared heteroatom (S and K)-doped polymeric C3N4, which exhibited an AQY at 450 nm approaching 100% for the H2O2 formation [174]. The synergistic association of K and S dopants in polymeric C3N4 was suggested to boost the separation of interlayer charge carriers and the polarization of trapped electrons, which is beneficial for the capture and reduction of O2 in ORR kinetics, as indicated by the spectroscopic analysis in spatiotemporal resolution and DFT calculations (Fig. 5d). In another study, Liu et al. doped polymeric C3N4 with dual-heteroatom (O and K), and verified that the as-prepared O/K-CN facilitated the electron transfer from the β spin-orbital to π* orbital of O2 compared to other C3N4, thereby optimizing the activation of O2 to enhance the formation of *OOH intermediate and lowering the energy barrier for the generation of H2O2 [176].
Creating surface defects in g-C3N4 is another important strategy to improve their photocatalytic activity. Two types of surface defects, C- and N-vacancies, in g-C3N4, have shown advantages in H2O2 production [181,184]. For instance, Wang et al. introduced C-vacancies on the surface of g-C3N4 via calcination treatment and investigated the effects of C-vacancies on the O2 reduction over g-C3N4 [181]. It is revealed that the C-vacancies significantly enhanced the activity of g-C3N4 in reducing O2 to generate H2O2 without the addition of any organic sacrificial agents. The authors observed that the adsorption strength of O2 on Cv-g-C3N4 was stronger than that on pristine g-C3N4 by temperature-programmed desorption spectra. Additionally, the nitrogen atoms of the amino group around C-vacancies acted as a Lewis base. Since O2 functions as a Lewis acid, these nitrogen atoms can interact with O2 through Lewis acid-base interactions, thereby facilitating electron transfer. As a result, the route for reducing O2 to product H2O2 changed from indirect two-step e– ORR to direct one-step 2e– ORR (Fig. 5e). In another study, Xie et al. prepared g-C3N4 with two types of cooperative N-vacancies (NHx and N2C) through simple one-pot potassium hydroxide assisted calcination [186]. The optimized photocatalyst exhibited a H2O2 yield of 152.6 µmol/h under simulated sunlight irradiation, which was 15-fold higher than that of pristine g-C3N4. Both the experimental and DFT results uncovered that NHx and N2C vacancies boosted the separation of photoinduced charge carriers as well as the activation of oxygen via an indirect two-step e– ORR pathway. Furthermore, Zheng and his co-authors designed a g-C3N4 with dual defect sites (–C≡N groups and N-vacancies) for the overall photosynthesis of H2O2 [179]. Based on the experimental results and DFT calculations, the authors discovered that the synergistic effect between –C≡N groups and N-vacancies enhanced the absorption of visible light and facilitated carrier separation. Specifically, the N-vacancies enhanced the adsorption and activation of O2, while the –C≡N groups facilitated the adsorption of H+, thereby synergistically promoting H2O2 production through the direct one-step 2e– ORR. However, it has recently been reported that constructing g-C3N4 with a high crystalline structure and eliminating defects can also significantly improve the photocatalytic activity of g-C3N4 [187]. For example, a highly crystalline g-C3N4 with extremely low defect concentrations was synthesized by a molten salt method. The highly crystalline g-C3N4 obtained by using thiourea as the precursor showed the highest H2O2 production rate (2.48 mmol g–1 h–1) and AQY of 22% at 400 nm. In another work, Shiraishi et al. explored the effect of surface defects of g-C3N4 on the photocatalytic activity of H2O2 synthesis [182]. It was found that the surface of mesopore g-C3N4 with a larger surface area has abundant primary amine groups and these defects served as active sites not only for the 4e– ORR to reduce the selectivity of H2O2, but also for decomposing the formed H2O2. Therefore, g-C3N4 with a relatively large surface area but extremely low defect concentrations is considered to promote efficient H2O2 production. Controlling defects on the g-C3N4 surface is therefore crucial for enhancing photocatalytic H2O2 production.
Constructing heterojunction is an effective approach to regulate the behaviour of charge carriers in g-C3N4, enhancing their spatial separation and migration. For example, Wang and his co-authors built a Z-scheme heterojunction PIx-NCN by assembling perylene imide (PI) on g-C3N4 nanosheets (NCN) [197]. The authors claimed that the photoexcited electrons transferred from the CB of perylene imide to the VB of NCN, promoting the charge separation and enabling more electrons for the reduction of O2 to generate H2O2. Most importantly, the perylene imide changed the photocatalytic H2O2 production from an indirect two-step e– ORR single approach to indirect two-step e– ORR and indirect two-step e– WOR hybrid approaches, thus significantly enhancing the H2O2 generation under visible light (Fig. 5f). In another study, Li et al. prepared a heptazine/triazine layer stacked g-C3N4 heterojunction with F/K dual sites (FKHTCN) [200]. It has been found that the formation of a heptazine/triazine heterostructure leads to a reduction in the interlayer distance of aromatic units, while the incorporation of F/K dual sites adjusts the layer-component ratio. As a result, the dissociation of interlayer exciton was promoted and the lifetime of charge carriers was extended. The introduced F/K dual sites were also found to enhance the adsorption of O2 and intermediate *OOH, optimizing the ORR pathway and consequently improving the photocatalytic production of H2O2 by 2e– ORR (Fig. 5g).
MOFs are a novel class of structurally defined porous organic polymers consisting of metal clusters (Ti, Zr, Ni, Cu, etc.) and organic ligands [207,208]. Since the first reported application of MOF-5 for photocatalytic reaction in 2007 [209], MOFs have been widely employed in photocatalytic elimination of organic contaminants [210], overall water splitting [211], CO2 reduction [212], and ammonia production [213] due to their high structural and functional tunability. In addition, some MOFs possess the properties of visible light response resulting from their amine-functionalized linkers [214-219], which greatly boost the utilization of solar energy. Therefore, MOFs are regarded as promising photocatalysts for H2O2 production.
The generation of H2O2 using MOF photocatalysts was first reported by Yamashita's group in 2018 [220]. The authors deposited NiO nanoparticles on MIL-125-NH2, a commonly used MOF composed of Ti8O8(OH)4 clusters and 2-aminoterephthalic acid ligands, to synthesize Ni/MIL-125-NH2. In the presence of electron donors (such as H2O, and benzyl alcohol), Ni/MIL-125-NH2 generated H2O2 via an indirect two-step e– ORR pathway, and the deposition of NiO accelerated the formation rate of H2O2. However, the photocatalytic activity of pristine MOFs is limited due to their short light adsorption edge (at 345 nm) [221].
Therefore, in pursuit of higher efficiency for H2O2 production, it is particularly important to develop visible-light-driven MOF-based photocatalysts that can both perform efficient H2O2 formation and suppress H2O2 decomposition. To this end, several strategies have been explored including surface modification, defect engineering, ion doping, and interfacial engineering to improve the photocatalytic activity of visible-light-driven MOFs for H2O2 production (Table S5 in Supporting information) [220,222-240].
To enhance the visible light absorption and photocatalytic activity of MIL-125-NH2 for H2O2 generation, Yan et al. synthesized Ce-doped MIL-125-NH2 by impregnation-coordination method [235]. The results demonstrated that the introduction of Ce optimized the electronic structure and band gap of MIL-125-NH2, and the formed Ce-O-Ti charge-transfer transitions caused a band-to-band red shift of absorption edge. Additionally, the Ce4+/Ce3+ and Ti4+/Ti3+ redox pairs acted as reaction sites and facilitated the transfer of charge carriers, thereby boosting the generation rate of H2O2 via indirect two-step e– ORR (Fig. 6a).
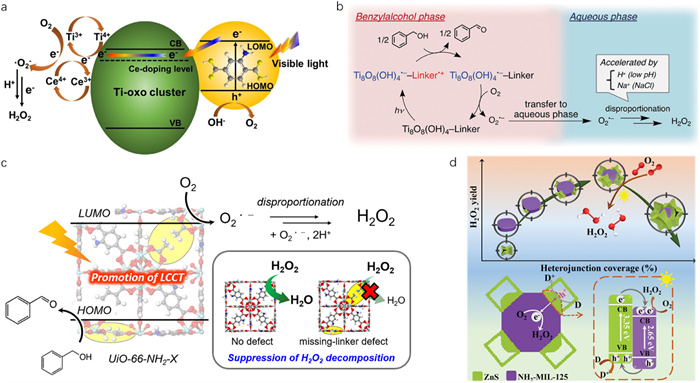
Recently, Yamashita and co-authors modified MIL-125-NH2 with various alkyl chains and achieved further-enhanced photocatalytic activity for H2O2 production [224,225]. It has been suggested that O2 was first reduced to O2•– radicals by TiⅢ species in the benzyl alcohol phase, and subsequently the formed O2•– radicals transferred to aqueous phase to generate H2O2. Besides, the production of H2O2 was accelerated in the presence of H+ and Na+ [224]. Therefore, by utilizing hydrophobic MOFs, spontaneous separation of products was achieved, thereby favouring the production of H2O2 (Fig. 6b).
In another example, Kondo et al. investigated the effect of missing-linker defects on the photocatalytic H2O2 production performance over UiO-66-NH2-X [236]. By adjusting the amount of acetic acid before solvothermal treatment, the concentration of missing linker defects introduced into the UiO-66-NH2 was altered. As a result, the formation of H2O2 via an indirect two-step e– ORR pathway was dramatically improved, while the decomposition of H2O2 was restrained (Fig. 6c).
Very recently, Liu et al. anchored ZnS on the corners of the NH2−MIL-125 body and constructed a NH2−MIL-125@ZnS heterojunction with tunable heterojunction coverage [238]. The obtained NH2−MIL-125@ZnS heterojunction with strong interface interactions is critical to photocatalytic H2O2 production. The heterojunction coverage serves as a vital parameter that affects the formation of e–-h+ pairs and charge separation. Additionally, it determines light absorption and the accessibility of photoinduced electrons. Consequently, the NH2−MIL-125@ZnS heterojunction, with an average heterojunction coverage of 45.1%, exhibited optimal photocatalytic activity, achieving a H2O2 production rate of 120 mM g–1 h–1 under visible light via indirect two-step e– ORR pathway (Fig. 6d).
Since their discovery in 2005, COFs have proven to be promising metal-free materials for their diverse structures and precise functionalities [241-243]. Of particular interest, 2D COFs structurally resemble graphite, with building blocks covalently bonded within the walls to form 2D planes and close π-π stacking between layers [244]. Such stacked 2D layered COFs form conductive columns, providing ideal channels for the rapid diffusion and transfer of photoinduced charge carriers [245].
In 2014, Stegbauer et al. first reported a hydrazone-based COF for the photocatalytic H2 evolution, with quantum efficiencies similar to g-C3N4 [246]. Subsequently, various COFs have been synthesized and used for a wide variety of photocatalytic reactions [26,247-250]. COFs emerged as extremely promising candidates for photocatalytic H2O2 production [251-253], exhibiting comparable photocatalytic activity to inorganic materials. In 2019, Xu's group utilized COFs for photocatalytic H2O2 production for the first time [251]. They designed three different 2D covalent triazine frameworks (CTFs), a branch of the COFs, by embedding acetylene or diacetylene moieties into CTFs (CTF-(1,1′-biphenyl)-4,4′-dicarbonitrile (CTF-BPDCN), CTF-4,4′-(ethyne-1,2-diyl)dibenzonitrile (CTF-EDDBN) and CTF-4,4′-(buta-1,3-diyne-1,4-diyl)dibenzonitrile (CTF-BDDBN)) to extend π-conjugation, thereby finely tuning the electronic structure of CTFs. The rotating disk electrode and EPR measurements results demonstrated all of these three CTFs reduced O2 into H2O2 by direct one-step 2e– ORR. The H218O isotopic labelling experiments identified that CTF-BPDCN oxidized H2O to O2 via 4e– WOR while CTF-EDDBN and CTF-BDDBN directly oxidized H2O to H2O2 through direct one-step 2e– WOR pathway. The authors claimed that acetylene and diacetylene units on CFTs significantly reduced the Gibbs free energy (ΔG) for the formation of OH* intermediates, enabling CTF-EDDBN and CTF-BDDBN to produce H2O2 from H2O via direct one-step 2e– WOR pathway. Therefore, different reaction pathways toward H2O2 photosynthesis using CTF-BPDCN, CTF-EDDBN, and CTF-BDDBN were proposed. Among them, CTF-BDDBN exhibited the highest photocatalytic H2O2 generation without any sacrificial agents. These acetylene and diacetylene functionalized CTFs realized 100% atom utilization efficiency, yet the SCC efficiency is still limited to 0.1%. Hence, much effort has been devoted to further advancing COFs and optimizing the photocatalytic generation of H2O2 (Table S6 in Supporting information) [30,46-48,50-52,251,252,254-294].
An effective strategy to functionalize COFs at a molecular level is to optimize their structures and properties by modifying the starting linker with functional groups, such as thiourea [261], piperazine [256], sulfone [262,295], thiophene [259] and diazine [260]. For example, Han and co-workers found that thiourea functionalized CTF (Bpt-CTF) functioned as an active photocatalyst for H2O2 production even without sacrificial agents [261]. By introducing the thiourea functional group into COFs, the charge separation/transport and proton transfer were greatly improved, resulting in a high H2O2 production rate (3268.1 µmol h–1 g–1) with an impressive AQY of 8.6% (at 400 nm). H2O2 was produced via coupled direct one-step 2e– ORR with the 4e– WOR pathway. In another example, Zhi et al. synthesized piperazine-linked porous CoPc-based COFs, which showed strong visible light absorption (at 400–1000 nm) and enhanced the separation/transfer efficiency of photoinduced charge carriers, thereby boosting the H2O2 production via indirect two-step e– ORR with a rate of 2096 µmol h–1 g–1 [256]. Luo et al. also achieved an optimized visible-light-driven photocatalytic H2O2 production yield of 3904.2 µmol h–1 g–1 without any sacrificial agents by grafting sulfone functional group onto COFs (FS-COFs) [262]. Based on experimental results and theoretical calculations, the authors found that modification of the sulfone functional group not only accelerated the separation of photoinduced charge carriers but also enhanced the protonation of COFs and boosted the adsorption of O2 molecular in a "side-on" Yeager-type. As a result, FS-COFs altered the photocatalytic ORR process from indirect two-step e– ORR to direct one-step 2e– ORR, achieving efficient H2O2 production (Fig. 7a). Moreover, Yue et al. designed and prepared two thiophene-containing COFs (TDCOF and TT-COF), which realized overall photosynthetic H2O2 production by indirect two-step e– ORR and direct one-step 2e– WOR dual-channel pathways in the absence of sacrificial agents [259]. By adjusting the N-heterocycle units (pyridine and triazine) in COFs, the generation of H2O2 was regulated. Using only air and water as the reactants without the addition of sacrificial agents, the photocatalytic H2O2 generation rates over TDCOF in natural seawater and deionized water were 3364 µmol g–1 h–1 and 4060 µmol g–1 h–1, respectively. The H2O2 yield in natural seawater is slightly lower than that in deionized water due to the presence of plenty of ions, which may interfere with the production or cause the decomposition of H2O2. In addition, theoretical calculations confirmed that thiophene was the primary photoreduction unit for ORR, while the benzene ring connected to thiophene through an imine bond was the main photooxidation unit for WOR. Very recently, Xi and co-workers synthesized a serious of COFs functionalized with different diazines, including pyridazine, pyrazine, and pyrimidine (denoted as TpDz, TpPz and TpMd) and in-depth studied the effects of relative nitrogen locations of diazines functionalized COFs on the photocatalytic production of H2O2 from O2 and pure water [260]. The intercalation of pyridazine in TpDz preferred to stabilize the endoperoxide intermediate compared to pyrimidine and pyrazine, leading to a more efficient direct one-step 2e– ORR pathway. In addition, TpDz possessed higher charge carrier separation efficiency and lower charge transfer resistance. Consequently, TpDz produced H2O2 via the direct one-step 2e– ORR and 4e– WOR pathways with a photocatalytic H2O2 yield of 7327 µmol h–1 g–1 and 0.62% SCC efficiency under visible light irradiation (λ > 420 nm).
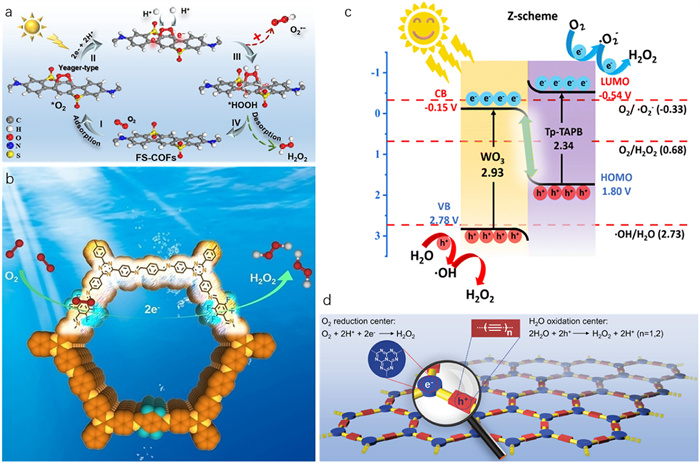
As another alternative strategy, ion doping is a traditional method to modulate the electronic band filling and the transport of charge carriers in organic polymers, thereby improving the photocatalytic activity for H2O2 production [296]. For instance, Wang et al. developed and designed a highly crystalline fluorine-doped COF (TF-COF) [283]. By partially fluorinating the edge of aromatic parts of 2D COF (TF50–COF), plenty of Lewis acid sites were generated, adjusting the electron distribution of adjacent C atoms. TF50–COF with AA stacking mode was found to exhibit the strongest π-π interactions and the highest crystallinity compared to non-fluorinated COF and TF-COF, thereby effectively promoting the separation and transfer of charge carriers. The UV–vis absorption spectra showed that the visible-light-responsive range of TF50–COF was also widened. Furthermore, the adsorption of O2 was significantly enhanced due to the formation of *OOH intermediates. Consequently, the TF50–COF exhibited remarkable photocatalytic H2O2 performance (1739 µmol h–1 g–1) with an AQY of 5.1% at 400 nm via indirect two-step e– ORR H2O2 production approach (Fig. 7b). Very recently, Guo's team rationally engineered F-containing COFs (TAPT-TFPA COFs) to strongly confine Pd metal-isolated clusters (ICs) for enhanced H2O2 photosynthesis [284]. The results showed that the strong electronegative F in TAPT-TFPA COFs@Pd ICs not only strengthened the metal-support interaction for improving the photochemical stability but also optimized the D-band centre of Pd ICs. Therefore, TAPT-TFPA COFs@Pd ICs exhibited an outstanding photocatalytic H2O2 production activity (2143 µmol h–1 g–1), spectacular SCC efficiency (0.82%), and high stability over 100 h in an aqueous solution containing 10% ethanol under simulated solar irradiation (AM 1.5). H2O2 was formed via an indirect two-step e– ORR pathway.
Additionally, the photocatalytic H2O2 production using COFs can be significantly boosted by the construction of heterojunctions. Yao and co-workers constructed ZnIn2S4/TpPa-1 heterostructure for photocatalytic H2O2 evolution in air [285]. Under visible light irradiation, this hybrid photocatalyst efficiently absorbed visible light and captured O2 in air, achieving effective H2O2 production via an indirect two-step e– ORR pathway. In another study, Yang et al. prepared WO3/Tp-TAPB heterojunction and observed a high H2O2 production rate in pure water, which was 72.3- and 2.8-fold than that of WO3 and Tp-TAPB, respectively [288]. The heterojunction structure possessed a large specific surface area, providing increased active sites. Additionally, it facilitated the separation and transfer of charge carriers and maintained a high redox potential. Ultimately, the WO3/Tp-TAPB achieved H2O2 formation with high efficiency via indirect two-step e– ORR and indirect two-step e– WOR two-channel pathways (Fig. 7c).
Construction donor-acceptor (D-A) structure is one of the common and valid approaches to affect the efficiency of photo-capture, and photoinduced excitons splitting in COFs [297,298]. This is because the existence of a polarization electric field facilitates the separation of photoinduced charge carriers in a D-A structure through intramolecular charge transfer. Additionally, this unique spatial separation feature greatly reduces the exciton binding energy, thereby inhibiting the recombination of the charge carriers [299]. Thus, in recent years, various donor and acceptor units, especially alternating donor and acceptor building blocks with planar structure, have been designed to build D-A configuration in COFs, achieving considerable photocatalytic activity in H2O2 production. For example, Xu and coworkers first reported heptazine-based COFs (CHF-DPA and CHF-DPDA) in 2021, which possessed spatially separated redox centres and efficiently produced H2O2 from H2O and O2 under visible light irradiation without additional sacrificial agents [46]. They used femtosecond time-resolved transient absorption and X-ray absorption near-edge structure (XANES) spectroscopy to study the photoexcited carrier dynamics and charge transfer process in these heptazine-based COFs and found that photoinduced e– was concentrated in s-heptazine units, whereas photoinduced h+ was accumulated in acetylene or diacetylene parts. Moreover, theoretical and experimental results confirmed that the C atoms in s-heptazine units acted as the reduction centres favouring the formation of *H and *OO* intermediates, leading to direct one-step 2e– ORR pathway, while the C atoms in acetylene or diacetylene groups most likely served as oxidation active sites, beneficial for the generation of OH* intermediates, resulting in direct one-step 2e– WOR pathway (Fig. 7d). As reported, the high-N-content s-heptazine and triazine moieties often serve as the O2 reduction centres for H2O2 generation due to their ability to stabilize suitable intermediates [261,290]. Besides, benzene groups were proven to be the reaction centre for the oxidation of H2O to generate O2 [300]. Inspired by this, Chen et al. designed and prepared a 2D s-heptazine-based COFs (HEP-TAPT-COF) with spatially separated redox centres at the molecular level for the production of H2O2 from O2 and pure water [48]. HEP-TAPB-COF, integrating dual O2 reduction centres of triazine and s-heptazine units, showed excellent photocatalytic performance in producing H2O2 with a 0.65% SCC efficiency and an AQY of 15.35% at 420 nm. The experimental and DFT investigations demonstrated that the spatially separated redox centres facilitated the separation of charge carriers in this conjugated structure. As a result, HEP-TAPB-COF reduced O2 toward H2O2 with high selectivity via a direct one-step 2e– ORR pathway and directly oxidised H2O into O2 by 4e– WOR pathway. Additionally, the H218O isotopic labelling experiments confirmed that the O2 generated from H2O oxidation in turn as a reactant to promote H2O2 generation. In another study, Chang et al. prepared an oxidation–reduction molecular junction TTF-BT-COF by covalent coupling of tetrathiafulvalene (TTF) and benzothiazole (BT) [52]. Interestingly, TTF groups served as oxidation sites and BT parts acted as reduction sites, achieving the full reaction of photosynthesis for H2O2 generation through direct one-step 2e– ORR and indirect two-step e– WOR two-channel pathways. Tong's group also designed and synthesized a novel D-A configuration COFs by optimizing the intramolecular polarity of COFs by introducing an appropriate amount of phenyl moieties as the electron donors, which significantly increased the direct photosynthesis of H2O2 from pure water and air [50]. Theoretical and experimental results revealed that appropriate N 2p and C 2p states with optimal intramolecular polarity in COF-N32 effectively lowered the energy barrier for H2O oxidation and O2 reduction, respectively. Without the use of any sacrificial agent, COF-N32 exhibited a high photocatalytic H2O2 production rate (605 µmol g–1 h–1), SCC efficiency of 0.31% and AQY of 6.2% at 459 nm. H2O2 was formed by coupling the indirect two-step e– ORR and indirect two-step e– WOR dual pathways. Very recently, Wang and coworkers prepared a benzotrithiophene-based D-A configuration COFs (TaptBtt) for H2O2 photosynthesis from H2O and O2 under visible light irradiation without additional sacrificial agents [30]. Due to the spatially separated redox centres, the reduction sites were predominantly centred on C atoms in the imine bond of TaptBtt, which regulated the direction of charge transfer and the energy difference of intramolecular D-A. This further reduced the Gibbs free energy of OH* and OOH* intermediates, facilitating the generation of H2O2 via the synchronous indirect two-step e– ORR and direct one-step 2e– WOR two-channel pathways.
As the global demand for H2O2 rises sharply, it is particularly important to develop stable and efficient photocatalysts for H2O2 production. In this review, we thoroughly outline the recent progress in designing 2D photocatalysts, including inorganic materials (e.g., metal oxides, metal chalcogenides and bismuth-based materials) and organic polymers (e.g., g-C3N4, MOFs and COFs), for the high-efficiency photocatalytic H2O2 production. Moreover, common methods for detecting and quantifying H2O2 are presented. We have also highlighted various rational and effective strategies to promote photocatalytic H2O2 production and discussed the related mechanisms. Specifically, strategies including surface modification, ion doping, defect engineering, and heterojunction construction are employed to expand the light absorption range, retain the high redox capacity, boost the separation/transfer of charge carriers and enhance the adsorption of O2 and redox sites entrenchment.
Although significant progress has been achieved in photocatalytic H2O2 production, practical and large-scale industrial production remains elusive, and several key challenges still need to be addressed.
Firstly, there is a need to gain a comprehensive understanding of the mechanisms underlying efficient and sustainable generation of H2O2 [27,301]. The interplay between oxygen reduction and water oxidation half-reactions plays a crucial role in governing the kinetics and efficiency of photocatalytic H2O2 production [293]. It is worth noting that previous studies have primarily focused on ORR, while the significance of WOR has often been overlooked, despite its equal importance [25,302-305]. Up to now, the high-efficiency photocatalytic ORR for producing H2O2 has been achieved, but it typically requires the addition of sacrificial agents like methanol, ethanol, benzyl alcohol, or isopropanol. However, the use of these sacrificial agents may lead to the generation of by-products and complicate the subsequent separation and purification of H2O2 [306]. Alternatively, exploring the coupling of ORR with other oxidation reactions to simultaneously generate value-added products could be considered [293]. Therefore, more attention should be given to WOR and non-sacrificial H2O2 photosynthesis in the future, even though water oxidation presents more challenges [17]. In this scenario, it is imperative to investigate both ORR and WOR to optimize the reaction process, particularly the meticulous integration of them, to achieve simultaneous ORR and WOR pathways for efficient photocatalytic H2O2 generation [307,308]. Furthermore, employing advanced time-resolved or space-resolved characterization methods (e.g., time-resolved photoluminescence, transient absorption spectroscopy, transient electron paramagnetic resonance, in situ X-ray photoelectron spectroscopy, and Kelvin probe force microscopy) is crucial to observe the charge transfer behaviour [309]. Additionally, conducting in situ monitoring of reaction intermediates (e.g., in situ diffuse reflectance infrared Fourier transform spectroscopy) during the photocatalytic H2O2 production process is beneficial [30,50]. If necessary, theoretical calculations should be combined with experimental observations to better guide the fabrication of photocatalysts for efficient H2O2 generation.
Secondly, there is a need to explore and develop 2D photocatalysts with high solar-to-H2O2 conversion efficiency. Despite the enormous variety of 2D photocatalysts reported to date, including metal oxides, metal chalcogenides, bismuth-based materials, g-C3N4, MOFs and COFs, many face challenges such as insufficient light absorption ability, rapid charge recombination, and kinetically sluggish WOR half-reaction (Table 2). As a result, their SCC efficiency generally remains lower than 0.5% [308]. One approach to address this challenge is to precisely engineer 2D photocatalysts with suitable band positions and bandgaps to ensure broad light absorption ranges, high efficiency in charge carrier separation/transfer, and strong redox capabilities [255,260]. In this context, the detection of the electronic band structures of 2D photocatalysts becomes imperative. Angle-resolved photoemission spectroscopy, an emerging energy- and momentum-resolved technique, directly visualizes electronic band structures and provides information on band gaps, band edge positions, Fermi surface, and spin polarization properties [310]. Additionally, it is crucial to rationally design and modify 2D photocatalysts to simultaneously accelerate both the oxygen reduction and water oxidation reactions [46]. This design strategy is grounded in a deep understanding of the mechanism underlying H2O2 production. Incorporating multiple strategies, including modification, doping, defect, and interfacial engineering, without limitation to a particular approach, is essential to simultaneously modulate the three key steps of light absorption, charge separation/transfer, and surface catalytic reactions in preparing photocatalysts for high-efficiency H2O2 photosynthesis [178,200].
Thirdly, accurately evaluating and reporting the activity of photocatalysts for H2O2 production is crucial. Despite recent progress in developing efficient 2D photocatalysts for H2O2 synthesis, the absence of a standardized evaluation method impedes meaningful comparisons between different catalyst systems and even across laboratories, posing a significant obstacle to the advancement of 2D photocatalysts [311,312]. Currently, in the study of photocatalytic H2O2 synthesis, the main evaluation parameter is the reaction rate [253]. However, the H2O2 production rate is influenced by factors such as light source intensity, incident light wavelength range, reactor type, and reaction temperature. Therefore, subsequent studies need to provide sufficient and accurate experimental details, including the AQY and SCC efficiency.
Fourthly, the stability of photocatalysts is paramount. Over extended periods of light exposure, photocatalysts often encounter issues such as photocorrosion, aggregation, or other stability concerns, especially given that the product, H2O2, is a potent oxidant [313]. Inorganic photocatalysts, such as metal oxides, may experience corrosion or metal leaching upon prolonged contact with accumulated H2O2 [17]. Additionally, radicals like O2•– generated through the indirect two-step e– ORR pathway and •OH produced by the indirect two-step e– WOR pathway may lead to the decomposition of polymer photocatalysts [314]. We believe that further exploration of photocatalysts with long-term stability will be beneficial for the practical applications of photocatalytic H2O2 production.
Fifthly, the photocatalytic H2O2 production process is often accompanied by in situ H2O2 decomposition [315]. The generated H2O2 absorbs UV light and directly decomposes under the illumination of sunlight (2H2O2 → 2H2O + O2) [33]. Furthermore, H2O2 can undergo redox to generate •OH radical (H2O2 + e– → •OH + OH–) and O2•– radical (H2O2 + h+ → O2•– + 2H+) [306]. Hence, future research should focus on developing photocatalytic systems driven by visible light and avoiding the decomposition of in situ formed H2O2 [231,233]. Alternatively, combining with Fenton technology to convert the formed H2O2 into reactive oxygen species [316], such as •OH and O2•– radicals, could enable rapid sterilization [317], photodegradation of volatile organic compounds [318], and cancer therapeutic [319].
The comprehensive summary and insightful discussion provided in this timely review on the photocatalytic production of H2O2 by 2D photocatalysts would contribute to advancing the future of efficient and sustainable H2O2 generation.
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Liyong Ding: Writing – review & editing, Writing – original draft. Zhenhua Pan: Conceptualization, Writing – review & editing. Qian Wang: Writing – review & editing, Writing – original draft, Supervision.
This work was supported by the National Natural Science Foundation of China (No. 22106087), the Joint Funds of the Zhejiang Provincial Natural Science Foundation of China (No. LZY22B070001), the Foundation of China Scholarship Council (No. 202208330186), the JST Fusion Oriented Research for disruptive Science and Technology Program (No. JPMJFR213D), and JSPS Leading Initiative for Excellent Young Researchers program. In addition, Liyong Ding thanked Yu Chen for her understanding and support.
Supplementary material associated with this article can be found, in the online version, at doi:
J. Campos-Martin, G. Blanco-Brieva, J. Fierro, Angew. Chem. Int. Ed. 45 (2006) 6962–6984. doi: 10.1002/anie.200503779
L. Pi, J. Cai, L. Xiong, et al., Chem. Eng. J. 389 (2020) 123420. doi: 10.1016/j.cej.2019.123420
Z. Lu, G. Chen, S. Siahrostami, et al., Nat. Catal. 1 (2018) 156–162. doi: 10.1038/s41929-017-0017-x
Research and Markets, Hydrogen Peroxide Global Market Report 2023,
P. Landon, P. Collier, A. Papworth, et al., Chem. Commun. (2002) 2058–2059.
J. Lunsford, J. Catal. 216 (2003) 455–460. doi: 10.1016/S0021-9517(02)00070-2
Y. Sun, L. Han, P. Strasser, Chem. Soc. Rev. 49 (2020) 6605–6631. doi: 10.1039/d0cs00458h
Y. Tian, D. Deng, L. Xu, et al., Nano-Micro Lett. 15 (2023) 122. doi: 10.1007/s40820-023-01067-9
S. Qu, H. Wu, Y. Ng, Adv. Energy Mater. 13 (2023) 2301047. doi: 10.1002/aenm.202301047
N. Kaynan, B. Berke, O. Hazut, R. Yerushalmi, J. Mater. Chem. A 2 (2014) 13822–13826. doi: 10.1039/C4TA03004D
T. Liu, Z. Pan, J. Vequizo, et al., Nat. Commun. 13 (2022) 1034. doi: 10.1038/s41467-022-28686-x
Y. Zhang, C. Pan, G. Bian, et al., Nat. Energy 8 (2023) 361–371. doi: 10.1038/s41560-023-01218-7
Y. Ye, J. Pan, F. Xie, et al., Proc. Natl. Acad. Sci. U. S. A. 118 (2021) e2103964118. doi: 10.1073/pnas.2103964118
H. Hou, X. Zeng, X. Zhang, Angew. Chem. Int. Ed. 59 (2020) 17356–17376. doi: 10.1002/anie.201911609
X. Zhang, H. Su, P. Cui, et al., Nat. Commun. 14 (2023) 7115. doi: 10.1038/s41467-023-42887-y
H. Huang, Q. Zhang, R. Shi, et al., Appl. Catal. B: Environ. 317 (2022) 121731. doi: 10.1016/j.apcatb.2022.121731
S. Wu, X. Quan, ACS ES&T Eng. 2 (2022) 1068–1079. doi: 10.1021/acsestengg.1c00456
L. Wang, J. Zhang, Y. Zhang, et al., Small 18 (2022) 2104561. doi: 10.1002/smll.202104561
L. Zhang, J. Zhang, H. Yu, J. Yu, Adv. Mater. 34 (2022) e2107668. doi: 10.1002/adma.202107668
X. Xu, Y. Sui, W. Chen, et al., Appl. Catal. B: Environ. 341 (2024) 123271. doi: 10.1016/j.apcatb.2023.123271
C. Feng, Z. Wu, K. Huang, et al., Adv. Mater. 34 (2022) 2200180. doi: 10.1002/adma.202200180
L. Wang, H. Xu, Prog. Poly. Sci. 145 (2023) 101734. doi: 10.1016/j.progpolymsci.2023.101734
Y. Zhou, Z. Wang, L. Huang, et al., Adv. Energy Mater. 11 (2021) 2003159. doi: 10.1002/aenm.202003159
Y. Guo, X. Tong, N. Yang, Nano-Micro Lett. 15 (2023) 77. doi: 10.1007/s40820-023-01052-2
X. Zeng, Y. Liu, X. Hu, X. Zhang, Green Chem. 23 (2021) 1466–1494. doi: 10.1039/d0gc04236f
Z. Yong, T. Ma, Angew. Chem. Int. Ed. 62 (2023) e202308980. doi: 10.1002/anie.202308980
H. Cheng, J. Cheng, L. Wang, H. Xu, Chem. Mater. 34 (2022) 4259–4273. doi: 10.1021/acs.chemmater.2c00936
J. Ma, X. Peng, Z. Zhou, et al., Chin. Chem. Lett. 34 (2023) 108784. doi: 10.1016/j.cclet.2023.108784
J. Tang, T. Zhao, D. Solanki, et al., Joule 5 (2021) 1432–1461. doi: 10.1016/j.joule.2021.04.012
C. Qin, X. Wu, L. Tang, et al., Nat. Commun. 14 (2023) 5238. doi: 10.1038/s41467-023-40991-7
E. Baur, C. Neuweiler, Helv. Chim. Acta 10 (1927) 901–907. doi: 10.1002/hlca.192701001113
T. Gill, X. Zheng, Chem. Mater. 32 (2020) 6285–6294. doi: 10.1021/acs.chemmater.0c02010
Y. Nosaka, A. Nosaka, Chem. Rev. 117 (2017) 11302–11336. doi: 10.1021/acs.chemrev.7b00161
G. Eisenberg, Ind. Eng. Chem. Anal. Ed. 15 (1943) 327–328. doi: 10.1021/i560117a011
V. Diesen, M. Jonsson, J. Phys. Chem. C 118 (2014) 10083–10087. doi: 10.1021/jp500315u
K. Sahel, L. Elsellami, I. Mirali, et al., Appl. Catal. B: Environ. 188 (2016) 106–112. doi: 10.1016/j.apcatb.2015.12.044
Y. Zhao, P. Zhang, Z. Yang, et al., Nat. Commun. 12 (2021) 3701. doi: 10.1038/s41467-021-24048-1
C. Marquette, L. Blum, Anal. Bioanal. Chem. 385 (2006) 546–554. doi: 10.1007/s00216-006-0439-9
T. Hirakawa, Y. Nosaka, J. Phys. Chem. C 112 (2008) 15818–15823. doi: 10.1021/jp8055015
C. Chu, Q. Zhu, Z. Pan, et al., Proc. Natl. Acad. Sci. U. S. A. 117 (2020) 6376–6382. doi: 10.1073/pnas.1913403117
A. Keston, R. Brandt, Anal. Biochem. 11 (1965) 1–5. doi: 10.1016/0003-2697(65)90034-5
C. Bernardini, G. Cappelletti, M. Dozzi, E. Selli, J. Photochem. Photobiol. A 211 (2010) 185–192. doi: 10.1016/j.jphotochem.2010.03.006
C. Huckaba, F. Keyes, J. Am. Chem. Soc. 70 (1948) 1640–1644. doi: 10.1021/ja01184a098
E. Jung, H. Shin, B. Lee, et al., Nat. Mater. 19 (2020) 436–442. doi: 10.1038/s41563-019-0571-5
X. Shi, S. Siahrostami, G. Li, et al., Nat. Commun. 8 (2017) 701. doi: 10.1038/s41467-017-00585-6
H. Cheng, H. Lv, J. Cheng, et al., Adv. Mater. 34 (2022) 2107480. doi: 10.1002/adma.202107480
G. Li, P. Fu, Q. Yue, et al., Chem. Catal. 2 (2022) 1734–1747. doi: 10.1016/j.checat.2022.05.002
D. Chen, W. Chen, Y. Wu, et al., Angew. Chem. Int. Ed. 62 (2023) e202217479. doi: 10.1002/anie.202217479
X. Shi, Y. Zhang, S. Siahrostami, X. Zheng, Adv. Energy Mater. 8 (2018) 1801158. doi: 10.1002/aenm.201801158
F. Liu, P. Zhou, Y. Hou, et al., Nat. Commun. 14 (2023) 4344. doi: 10.1038/s41467-023-40007-4
Y. Mou, X. Wu, C. Qin, et al., Angew. Chem. Int. Ed. 62 (2023) e202309480. doi: 10.1002/anie.202309480
J. Chang, Q. Li, J. Shi, et al., Angew. Chem. Int. Ed. 62 (2023) e202218868. doi: 10.1002/anie.202218868
G. Moon, W. Kim, A. Bokare, et al., Energy Environ. Sci. 7 (2014) 4023–4028. doi: 10.1039/C4EE02757D
R. Nogueira, M. Oliveira, W. Paterlini, Talanta 66 (2005) 86–91. doi: 10.1016/j.talanta.2004.10.001
F. Kuttassery, Y. Ohsaki, A. Thomas, et al., Angew. Chem. Int. Ed. 62 (2023) e202308956. doi: 10.1002/anie.202308956
V. Sanz, S. Marcos, J. Castillo, J. Galbán, J. Am. Chem. Soc. 127 (2005) 1038–1048. doi: 10.1021/ja046830k
M. Zhou, Z. Diwu, N. Panchuk-Voloshina, R. Haugland, Anal. Biochem. 253 (1997) 162–168. doi: 10.1006/abio.1997.2391
Z. Wu, M. Liu, Z. Liu, Y. Tian, J. Am. Chem. Soc. 142 (2020) 7532–7541. doi: 10.1021/jacs.0c00771
S. Du, J. Lian, F. Zhang, Trans. Tianjin Univ. 28 (2021) 33–52.
J. Harbour, J. Tromp, M. Hair, Can. J. Chem. 63 (1985) 204–208. doi: 10.1139/v85-032
V. Maurino, C. Minero, G. Mariella, E. Pelizzetti, Chem. Commun. (2005) 2627–2629. doi: 10.1039/b418789j
M. Teranishi, S. Naya, H. Tada, J. Am. Chem. Soc. 132 (2010) 7850–7851. doi: 10.1021/ja102651g
E. Goliaei, N. Seriani, J. Phys. Chem. C 123 (2019) 2855–2863. doi: 10.1021/acs.jpcc.8b09300
Y. Liu, X. Zeng, X. Hu, et al., Catal. Today 335 (2019) 243–251. doi: 10.1016/j.cattod.2018.11.053
Y. Liu, X. Zeng, C. Easton, et al., Nanoscale 12 (2020) 8775–8784. doi: 10.1039/d0nr01611j
B. Liu, C. Bie, Y. Zhang, et al., Langmuir 37 (2021) 14114–14124. doi: 10.1021/acs.langmuir.1c02360
C. Zhu, M. Zhu, Y. Sun, et al., ACS Appl. Energy Mater. 2 (2019) 8737–8746. doi: 10.1021/acsaem.9b01712
C. Fu, L. Liu, Z. Li, et al., J. Phys. Chem. Lett. 14 (2023) 7690–7696. doi: 10.1021/acs.jpclett.3c01865
Y. Wang, Y. Wang, J. Zhao, et al., Appl. Catal. B: Environ. 284 (2021) 119691. doi: 10.1016/j.apcatb.2020.119691
W. Xie, Z. Huang, R. Wang, et al., J. Mater. Sci. 55 (2020) 11829–11840. doi: 10.1007/s10853-020-04837-7
D. Tsukamoto, A. Shiro, Y. Shiraishi, et al., ACS Catal. 2 (2012) 599–603. doi: 10.1021/cs2006873
M. Teranishi, R. Hoshino, S. Naya, H. Tada, Angew. Chem. Int. Ed. 55 (2016) 12773–12777. doi: 10.1002/anie.201606734
S. Cao, T. Chan, Y. Lu, et al., Nano Energy 67 (2020) 104287. doi: 10.1016/j.nanoen.2019.104287
X. Zeng, Z. Wang, N. Meng, et al., Appl. Catal. B: Environ. 202 (2017) 33–41. doi: 10.1016/j.apcatb.2016.09.014
Y. Yang, Q. Wang, X. Zhang, et al., J. Mater. Chem. A 11 (2023) 1991–2001. doi: 10.1039/d2ta08145h
J. Wang, J. Wang, S. Zuo, et al., Chin. Chem. Lett. 34 (2023) 108157. doi: 10.1016/j.cclet.2023.108157
B. He, Z. Wang, P. Xiao, et al., Adv. Mater. 34 (2022) 2203225. doi: 10.1002/adma.202203225
X. Dang, S. Wu, H. Zhao, ACS Sustain. Chem. Eng. 10 (2022) 4161–4172. doi: 10.1021/acssuschemeng.1c07985
G. Han, F. Xu, B. Cheng, et al., Acta Phys. Chim. Sin. 38 (2022) 2112037. doi: 10.3866/pku.whxb202112037
X. Meng, P. Zong, L. Wang, et al., Catal. Commun. 134 (2020) 105860. doi: 10.1016/j.catcom.2019.105860
Z. Jiang, B. Cheng, Y. Zhang, et al., J. Mater. Sci. Technol. 124 (2022) 193–201. doi: 10.1016/j.jmst.2022.01.029
B. Mishra, L. Biswal, S. Das, et al., Langmuir 39 (2023) 957–971. doi: 10.1021/acs.langmuir.2c02315
X. Chen, W. Zhang, L. Zhang, et al., J. Mater. Chem. A 8 (2020) 18816–18825. doi: 10.1039/d0ta05753c
L. Feng, B. Li, Y. Xiao, et al., Catal. Commun. 155 (2021) 106315. doi: 10.1016/j.catcom.2021.106315
X. Yan, G. Yu, C. Xing, et al., Catal. Sci. Technol. 13 (2023) 3094–3105. doi: 10.1039/d3cy00235g
X. Chen, W. Zhang, L. Zhang, et al., ACS Appl. Mater. Interfaces 13 (2021) 25868–25878. doi: 10.1021/acsami.1c02953
S. Mansingh, K. Das, N. Priyadarshini, et al., Energy Fuels 37 (2023) 9873–9894. doi: 10.1021/acs.energyfuels.3c00717
J. Low, J. Yu, M. Jaroniec, et al., Adv. Mater. 29 (2017) 1601694. doi: 10.1002/adma.201601694
C. Pan, Z. Mao, X. Yuan, et al., Adv. Sci. 9 (2022) 2105747. doi: 10.1002/advs.202105747
X. Yue, J. Fan, Q. Xiang, Adv. Funct. Mater. 32 (2021) 2110258.
X. Mao, X. Lang, Z. Wang, et al., J. Phys. Chem. Lett. 4 (2013) 3839–3844. doi: 10.1021/jz402053p
W. Wang, Q. Song, Q. Luo, et al., Nat. Commun. 14 (2023) 2493. doi: 10.1002/ps.7431
F. Haque, T. Daeneke, K. Kalantar-zadeh, J. Ou, Nano-Micro Lett. 10 (2018) 23. doi: 10.1007/s40820-017-0176-y
H. Song, L. Wei, C. Chen, et al., J. Catal. 376 (2019) 198–208. doi: 10.1016/j.jcat.2019.06.015
G. Jiang, X. You, B. An, et al., Appl. Surf. Sci. 618 (2023) 156656. doi: 10.1016/j.apsusc.2023.156656
Y. Yang, H. Guo, D. Huang, et al., Chem. Eng. J. 479 (2024) 147863. doi: 10.1016/j.cej.2023.147863
A. Tikoo, N. Lohia, S. Kondeti, P. Meduri, J. Mater. Chem. A 11 (2023) 14887–14899. doi: 10.1039/d3ta02875e
W. Fang, L. Wang, X. Meng, C. Li, J. Alloy. Compd. 947 (2023) 169606. doi: 10.1016/j.jallcom.2023.169606
Y. Li, Y. Zhao, H. Nie, et al., J. Mater. Chem. A 9 (2021) 515–522. doi: 10.1039/d0ta10231h
R. Li, D. Zhang, Y. Shi, et al., J. Catal. 416 (2022) 322–331. doi: 10.1016/j.jcat.2022.11.016
Y. Yang, H. Yu, M. Wu, et al., Appl. Catal. B: Environ. 325 (2023) 122307. doi: 10.1016/j.apcatb.2022.122307
Y. Li, R. Jia, H. Lin, et al., Adv. Funct. Mater. 31 (2020) 2008420.
S. Thakur, T. Kshetri, N. Kim, J. Lee, J. Catal. 345 (2017) 78–86. doi: 10.5958/2455-7129.2017.00011.5
Z. Tian, C. Han, Y. Zhao, et al., Nat. Commun. 12 (2021) 2039. doi: 10.1038/s41467-021-22394-8
B. Jiang, D. Chen, N. Li, et al., Ind. Eng. Chem. Res. 62 (2023) 12974–12984. doi: 10.1021/acs.iecr.3c01800
A. Tikoo, A. Koushik, P. Meduri, ACS Appl. Eng. Mater. 1 (2023) 1397–1407. doi: 10.1021/acsaenm.3c00077
H. Chen, Y. Xing, S. Liu, et al., Chem. Eng. J. 462 (2023) 142038. doi: 10.1016/j.cej.2023.142038
Y. Yang, B. Cheng, J. Yu, et al., Nano Res. 16 (2021) 4506–4514. doi: 10.3390/app11104506
Z. Wang, B. Mi, Environ. Sci. Technol. 51 (2017) 8229–8244. doi: 10.1021/acs.est.7b01466
K. Mak, C. Lee, J. Hone, et al., Phys. Rev. Lett. 105 (2010) 136805. doi: 10.1103/PhysRevLett.105.136805
M. Bernardi, M. Palummo, J. Grossman, Nano Lett. 13 (2013) 3664–3670. doi: 10.1021/nl401544y
U. Maitra, U. Gupta, M. De, et al., Angew. Chem. Int. Ed. 52 (2013) 13057–13061. doi: 10.1002/anie.201306918
F. Bussolotti, J. Yang, H. Kawai, et al., ACS Nano 15 (2021) 2686–2697. doi: 10.1021/acsnano.0c07982
L. Ding, R. Wei, H. Chen, et al., Appl. Catal. B: Environ. 172 (2015) 91–99.
L. Ding, M. Li, Y. Zhao, et al., Appl. Catal. B: Environ. 266 (2020) 118634. doi: 10.1016/j.apcatb.2020.118634
X. Wu, H. Tan, C. Zhang, et al., Prog. Mater. Sci. 133 (2023) 101047. doi: 10.1016/j.pmatsci.2022.101047
P. Chen, H. Liu, W. Cui, et al., EcoMat 2 (2020) e12047. doi: 10.1002/eom2.12047
H. Hirakawa, S. Shiota, Y. Shiraishi, et al., ACS Catal. 6 (2016) 4976–4982. doi: 10.1021/acscatal.6b01187
K. Wang, M. Wang, J. Yu, et al., ACS Appl. Nano Mater. 4 (2021) 13158–13166. doi: 10.1021/acsanm.1c02688
H. Shi, Y. Li, K. Wang, et al., Chem. Eng. J. 443 (2022) 136429. doi: 10.1016/j.cej.2022.136429
H. Shi, Y. Li, X. Wang, et al., Appl. Catal. B: Environ. 297 (2021) 120414. doi: 10.1016/j.apcatb.2021.120414
K. Wang, Y. Li, H. Shi, et al., Adv. Sustain. Syst. 6 (2022) 2200144. doi: 10.1002/adsu.202200144
K. Fuku, R. Takioka, K. Iwamura, et al., Appl. Catal. B: Environ. 272 (2020) 119003. doi: 10.1016/j.apcatb.2020.119003
D. Dai, X. Bao, Q. Zhang, et al., Chem. Eur. J. 29 (2023) e202203765. doi: 10.1002/chem.202203765
M. Sun, X. Wang, H. Pan, et al., J. Colloid Interf. Sci. 629 (2023) 215–224. doi: 10.1016/j.jcis.2022.08.142
L. Wei, S. Liu, H.P. Wang, ACS Appl. Nano Mater. 5 (2022) 15378–15388. doi: 10.1021/acsanm.2c03420
Y. Zeng, H. Fu, L. Liu, et al., ChemistrySelect 8 (2023) e202204286. doi: 10.1002/slct.202204286
Y. Xu, H. Fu, L. Zhao, et al., New J. Chem. 45 (2021) 3335–3342. doi: 10.1039/d0nj05506a
Y. Gong, Y. Yuan, B. Wang, et al., Mater. Lett. 351 (2023) 135122. doi: 10.1016/j.matlet.2023.135122
W. Wang, X. Li, F. Deng, et al., Chin. Chem. Lett. 33 (2022) 5200–5207. doi: 10.1016/j.cclet.2022.01.058
X. Li, J. Xiong, X. Gao, et al., J. Hazard. Mater. 387 (2020) 121690. doi: 10.1016/j.jhazmat.2019.121690
S. Shen, X. Li, Y. Zhou, et al., J. Mater. Sci. Technol. 155 (2023) 148–159. doi: 10.1016/j.jmst.2023.03.006
H. Zhang, J. Liu, Y. Zhang, et al., J. Mater. Sci. Technol. 166 (2023) 241–249. doi: 10.1016/j.jmst.2023.05.030
X. Zhao, Y. You, S. Huang, et al., Appl. Catal. B: Environ. 278 (2020) 119251. doi: 10.1016/j.apcatb.2020.119251
R. An, Y. Zhao, H. Bai, et al., J. Solid State Chem. 306 (2022) 122722. doi: 10.1016/j.jssc.2021.122722
J. Wang, Y. Gong, M. Gao, et al., ACS Appl. Nano Mater. 7 (2023) 1067–1077.
A. Kudo, K. Omori, H. Kato, J. Am. Chem. Soc. 121 (1999) 11459–11467. doi: 10.1021/ja992541y
Z. Xing, J. Hu, M. Ma, et al., J. Am. Chem. Soc. 141 (2019) 19715–19727. doi: 10.1021/jacs.9b08651
J. Sheng, X. Li, Y. Xu, ACS Catal. 4 (2014) 732–737. doi: 10.1021/cs400927w
Y. Ding, S. Maitra, S. Halder, et al., Matter 5 (2022) 2119–2167. doi: 10.1016/j.matt.2022.05.011
J. Li, H. Li, G. Zhan, L. Zhang, Acc. Chem. Res. 50 (2017) 112–121. doi: 10.1021/acs.accounts.6b00523
H. Li, J. Li, Z. Ai, et al., Angew. Chem. Int. Ed. 57 (2018) 122–138. doi: 10.1002/anie.201705628
L. Ding, H. Chen, Q. Wang, et al., Chem. Commun. 52 (2016) 994–997. doi: 10.1039/C5CC08146G
X. Xiong, L. Ding, Q. Wang, et al., Appl. Catal. B: Environ. 188 (2016) 283–291. doi: 10.1016/j.apcatb.2016.02.018
J. Li, L. Cai, J. Shang, et al., Adv. Mater. 28 (2016) 4059–4064. doi: 10.1002/adma.201600301
Z. Zhao, Y. Zhou, F. Wang, et al., ACS Appl. Mater. Interfaces 7 (2015) 730–737. doi: 10.1021/am507089x
Z. Ni, Y. Sun, Y. Zhang, F. Dong, Appl. Surf. Sci. 365 (2016) 314–335. doi: 10.1016/j.apsusc.2015.12.231
H. Ou, P. Yang, L. Lin, et al., Angew. Chem. Int. Ed. 56 (2017) 10905–10910. doi: 10.1002/anie.201705926
Z. Wei, M. Liu, Z. Zhang, et al., Energy Environ. Sci. 11 (2018) 2581–2589. doi: 10.1039/c8ee01316k
B. Feng, Y. Liu, K. Wan, et al., Angew. Chem. Int. Ed. 63 (2024) e202401884. doi: 10.1002/anie.202401884
Y. Shiraishi, S. Kanazawa, Y. Sugano, et al., ACS Catal. 4 (2014) 774–780. doi: 10.1021/cs401208c
J. Fu, J. Yu, C. Jiang, B. Cheng, Adv. Energy Mater. 8 (2017) 1701503.
R. Du, K. Xiao, B. Li, et al., Chem. Eng. J. 441 (2022) 135999. doi: 10.1016/j.cej.2022.135999
Y. Shiraishi, S. Kanazawa, Y. Kofuji, et al., Angew. Chem. Int. Ed. 53 (2014) 13454–13459. doi: 10.1002/anie.201407938
Y. Kofuji, S. Ohkita, Y. Shiraishi, et al., ACS Catal. 6 (2016) 7021–7029. doi: 10.1021/acscatal.6b02367
Y. Kofuji, S. Ohkita, Y. Shiraishi, et al., ACS Sustain. Chem. Eng. 5 (2017) 6478–6485. doi: 10.1021/acssuschemeng.7b00575
Y. Kofuji, Y. Isobe, Y. Shiraishi, et al., J. Am. Chem. Soc. 138 (2016) 10019–10025. doi: 10.1021/jacs.6b05806
Y. Kofuji, Y. Isobe, Y. Shiraishi, et al., ChemCatChem 10 (2018) 2070–2077. doi: 10.1002/cctc.201701683
X. Zeng, Y. Liu, Y. Kang, et al., ACS Catal. 10 (2020) 3697–3706. doi: 10.1021/acscatal.9b05247
S. Yao, T. Tang, Y. Shen, et al., Sci. China Mater. 66 (2022) 672–678.
H. Che, X. Gao, J. Chen, et al., Angew. Chem. Int. Ed. 60 (2021) 25546–25550. doi: 10.1002/anie.202111769
Y. Peng, L. Wang, Y. Liu, et al., Eur. J. Inorg. Chem. 2017 (2017) 4797–4802. doi: 10.1002/ejic.201700930
P. Ren, T. Zhang, N. Jain, et al., J. Am. Chem. Soc. 145 (2023) 16584–16596. doi: 10.1021/jacs.3c03785
H. Liang, A. Wang, R. Cheng, et al., Small 19 (2023) 2303813. doi: 10.1002/smll.202303813
Y. Ye, C. Wen, J. Pan, et al., Appl. Catal. B: Environ. 285 (2021) 119726. doi: 10.1016/j.apcatb.2020.119726
S. Kim, G. Moon, H. Kim, et al., J. Catal. 357 (2018) 51–58. doi: 10.1016/j.jcat.2017.10.002
P. Sha, L. Huang, J. Zhao, et al., ACS Catal. 13 (2023) 10474–10486. doi: 10.1021/acscatal.3c02118
J. Cai, J. Huang, S. Wang, et al., Adv. Mater. 31 (2019) 1806314. doi: 10.1002/adma.201806314
G. Moon, M. Fujitsuka, S. Kim, et al., ACS Catal. 7 (2017) 2886–2895. doi: 10.1021/acscatal.6b03334
Y. Zhao, Y. Liu, J. Cao, et al., Appl. Catal. B: Environ. 278 (2020) 119289. doi: 10.1016/j.apcatb.2020.119289
Y. Yang, G. Zeng, D. Huang, et al., Appl. Catal. B: Environ. 272 (2020) 118970. doi: 10.1016/j.apcatb.2020.118970
Z. Teng, Q. Zhang, H. Yang, et al., Nat. Catal. 4 (2021) 374–384. doi: 10.1038/s41929-021-00605-1
L. Zhou, J. Feng, B. Qiu, et al., Appl. Catal. B: Environ. 267 (2020) 118396. doi: 10.1016/j.apcatb.2019.118396
P. Zhang, Y. Tong, Y. Liu, et al., Angew. Chem. Int. Ed. 59 (2020) 16209–16217. doi: 10.1002/anie.202006747
L. Chen, C. Chen, Z. Yang, et al., Adv. Funct. Mater. 31 (2021) 2105731. doi: 10.1002/adfm.202105731
W. Liu, P. Wang, J. Chen, et al., Adv. Funct. Mater. 32 (2022) 2205119. doi: 10.1002/adfm.202205119
D. Li, C. Wen, J. Huang, et al., Appl. Catal. B: Environ. 307 (2022) 121099. doi: 10.1016/j.apcatb.2022.121099
S. Wu, H. Yu, S. Chen, X. Quan, ACS Catal. 10 (2020) 14380–14389. doi: 10.1021/acscatal.0c03359
X. Zhang, P. Ma, C. Wang, et al., Energy Environ. Sci. 15 (2022) 830–842. doi: 10.1039/d1ee02369a
Z. Zhu, H. Pan, M. Murugananthan, et al., Appl. Catal. B: Environ. 232 (2018) 19–25. doi: 10.1504/ijipt.2018.10012654
S. Li, G. Dong, R. Hailili, et al., Appl. Catal. B: Environ. 190 (2016) 26–35. doi: 10.1016/j.apcatb.2016.03.004
Y. Shiraishi, Y. Kofuji, H. Sakamoto, et al., ACS Catal. 5 (2015) 3058–3066. doi: 10.1021/acscatal.5b00408
L. Shi, L. Yang, W. Zhou, et al., Small 14 (2018) 1703142. doi: 10.1002/smll.201703142
X. Li, J. Zhang, F. Zhou, et al., Chin. J. Catal. 39 (2018) 1090–1098. doi: 10.1016/S1872-2067(18)63046-3
Q. He, J. Ding, H. Tsai, et al., J. Colloid. Interf. Sci. 651 (2023) 18–26. doi: 10.1016/j.jcis.2023.07.168
Y. Xie, Y. Li, Z. Huang, et al., Appl. Catal. B: Environ. 265 (2020) 118581. doi: 10.1016/j.apcatb.2019.118581
J. Chen, W. Gao, Y. Lu, et al., ACS Appl. Nano Mater. 6 (2023) 3927–3935. doi: 10.1021/acsanm.3c00092
Y. Wang, D. Meng, X. Zhao, Appl. Catal. B: Environ. 273 (2020) 119064. doi: 10.1016/j.apcatb.2020.119064
H. Fattahimoghaddam, T. Mahvelati-Shamsabadi, B. Lee, ACS Sustain. Chem. Eng. 9 (2021) 4520–4530. doi: 10.1021/acssuschemeng.0c08884
Y. Zheng, Y. Luo, Q. Ruan, et al., Appl. Catal. B: Environ. 311 (2022) 121372. doi: 10.1016/j.apcatb.2022.121372
J. Yang, Z. Ji, S. Zhang, ACS Appl. Energy Mater. 6 (2023) 3401–3412. doi: 10.1021/acsaem.2c04124
J. Lei, B. Chen, W. Lv, et al., ACS Sustain. Chem. Eng. 7 (2019) 16467–16473. doi: 10.1021/acssuschemeng.9b03678
H. Xie, Y. Zheng, X. Guo, et al., ACS Sustain. Chem. Eng. 9 (2021) 6788–6798. doi: 10.1021/acssuschemeng.1c01012
Q. Li, Y. Jiao, Y. Tang, et al., J. Am. Chem. Soc. 145 (2023) 20837–20848. doi: 10.1021/jacs.3c05234
Y. Zhu, X. Liu, H. Liu, et al., SusMat 2 (2022) 617–629. doi: 10.1002/sus2.83
R. Li, Q. Deng, A. Chen, et al., ChemSusChem 16 (2023) e202300763. doi: 10.1002/cssc.202300763
L. Yang, G. Dong, D. Jacobs, et al., J. Catal. 352 (2017) 274–281. doi: 10.1016/j.jcat.2017.05.010
X. Zhang, J. Yu, W. Macyk, et al., Adv. Sustain. Syst. 7 (2022) 2200113.
X. Li, J. Liu, J. Huang, et al., Acta Phys. Chim. Sin. 37 (2020) 2010030. doi: 10.3866/pku.whxb202010030
B. Li, Z. Guo, Y. Feng, et al., ACS Appl. Mater. Interfaces 14 (2022) 43328–43338. doi: 10.1021/acsami.2c12038
X. Wang, Z. Han, L. Yu, et al., ACS Sustain. Chem. Eng. 6 (2018) 14542–14553. doi: 10.1021/acssuschemeng.8b03171
W. Zhang, S. Zhao, Y. Xing, et al., Chem. Eng. J. 442 (2022) 136151. doi: 10.1016/j.cej.2022.136151
K. Das, S. Mansingh, R. Mohanty, et al., J. Phys. Chem. C 127 (2022) 22–40.
P. Zhou, M. Luo, S. Guo, Nat. Rev. Chem. 6 (2022) 823–838. doi: 10.1038/s41570-022-00434-1
P. Zhou, H. Chen, Y. Chao, et al., Nat. Commun. 12 (2021) 4412. doi: 10.1038/s41467-021-24702-8
Z. Teng, W. Cai, W. Sim, et al., Appl. Catal. B: Environ. 282 (2021) 119589. doi: 10.1016/j.apcatb.2020.119589
G. Chakraborty, I. Park, R. Medishetty, J. Vittal, Chem. Rev. 121 (2021) 3751–3891. doi: 10.1021/acs.chemrev.0c01049
J. Choi, B. Check, X. Fang, et al., J. Am. Chem. Soc. 146 (2024) 11319–11327.
M. Alvaro, E. Carbonell, B. Ferrer, et al., Chem. Eur. J. 13 (2007) 5106–5112. doi: 10.1002/chem.200601003
S. Rojas, P. Horcajada, Chem. Rev. 120 (2020) 8378–8415. doi: 10.1021/acs.chemrev.9b00797
S. Navalon, A. Dhakshinamoorthy, M. Alvaro, et al., Chem. Rev. 123 (2023) 445–490. doi: 10.1021/acs.chemrev.2c00460
K. Sun, Y. Qian, H. Jiang, Angew. Chem. Int. Ed. 62 (2023) e202217565. doi: 10.1002/anie.202217565
W. Huang, M. Chai, R. Lin, et al., Ind. Eng. Chem. Res. 62 (2023) 14130–14143. doi: 10.1021/acs.iecr.3c02177
Y. Fu, D. Sun, Y. Chen, et al., Angew. Chem. Int. Ed. 51 (2012) 3364–3367. doi: 10.1002/anie.201108357
M. Wen, K. Mori, Y. Kuwahara, H. Yamashita, ACS Energy Lett. 2 (2016) 1–7.
Y. Horiuchi, T. Toyao, M. Saito, et al., J. Phys. Chem. C 116 (2012) 20848–20853. doi: 10.1021/jp3046005
C. Hendon, D. Tiana, M. Fontecave, et al., J. Am. Chem. Soc. 135 (2013) 10942–10945. doi: 10.1021/ja405350u
T. Wang, M. Chen, Y. Lai, et al., ACS Sustain. Chem. Eng. 11 (2023) 6465–6473. doi: 10.1021/acssuschemeng.3c00742
Q. Lan, S. Jin, B. Yang, et al., Trans. Tianjin Univ. 28 (2022) 214–225. doi: 10.1007/s12209-022-00324-z
Y. Isaka, Y. Kondo, Y. Kawase, et al., Chem. Commun. 54 (2018) 9270–9273. doi: 10.1039/c8cc02679c
K. Yue, X. Zhang, S. Jiang, et al., J. Mol. Liq. 335 (2021) 116108. doi: 10.1016/j.molliq.2021.116108
X. Chen, Y. Kondo, Y. Kuwahara, et al., Phys. Chem. Chem. Phys. 22 (2020) 14404–14414. doi: 10.1039/d0cp01759k
Y. Kondo, Y. Kuwahara, K. Mori, H. Yamashita, Chem 8 (2022) 2924–2938. doi: 10.1016/j.chempr.2022.10.007
Y. Isaka, Y. Kawase, Y. Kuwahara, et al., Angew. Chem. Int. Ed. 58 (2019) 5402–5406. doi: 10.1002/anie.201901961
Y. Kawase, Y. Isaka, Y. Kuwahara, et al., Chem. Commun. 55 (2019) 6743–6746. doi: 10.1039/c9cc02380a
L. Yuan, Y. Zou, L. Zhao, et al., Appl. Catal. B: Environ. 318 (2022) 121859. doi: 10.1016/j.apcatb.2022.121859
Y. Li, Y. Liu, Z. Wang, et al., Chin. J. Catal. 45 (2023) 132–140. doi: 10.3390/cryst13010132
X. Chen, Y. Kuwahara, K. Mori, et al., J. Mater. Chem. A 9 (2021) 2815–2821. doi: 10.1039/d0ta10944d
X. Chen, Y. Kondo, S. Li, et al., J. Mater. Chem. A 9 (2021) 26371–26380. doi: 10.1039/d1ta08036a
Y. Hao, L. Chen, J. Li, et al., Nat. Commun. 12 (2021) 2682. doi: 10.1038/s41467-021-22991-7
Y. Li, Y. Guo, D. Luan, et al., Angew. Chem. Int. Ed. 62 (2023) e202310847. doi: 10.1002/anie.202310847
X. Chen, Y. Kuwahara, K. Mori, et al., J. Mater. Chem. A 8 (2020) 1904–1910. doi: 10.1039/C9TA11120D
X. Chen, Y. Kuwahara, K. Mori, et al., ACS Appl. Energy Mater. 4 (2021) 4823–4830. doi: 10.1021/acsaem.1c00371
Y. Li, F. Ma, L. Zheng, et al., Mater. Horiz. 8 (2021) 2842–2850. doi: 10.1039/d1mh00869b
B. Yan, S. Hu, C. Bu, et al., ChemNanoMat 9 (2023) e202300079. doi: 10.1002/cnma.202300079
Y. Kondo, Y. Kuwahara, K. Mori, H. Yamashita, J. Phys. Chem. C 125 (2021) 27909–27918. doi: 10.1021/acs.jpcc.1c07735
Y. Kondo, K. Honda, Y. Kuwahara, et al., ACS Catal. 12 (2022) 14825–14835. doi: 10.1021/acscatal.2c04940
C. Liu, T. Bao, L. Yuan, et al., Adv. Funct. Mater. 32 (2022) 2111404. doi: 10.1002/adfm.202111404
R. Bariki, S. Pradhan, S. Panda, et al., Langmuir 39 (2023) 7707–7722. doi: 10.1021/acs.langmuir.3c00519
Y. Wu, X. Li, Q. Yang, et al., Chem. Eng. J. 390 (2020) 124519. doi: 10.1016/j.cej.2020.124519
A. Côté, A. Benin, N. Ockwig, et al., Science 310 (2005) 1166–1170. doi: 10.1126/science.1120411
S. Wang, Z. Xie, D. Zhu, et al., Nat. Commun. 14 (2023) 6891. doi: 10.1038/s41467-023-42720-6
P. Zhou, Y. Li, T. Zeng, et al., Angew. Chem. Int. Ed. 63 (2024) e202402911. doi: 10.1002/anie.202402911
S. Qi, R. Guo, Z. Bi, et al., Small 19 (2023) 2303632. doi: 10.1002/smll.202303632
Z. Li, J. Wang, S. Ma, et al., Appl. Catal. B: Environ. 310 (2022) 121335. doi: 10.1016/j.apcatb.2022.121335
L. Stegbauer, K. Schwinghammer, B. Lotsch, Chem. Sci. 5 (2014) 2789–2793. doi: 10.1039/C4SC00016A
H. Chen, H. Jena, X. Feng, et al., Angew. Chem. Int. Ed. 61 (2022) e202204938. doi: 10.1002/anie.202204938
R. Freund, O. Zaremba, G. Arnauts, et al., Angew. Chem. Int. Ed. 60 (2021) 23975–24001. doi: 10.1002/anie.202106259
T. Banerjee, F. Podjaski, J. Kröger, et al., Nat. Rev. Mater. 6 (2020) 168–190. doi: 10.1038/s41578-020-00254-z
K. Geng, T. He, R. Liu, et al., Chem. Rev. 120 (2020) 8814–8933. doi: 10.1021/acs.chemrev.9b00550
L. Chen, L. Wang, Y. Wan, et al., Adv. Mater. 32 (2020) 1904433. doi: 10.1002/adma.201904433
C. Krishnaraj, H. Jena, L. Bourda, et al., J. Am. Chem. Soc. 142 (2020) 20107–20116. doi: 10.1021/jacs.0c09684
D. Tan, R. Zhuang, R. Chen, et al., Adv. Funct. Mater. 34 (2024) 2311655. doi: 10.1002/adfm.202311655
Y. Cong, X. Li, S. Zhang, et al., ACS Appl. Mater. Interfaces 15 (2023) 43799–43809. doi: 10.1021/acsami.3c09039
M. Kou, Y. Wang, Y. Xu, et al., Angew. Chem. Int. Ed. 61 (2022) e202200413. doi: 10.1002/anie.202200413
Q. Zhi, W. Liu, R. Jiang, et al., J. Am. Chem. Soc. 144 (2022) 21328–21336. doi: 10.1021/jacs.2c09482
W. Zhao, P. Yan, B. Li, et al., J. Am. Chem. Soc. 144 (2022) 9902–9909. doi: 10.1021/jacs.2c02666
G. Pan, X. Hou, Z. Liu, et al., ACS Catal. 12 (2022) 14911–14917. doi: 10.1021/acscatal.2c03878
J. Yue, L. Song, Y. Fan, et al., Angew. Chem. Int. Ed. 62 (2023) e202309624. doi: 10.1002/anie.202309624
Q. Liao, Q. Sun, H. Xu, et al., Angew. Chem. Int. Ed. 62 (2023) e202310556. doi: 10.1002/anie.202310556
C. Wu, Z. Teng, C. Yang, et al., Adv. Mater. 34 (2022) 2110266. doi: 10.1002/adma.202110266
Y. Luo, B. Zhang, C. Liu, et al., Angew. Chem. Int. Ed. 62 (2023) e202305355. doi: 10.1002/anie.202305355
X. Di, X. Lv, H. Wang, et al., Chem. Eng. J. 455 (2023) 140124. doi: 10.1016/j.cej.2022.140124
P. Das, G. Chakraborty, J. Roeser, et al., J. Am. Chem. Soc. 145 (2023) 2975–2984. doi: 10.1021/jacs.2c11454
P. Das, J. Roeser, A. Thomas, Angew. Chem. Int. Ed. 62 (2023) e202304349. doi: 10.1002/anie.202304349
P. Gao, C. Wu, S. Wang, et al., J. Colloid. Interf. Sci. 650 (2023) 40–46. doi: 10.1016/j.jcis.2023.06.186
H. Hu, Y. Tao, D. Wang, et al., J. Colloid. Interf. Sci. 629 (2023) 750–762. doi: 10.1016/j.jcis.2022.09.111
C. Shao, Q. He, M. Zhang, et al., Chin. J. Catal. 46 (2023) 28–35. doi: 10.1016/S1872-2067(22)64205-0
L. Li, L. Xu, Z. Hu, J. Yu, Adv. Funct. Mater. 31 (2021) 2106120. doi: 10.1002/adfm.202106120
Q. Shang, Y. Liu, J. Ai, et al., J. Mater. Chem. A 11 (2023) 21109–21122. doi: 10.1039/d3ta03966h
S. Zhou, H. Hu, H. Hu, et al., Sci. China Mater. 66 (2023) 1837–1846. doi: 10.1007/s40843-022-2337-7
Y. Yang, J. Kang, Y. Li, et al., New J. Chem. 46 (2022) 21605–21614. doi: 10.1039/d2nj03744k
M. Deng, J. Sun, A. Laemont, et al., Green Chem. 25 (2023) 3069–3076. doi: 10.1039/d2gc04459e
Y. Liu, W. Han, W. Chi, et al., Appl. Catal. B: Environ. 331 (2023) 122691. doi: 10.1016/j.apcatb.2023.122691
J. Chu, Z. Liu, T. Yang, A. Kong, Appl. Surf. Sci. 611 (2023) 155717. doi: 10.1016/j.apsusc.2022.155717
T. Yang, Y. Wang, Y. Chen, et al., CrystEngComm 25 (2023) 4511–4520. doi: 10.1039/d3ce00607g
J. Sun, H. Jena, C. Krishnaraj, et al., Angew. Chem. Int. Ed. 62 (2023) e202216719. doi: 10.1002/anie.202216719
L. Wang, J. Sun, M. Deng, et al., Catal. Sci. Technol. 13 (2023) 6463–6471. doi: 10.1039/D3CY01175E
T. Yang, Y. Chen, Y. Wang, et al., ACS Appl. Mater. Interfaces 15 (2023) 8066–8075. doi: 10.1021/acsami.2c20506
F. Hao, C. Yang, X. Lv, et al., Angew. Chem. Int. Ed. 62 (2023) e202315456. doi: 10.1002/anie.202315456
Z. Zhou, M. Sun, Y. Zhu, et al., App. Catal. B: Environ. 334 (2023) 122862. doi: 10.1016/j.apcatb.2023.122862
X. Yu, B. Viengkeo, Q. He, et al., Adv. Sustain. Syst. 5 (2021) 2100184. doi: 10.1002/adsu.202100184
H. Wang, C. Yang, F. Chen, et al., Angew. Chem. Int. Ed. 61 (2022) e202202328. doi: 10.1002/anie.202202328
Y. Liu, L. Li, H. Tan, et al., J. Am. Chem. Soc. 145 (2023) 19877–19884. doi: 10.1021/jacs.3c05914
G. Xia, J. Qiu, L. Zhang, et al., Colloid Surf. A: Physicochem. Eng. Asp. 664 (2023) 131124. doi: 10.1016/j.colsurfa.2023.131124
Y. Yang, J. Liu, M. Gu, et al., Appl. Catal. B: Environ. 333 (2023) 122780. doi: 10.1016/j.apcatb.2023.122780
Y. Zhang, J. Qiu, B. Zhu, et al., Chem. Eng. J. 444 (2022) 136584. doi: 10.1016/j.cej.2022.136584
Y. Yang, Y. Li, X. Ma, et al., Catal. Sci. Technol. 13 (2023) 5599–5609. doi: 10.1039/d3cy00878a
H. Chen, S. Gao, G. Huang, et al., Appl. Catal. B: Environ. 343 (2024) 123545. doi: 10.1016/j.apcatb.2023.123545
L. Zhai, Z. Xie, C. Cui, et al., Chem. Mater. 34 (2022) 5232–5240. doi: 10.1021/acs.chemmater.2c00910
S. Chai, X. Chen, X. Zhang, et al., Environ. Sci. 9 (2022) 2464–2469 Nano. doi: 10.1039/d2en00135g
X. Zhang, J. Zhang, J. Miao, et al., Chem. Eng. J. 466 (2023) 143085. doi: 10.1016/j.cej.2023.143085
J. Chang, J. Shi, Q. Li, et al., Angew. Chem. Int. Ed. 62 (2023) e202303606. doi: 10.1002/anie.202303606
R. Liu, Y. Chen, H. Yu, et al., Nat. Catal. 7 (2024) 195–206. doi: 10.1038/s41929-023-01102-3
C. Shu, X. Yang, L. Liu, et al., Angew. Chem. Int. Ed. 63 (2024) e202403926. doi: 10.1002/anie.202403926
H. Li, H. Li, S. Xun, J. Brédas, Chem. Mater. 32 (2020) 9228–9237. doi: 10.1021/acs.chemmater.0c02913
Z. Lan, M. Wu, Z. Fang, et al., Angew. Chem. Int. Ed. 60 (2021) 16355–16359. doi: 10.1002/anie.202103992
Z. Luo, X. Chen, Y. Hu, et al., Angew. Chem. Int. Ed. 62 (2023) e202304875. doi: 10.1002/anie.202304875
Y. Qian, Y. Han, X. Zhang, et al., Nat. Commun. 14 (2023) 3083. doi: 10.1038/s41467-023-38884-w
Y. Wan, L. Wang, H. Xu, et al., J. Am. Chem. Soc. 142 (2020) 4508–4516. doi: 10.1021/jacs.0c00564
X. Wang, X. Yang, C. Zhao, et al., Angew. Chem. Int. Ed. 62 (2023) e202302829. doi: 10.1002/anie.202302829
L. Liu, F. Chen, J. Wu, et al., Proc. Natl. Acad. Sci. U. S. A. 120 (2023) e2215305120. doi: 10.1073/pnas.2215305120
A. Gopakumar, P. Ren, J. Chen, et al., J. Am. Chem. Soc. 144 (2022) 2603–2613. doi: 10.1021/jacs.1c10786
Y. Zheng, Z. Yu, H. Ou, et al., Adv. Funct. Mater. 28 (2018) 1705407. doi: 10.1002/adfm.201705407
B. Sheng, Y. Xie, Q. Zhao, et al., Energy Environ. Sci. 16 (2023) 4612–4619. doi: 10.1039/d3ee02200e
T. Freese, J. Meijer, B. Feringa, S. Beil, Nat. Catal. 6 (2023) 553–558. doi: 10.1038/s41929-023-00980-x
H. Tan, P. Zhou, M. Liu, et al., Nat. Synth. 2 (2023) 557–563. doi: 10.1038/s44160-023-00272-z
C. Zhao, X. Wang, Y. Yin, et al., Angew. Chem. Int. Ed. 62 (2023) e202218318. doi: 10.1002/anie.202218318
L. Wang, L. Liu, Y. Li, et al., Adv. Energy Mater. 14 (2023) 2303346.
R. Yang, Y. Fan, J. Hu, et al., Chem. Soc. Rev. 52 (2023) 7687–7706. doi: 10.1039/d2cs00205a
Z. Wang, C. Li, K. Domen, Chem. Soc. Rev. 48 (2019) 2109–2125. doi: 10.1039/C8CS00542G
S. Cao, L. Piao, Angew. Chem. Int. Ed. 59 (2020) 18312–18320. doi: 10.1002/anie.202009633
L. Liu, M. Gao, H. Yang, et al., J. Am. Chem. Soc. 143 (2021) 19287–19293. doi: 10.1021/jacs.1c09979
Q. Wang, X. Kong, Y. Wang, et al., ChemSusChem 15 (2022) e202201514. doi: 10.1002/cssc.202201514
H. Zhao, Q. Jin, M. Khan, et al., Chem. Catal. 2 (2022) 1720–1733. doi: 10.1016/j.checat.2022.04.015
Y. Li, S. Ouyang, H. Xu, et al., J. Am. Chem. Soc. 138 (2016) 13289–13297. doi: 10.1021/jacs.6b07272
Q. Lian, Z. Liang, X. Guan, et al., Appl. Catal. B: Environ. 331 (2023) 122724. doi: 10.1016/j.apcatb.2023.122724
S. Zhou, R. He, J. Pei, et al., Environ. Sci. Technol. 56 (2022) 10474–10482. doi: 10.1021/acs.est.2c02067
Z. Chu, J. Yang, W. Zheng, et al., Coord. Chem. Rev. 481 (2023) 215049. doi: 10.1016/j.ccr.2023.215049

Figure 1 Schematic illustration of (a) photocatalytic processes for H2O2 production and (b) corresponding energy diagrams. (c) Reaction pathways and corresponding energy diagrams for ORR and WOR pathways.

Figure 2 (a) Photocatalytic H2O2 production over HT-CN/TiO2 featuring the Z-scheme charge transfer mechanism. Reproduced with permission [64]. Copyright 2019, Elsevier. (b) The photocatalytic production of H2O2 pathway over In2S3@In2O3 and In2S3@Ov/In2O3 composites. Reproduced with permission [86]. Copyright 2021, American Chemical Society. (c) Schematic diagram of the reaction mechanism over Au/WO3. Reproduced with permission [69]. Copyright 2021, Elsevier. (d) Mechanism of Gd3+ doped WO3 photocatalysts for photocatalytic H2O2 production. Reproduced with permission [76]. Copyright 2023, Elsevier.

Figure 3 (a) Mn2+ doping and Au0 modification on MoS2 nanosheets, and in-situ-Fenton process using photogenerated H2O2. Reproduced with permission [94]. Copyright 2019, Elsevier. (b) Mechanism of Sv−MoS2/BN heterojunction toward sacrificial agent-free photocatalytic H2O2 production for in situ water disinfection. Reproduced with permission [96]. Copyright 2024, Elsevier. (c) Schematic diagram of H2O2 production over CBO@MoS2 Z-scheme heterojunction Reproduced with permission [97]. Copyright 2023, Royal Society of Chemistry. (d). Mechanism of CDs modified SnS2/In2S3 type Ⅱ heterostructure for photocatalytic H2O2 production by indirect two-step e– ORR and 4e– WOR. Reproduced with permission [99]. Copyright 2021, Royal Society of Chemistry.

Figure 4 (a) Schematic diagram of efficient photocatalytic H2O2 production by Cu@Au/BiVO4. Reproduced with permission [119]. Copyright 2021, American Chemical Society. (b) Schematic illustration for the H2O2 production mechanism of Y-doped BiVO4 photocatalyst. Reproduced with permission [124]. Copyright 2023, John Wiley and Sons. (c) H2O2 production on defective BiVO4 via the direct one-step 2e– ORR pathway. Reproduced with permission [125]. Copyright 2023, Elsevier. (d) Energy diagram for NiCo2O4/BiVO4 heterojunction depicting the production of H2O2 and degradation of organic pollutants, simultaneously. Reproduced with permission [126]. Copyright 2022, American Chemical Society.

Figure 5 (a) Mechanism of PDI-modified g-C3N4 for photocatalytic H2O2 production from H2O and O2. Reproduced with permission [154]. Copyright 2014, John Wiley and Sons. (b) The different adsorption modes of O2 adsorption on metal surfaces and the mechanism of photocatalytic H2O2 production. Reproduced with permission [172]. Copyright 2021, Springer Nature. (c) Mechanism of an aryl amino-substituted g-C3N4 photocatalyst with atomically dispersed Mn for photocatalytic H2O2 production from seawater. Reproduced with permission [163]. Copyright 2023, American Chemical Society. (d) Mechanism of heteroatom doped g-C3N4 (AKMT) for photocatalytic H2O2 production. Reproduced with permission [174]. Copyright 2020, John Wiley and Sons. (e) The presence of C-vacancies changed the photocatalytic H2O2 production from the indirect two-step e– ORR pathway to the direct one-step 2e– ORR pathway. Reproduced with permission [181]. Copyright 2016, Elsevier. (f) Mechanism of PIx-NCN heterojunction for photocatalytic H2O2 production. Reproduced with permission [197]. Copyright 2017, Elsevier. (g) Mechanism of heptazine/triazine layer stacked g-C3N4 heterojunction with F/K dual sites modified for photocatalytic H2O2 production. Reproduced with permission [200]. Copyright 2022, American Chemical Society.

Figure 6 (a) Proposed reaction mechanism for the generation of H2O2 over Ce-doped MIL-125-NH2. Reproduced with permission [235]. Copyright 2023, John Wiley and Sons. (b) The proposed mechanism of H2O2 production over alkylated MIL-125-R7 in a two-phase system. Reproduced with permission [224]. Copyright 2019, John Wiley and Sons. (c) Reaction mechanism of H2O2 production over missing-linker defective UiO-66-NH2-X photocatalyst. Reproduced with permission [236]. Copyright 2021, American Chemical Society. (d) The relationship between the yield of H2O2 production and heterojunction coverage, as well as the proposed mechanism over NH2−MIL-125@ZnS. Reproduced with permission [238]. Copyright 2022, John Wiley and Sons.

Figure 7 (a) Reaction mechanism of photocatalytic H2O2 production over sulfone functional group modified COF (FS-COFs). Reproduced with permission [262]. Copyright 2023, John Wiley and Sons. (b) Proposed reaction mechanism for the generation of H2O2 over fluorine-doped COF (TF50–COF). Reproduced with permission [283]. Copyright 2022, John Wiley and Sons. (c) Schematic illustration of photocatalytic reaction pathways toward H2O2 production over Z-scheme WO3/COF heterojunction. Reproduced with permission [288]. Copyright 2023, Royal Society of Chemistry. (d) The schematic diagram of photocatalytic H2O2 production over CHF-DPA and CHF-DPDA with separated redox centres. Reproduced with permission [46]. Copyright 2022, John Wiley and Sons.

 扫一扫看文章
扫一扫看文章

扫一扫关注我们
 DownLoad:
DownLoad:


 下载:
下载:
 下载:
下载: